针对测序平台特征的碱基质量值矫正方法、装置、电子设备和存储介质
文献发布时间:2023-06-19 18:32:25
技术领域
本申请属于基因测序技术领域,具体涉及针对测序平台特征的碱基质量值矫正方法、装置、电子设备和存储介质。
背景技术
随着高通量测序技术(NGS)普遍应用于肿瘤的伴随诊断领域,基于液体活检(cfDNA)的早筛和微小残留病灶(MRD)检测也得到越来越多的关注和认可。虽然随着NGS测序平台的不断发展,提供了更高的通量和测序准确性,但是对于识别来自肿瘤组织的DNA(ctDNA),测序数据的质量仍是一个关键的技术瓶颈。例如,识别cfDNA样本中的体细胞突变,往往需要达到低于0.1%的水平,这一检测要求低于目前测序平台普遍的错误率。如何有效降低测序错误,识别真实的低频变异,是提高检测产品性能的关键。
NGS测序仪产出测序读长(reads)普遍的格式为fastq,其中每条read由四行组成,reads的长度由测序模式决定,每个位置都包含碱基序列(ATCGN)信息和其对应的碱基质量值(quality score,Q-score)信息。碱基质量值是碱基识别出错概率的整数映射,Q=-10×lg(P),其中P为碱基识别出错的概率。碱基质量值代表测序仪对于碱基序列信号的可信度评估,是用来评估测序错误的直接指标。例如碱基质量值Q20,代表该碱基是正确测序结果的可信度达到99%。然而,测序仪产生的碱基质量值受各种因素的影响,其中系统性的偏好,往往会导致高估或低估数据中的碱基质量值,从而产生批次效应。因此,为了消除不同平台和批次的测序错误偏好,提高检测技术和方法的适用性,对测序数据的碱基质量值进行矫正尤为重要。
目前,碱基质量值的矫正方式普遍采用GATK-BQSR,其原理是将物种突变数据库以外的与参考基因组错配的碱基都作为测序错误,对于尚未有完善群体突变数据库的物种,该方法无法进行质量值矫正。此外,GATK-BQSR的测序错误包含了取样和建库等测序前引入的错配、生物学体细胞突变以及真正的测序错误。对于当前追求高灵敏度和准确性的疾病诊断的液体活检所运用的cfDNA数据,样本特异性的体细胞变异和克隆性造血产生的突变也是不容忽视的存在,GATK-BQSR质量值矫正方式不能区分这些样本特异性的生物学变异和真正的测序错误(即碱基识别错误),会对矫正结果产生影响,尤其对于高TMB的肿瘤样本可能会产生非常严重的偏差,从而对数据检测的灵敏度和准确度产生较大影响。GATK-BQSR在样本水平的建模并矫正,对于高深度大数据量的数据需要消耗较多的时间和计算资源,会延长产品数据分析周期。
发明内容
1. 要解决的问题
本申请针对上述采用GATK-BQSR进行碱基质量值矫正中存在的问题之一,提供一种针对测序平台特征的碱基质量值矫正方法、装置、电子设备和存储介质,该方法以原始双端测序数据为基础,提取read1和read2的重叠碱基,利用重叠碱基区分测序错误碱基和非测序错误碱基,并根据测序错误碱基所在的read朝向(read1 or read2)、测序循环数(cycle)、测序方向的dinucleotide及测序仪给定的碱基质量值(report quality score,RQS),将提取的重叠区域的碱基划分成不同的bins,统计各特征bins下测序错误碱基,并计算经验质量值(empricial quality score,EQS),采用局部加权回归模型(locallyweighted regression,lowess)对特征bins内的RQS和EQS进行多项式拟合建模,并利用建立的模型对原始碱基质量值进行矫正。本申请能够区分测序错误来源碱基和非测序错误来源碱基,能够更准确的反映测序仪偏好,在此基础上评估测序仪给定碱基质量值的准确性并且建模矫正,能够全面提高碱基质量值的可信度。
2. 技术方案
为了解决上述问题,本申请所采用的技术方案如下:
作为本申请的第一方面,本申请提供了一种针对测序平台特征的碱基质量值矫正方法,该方法包括如下步骤:
S1:将原始双端测序数据预处理后比对至参考基因组生成Bam文件,预处理包括去除接头序列和低质量序列;
S2:以S1中生成的Bam文件为输入文件,构建参考基因组一致性序列并进行reads水平过滤,但不对read1和read2重叠部分的碱基依据碱基质量值过滤,即保留read1和read2重叠部分的碱基;
S3:从S2中过滤后的Bam文件中提取reads对(即read1和read2)的所有重叠碱基,即比对到参考基因组同一个位置的分别位于read1和read2的两个碱基,将重叠碱基不配对且其中与参考基因组一致性序列碱基不匹配的碱基标记为测序错误碱基,即测序仪识别出错的碱基;将重叠碱基配对且都与参考基因组一致性序列碱基不匹配的碱基标记为非测序错误碱基,即非测序仪识别出错的碱基,可能源于取样和建库等测序前引入的错配、生物学体细胞突变等;提取重叠区域碱基的测序特征,包括read朝向(即位于read1或read2)、测序循环数(cycle)、与测序扩增方向的前一个碱基组成的二核苷酸(dinucleotide)及仪器报告的碱基质量值(report quality score,RQS);
S4:根据S3中提取的碱基的测序特征,将提取的重叠碱基划分到不同的分组形成特征bins,即将测序特征相同的碱基划分至一个小组生成一个特征bin;若特征bin内碱基数目小于30000,则以步长为1沿cycle上下滑窗,直到特征bin内碱基数目大于等于30000;碱基质量值Q是碱基识别出错概率的整数映射,即Q=-10×lg(P),其中P为碱基识别出错的概率,当Q=40时,P=0.01%,因此至少统计10000个碱基以上,才有可能比较准确计算Q=40的碱基,本申请中设置30000,以便有充足的数据,使得计算的经验质量值更准确;
S5:统计S4中各特征bins中测序错误碱基和总碱基数目,根据式(1)计算各bin的经验质量值(empricial quality score,EQS):
其中,empricial quality代表bin的经验质量值(empricial quality score,EQS),mismatches代表bin的内测序错误碱基数目,bases代表bin内所有碱基数目;
S6:采用局部加权回归模型(locally weighted regression,lowess)对仪器报告的碱基质量值(report quality score,RQS)和经验质量值(empricial quality score,EQS)进行多项式拟合建模,基于该模型对仪器报告的碱基质量值进行矫正,获得矫正后的碱基质量值。
进一步地,上述S1中,原始双端测序数据为FASTQ文件的测序数据。
进一步地,上述S1中,去除低质量序列,包括:去除长度小于51 bp的reads;以窗口大小为1个碱基从read前到后进行滑窗,如果窗口内平均碱基质量值≤1,则过滤掉窗口右边的碱基。
进一步地,上述S1中,去除接头序列和低质量序列,利用去接头软件Trimmomatic去除,得到预处理后的FASTQ文件。
进一步地,上述S1中,比对至参考基因组生成Bam文件,包括将预处理后的FASTQ文件比对到hg19人类参考基因组序列并生成Bam文件。
进一步地,上述S1中,比对至参考基因组生成Bam文件,利用基因组比对工具BWAmem-M完成。
进一步地,上述S2中,构建参考基因组一致性序列即将突变频率≥99%的碱基作为该位点的真实碱基类型。
进一步地,上述S2中,reads水平过滤包括:过滤掉只有一条read比对到参考基因组的reads对、没有比对到参考基因组的reads对、非主要比对的reads、平台/供应商质量检查失败的reads和嵌和比对但不是主要代表性比对的reads。
进一步地,上述S6中,局部加权回归模型的构建是采用python的sm.nonparametric模块的lowess方法建立,参数frac设置为0.1。
作为本申请的第二方面,本申请提供了一种针对测序平台特征的碱基质量值矫正装置,包括:
数据输入模块:用于输入原始双端测序数据,如原始的fastq测序数据;
去接头模块(trim模块):用于去除原始双端测序数据中接头序列,防止接头序列对后续提取碱基,区分碱基错误来源造成影响;本模块中可包含商业软件,如fastp软件,对输入的原始双端测序数据去除接头;
比对模块(mapping模块):用于将输入的去接头的测序数据比对到参考基因组,生成比对到参考基因组的Bam文件;本模块中可包含商业软件,如samtools view,去接头的测序数据比对到参考基因组;
过滤模块(filtering模块):用于对输入的生成比对到参考基因组的Bam文件进行构建参考基因组一致性序列并进行reads水平过滤,进行构建参考基因组一致性序列,即将突变频率大于等于99%的碱基作为该位点的真实碱基类型;reads水平过滤包括过滤掉只有一条read比对到参考基因组的reads对、没有比对到参考基因组的reads对、非主要比对的reads、平台/供应商质量检查失败的reads和嵌和比对但不是主要代表性比对的reads,但不对read1和read2重叠部分的碱基依据碱基质量值过滤,即保留read1和read2重叠部分的碱基;本模块中可包含商业软件,如samtools软件和python的pysam模块,将比对模块(mapping模块)输出的比对后文件进行过滤,过滤掉只有一条read比对上的reads对(SAM标记是4或者8),过滤掉非主要比对的reads(not primary alignment,supplementaryalignment,SAM标记为256和2048),过滤掉平台或者供应商质量检查失败的reads(SAM 标记512);
重叠碱基提取和区分错误来源模块(overlapping模块):用于从过滤后的Bam文件中提取出read1和read2所有重叠的碱基,按照在相同的参考基因组一致性序列上的坐标位置,read1的碱基与read2的碱基是否互补配对,与参考基因组一致性序列碱基是否匹配区分错误来源,将read1的碱基与read2的碱基不配对,且其中与参考基因组一致性序列碱基不匹配的碱基标记为来源于测序错误的碱基;将read1的碱基与read2的碱基配对,且都与参考基因组一致性序列碱基不匹配的碱基标记为来源于非测序错误的碱基;本模块中可包含商业软件,如python的pysam模块,用于重叠碱基提取和区分错误来源;
错误率统计模块(feature_bin模块):用于提取重叠碱基的特征,包括read朝向(即位于read1或read2)、测序循环数(cycle)、与测序扩增方向的前一个碱基组成的二核苷酸(dinucleotide)及仪器报告的碱基质量值(report quality score,RQS),并按照特征将碱基分组(bin)统计,统计各bin中的碱基错误率;基于bin内测序错误碱基照式(1)计算经验质量值(empricial quality score,EQS);
其中,empricial quality代表bin的经验质量值(empricial quality score,EQS),mismatches代表bin的内测序错误碱基数目,bases代表bin内所有碱基数目;
矫正模型建模模块(modeling模块):用于根据错误率统计模块(feature_bin模块)计算的经验质量值,使用局部加权回归(locally weighted regression,lowess)对不同特征下仪器报告的碱基质量值(RQS)和经验质量值(EQS)进行拟合建模;
矫正模块(correction模块):用于根据矫正模型建模模块(modeling模块)建立的模型对仪器报告的碱基质量值进行矫正,并输出矫正后的碱基质量值。
作为本申请的第三方面,本申请提供了一种电子设备,包括:一个或多个处理器;存储装置,其上存储有一个或多个程序,当一个或多个程序被一个或多个处理器执行,使得一个或多个处理器实现上述第一方面任一针对测序平台特征的碱基质量值矫正方法。
作为本申请的第四方面,本申请提供了一种计算机存储介质,其上存储有计算机程序,其中,程序被处理器执行时实现上述第一方面任一针对测序平台特征的碱基质量值矫正方法。
3. 有益效果
本申请与现有技术相比,其有益效果在于:
(1)本申请提供的一种针对测序平台特征的碱基质量值矫正方法,利用read1和read2的重叠部分的碱基来区分测序错误碱基和非测序错误碱基,能够更准确的描述测序错误,反映测序仪的系统性偏好和测序批次的差异;此外,在不同的测序特征,包括位于read1或read2、cycle、dinucleotide特征和测序仪报告的碱基质量值下分析统计测序错误,并以此为建模对象,建立矫正模型用于矫正碱基质量值,避免了现有技术中用于矫正的测序错误可能包括取样建库以及样本特异性体细胞突变等,弥补了BQSR对cfDNA数据的尤其是高肿瘤突变负荷(TMB)样本上的缺陷,能够更准确的反映测序仪偏好,全面提高碱基质量值的可信度。
(2)本申请提供的一种针对测序平台特征的碱基质量值矫正方法,克服现有的矫正方法对于物种突变数据库的依赖和应用范围的局限,无需依赖物种已知突变数据库,不仅适用于胚系突变分析的测序样本,对体系突变的测序样本同样适用,不受样本突变数目的影响,高低TMB的肿瘤样本都适用,适用性更广。
(3)本申请提供的一种针对测序平台特征的碱基质量值矫正方法,能够明确区分测序错误和非测序错误,准确的度量测序错误,为提高数据检测灵敏度和准确度提供了一种可靠的方法。
(4)本申请提供的一种针对测序平台特征的碱基质量值矫正方法,利用重叠碱基能够准确的反映测序批次和测序仪的系统性偏好,因此同一个测序批次不同样本可以共用一个矫正模型,而不需要构建样本特异的矫正模型,这种方式能够大幅提升矫正碱基质量的效率。
附图说明
图1是NovaSeq 6000测序平台在不同dinucleotide下的测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值。
图2是NovaSeq 6000测序平台在不同read和cycle下的测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值。
图3是NovaSeq 6000测序平台在不同dinucleotide下的非测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值。
图4是NovaSeq 6000测序平台在不同read和cycle下的非测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值。
图5是NovaSeq 6000测序平台在不同dinucleotide和cycle下碱基质量值的准确性分布图,其中横轴为特征类型,纵轴为碱基质量值的准确性度量,即经验质量值-仪器报告碱基质量值。
图6是NovaSeq 6000测序平台矫正前后碱基质量值与经验质量值关系,其中Report为矫正前碱基质量值,Predict为矫正后碱基质量值。
图7是NovaSeq 6000测序平台在不同dinucleotide和cycle下矫正后碱基质量值的准确性分布图,其中横轴为特征类型,纵轴为碱基质量值的准确性度量,即经验质量值-矫正后碱基质量值。
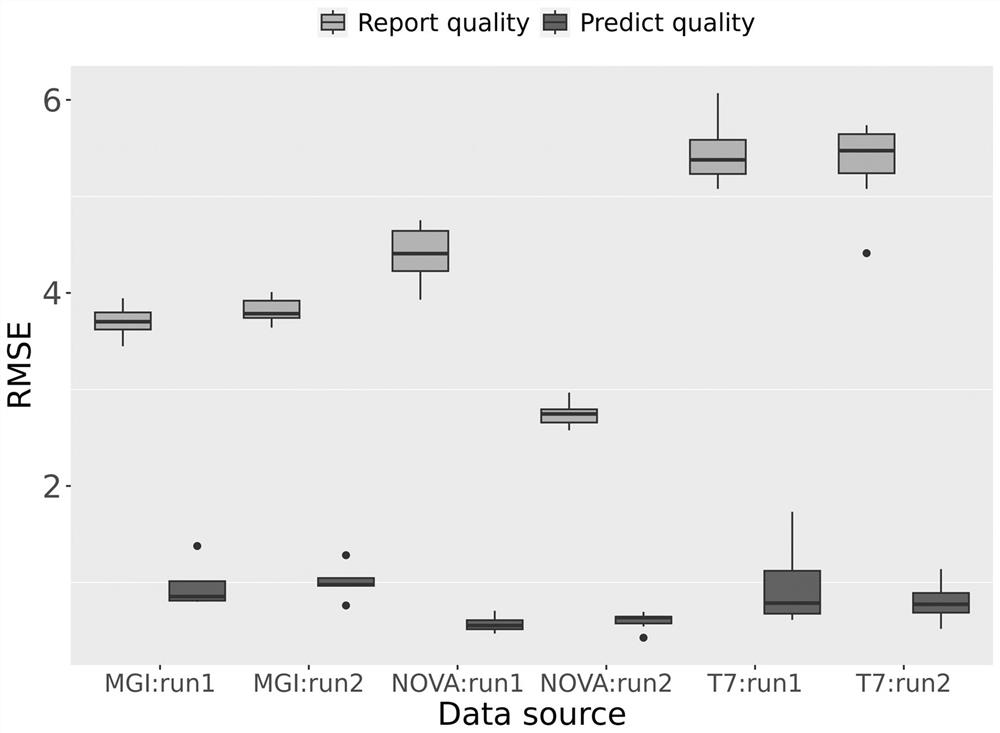
图8是NovaSeq 6000、MGISEQ-2000、MGISEQ-T7三个测序平台两个测序批次(run1和run2)的碱基质量值矫正前后的RMSE分布图。
图9是NovaSeq 6000测序平台以Q30为阈值在不同dinucleotide下矫正前后测序错误率的分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图10是NovaSeq 6000测序平台以Q30为阈值在不同read和cycle下矫正前后测序错误率的分布图,其中横轴为特征类型,纵轴为突变类型,颜色代表测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图11是NovaSeq 6000测序平台以Q30为阈值在不同dinucleotide下矫正前后非测序错误率的分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图12是NovaSeq 6000测序平台以Q30为阈值在不同read和cycle下矫正前后非测序错误率的分布图,其中横轴为特征类型,纵轴为突变类型,颜色代表非测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图13是MGISEQ-2000测序平台在不同dinucleotide下的测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值。
图14是MGISEQ-2000测序平台在不同read和cycle下的测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值。
图15是MGISEQ-2000测序平台在不同dinucleotide下的非测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值。
图16是MGISEQ-2000测序平台在不同read和cycle下的非测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值。
图17是MGISEQ-2000测序平台在不同dinucleotide和cycle下碱基质量值的准确性分布图,其中横轴为特征类型,纵轴为碱基质量值的准确性度量,即经验质量值-仪器报告碱基质量值。
图18是MGISEQ-2000测序平台矫正前后碱基质量值与经验质量值关系,其中Report为矫正前碱基质量值,Predict为矫正后碱基质量值。
图19是MGISEQ-2000测序平台在不同dinucleotide和cycle下矫正后碱基质量值的准确性分布图,其中横轴为特征类型,纵轴为碱基质量值的准确性度量,即经验质量值-矫正后碱基质量值。
图20是MGISEQ-2000测序平台以Q30为阈值在不同dinucleotide下矫正前后测序错误率的分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图21是MGISEQ-2000测序平台以Q30为阈值在不同read和cycle下矫正前后测序错误率的分布图,其中横轴为特征类型,纵轴为突变类型,颜色代表测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图22是MGISEQ-2000测序平台以Q30为阈值在不同dinucleotide下矫正前后非测序错误率的分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图23是MGISEQ-2000测序平台以Q30为阈值在不同read和cycle下矫正前后非测序错误率的分布图,其中横轴为特征类型,纵轴为突变类型,颜色代表非测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图24是MGISEQ-T7测序平台在不同dinucleotide下的测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值。
图25是MGISEQ-T7测序平台在不同read和cycle下的测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值。
图26是MGISEQ-T7测序平台在不同dinucleotide下的非测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值。
图27是MGISEQ-T7测序平台在不同read和cycle下的非测序错误率分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值。
图28是MGISEQ-T7测序平台在不同dinucleotide和cycle下碱基质量值的准确性分布图,其中横轴为特征类型,纵轴为碱基质量值的准确性度量,即经验质量值-仪器报告碱基质量值。
图29是MGISEQ-T7测序平台矫正前后碱基质量值与经验质量值关系,其中Report为矫正前碱基质量值,Predict为矫正后碱基质量值。
图30是MGISEQ-T7测序平台在不同dinucleotide和cycle下矫正后碱基质量值的准确性分布图,其中横轴为特征类型,纵轴为碱基质量值的准确性度量,即经验质量值-矫正后碱基质量值。
图31是MGISEQ-T7测序平台以Q30为阈值在不同dinucleotide下矫正前后测序错误率的分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图32是MGISEQ-T7测序平台以Q30为阈值在不同read和cycle下矫正前后测序错误率的分布图,其中横轴为特征类型,纵轴为突变类型,颜色代表测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图33是MGISEQ-T7测序平台以Q30为阈值在不同dinucleotide下矫正前后非测序错误率的分布图,其中横轴为突变类型,纵轴为特征类型,颜色代表非测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
图34是MGISEQ-T7测序平台以Q30为阈值在不同read和cycle下矫正前后非测序错误率的分布图,其中横轴为特征类型,纵轴为突变类型,颜色代表非测序错误率数值,其中Report quality为矫正前碱基质量值,Predict quality为矫正后碱基质量值。
具体实施方式
下面结合具体实施例对本申请进一步进行描述。
需要说明的是,本说明书中所引用的如“上”、“下”、“左”、“右”、“中间”等用语,亦仅为便于叙述的明了,而非用以限定可实施的范围,其相对关系的改变或调整,在无实质变更技术内容下,当亦视为本发明可实施的范畴。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同;本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
如本文所使用,术语“约”用于提供与给定术语、度量或值相关联的灵活性和不精确性。本领域技术人员可以容易地确定具体变量的灵活性程度。
如本文所使用,术语“......中的至少一个”旨在与“......中的一个或多个”同义。例如,“A、B和C中的至少一个”明确包括仅A、仅B、仅C以及它们各自的组合。
浓度、量和其他数值数据可以在本文中以范围格式呈现。应当理解,这样的范围格式仅是为了方便和简洁而使用,并且应当灵活地解释为不仅包括明确叙述为范围极限的数值,而且还包括涵盖在所述范围内的所有单独的数值或子范围,就如同每个数值和子范围都被明确叙述一样。例如,约1至约4.5的数值范围应当被解释为不仅包括明确叙述的1至约4.5的极限值,而且还包括单独的数字(诸如2、3、4)和子范围(诸如1至3、2至4等)。相同的原理适用于仅叙述一个数值的范围,诸如“小于约4.5”,应当将其解释为包括所有上述的值和范围。此外,无论所描述的范围或特征的广度如何,都应当适用这种解释。
本申请的原始测序数据FASTQ文件分别来源于NovaSeq 6000、MGISEQ-2000和MGISEQ-T7三个测序平台,具体包括:使用MagMAX™游离DNA分离试剂盒(ThermoFisher)提取血浆样本中cfDNA;使用KAPA HyperPrep文库构建试剂盒(Roche)构建血浆文库;使用Panel_P探针(IDT合成)富集血浆文库的目标区域,其中Panel_P探针覆盖基因组2.1 Mb的区间,覆盖了207个肿瘤发生发展相关的重要通路的共769个基因;利用NovaSeq 6000、MGISEQ-2000和MGISEQ-T7三个测序平台分别进行测序获得原始测序数据FASTQ文件。
实施例1
本实施例提供NovaSeq 6000测序平台的碱基质量值矫正方法,具体包括如下步骤:
S1:原始测序数据FASTQ文件进行预处理并比对至参考基因组生成Bam文件。其中,所述预处理包括去除接头序列和低质量碱基,去除低质量碱基包括:1)去除长度小于51的reads;2)以窗口大小为1个碱基从read前到后进行滑窗,如果窗口内平均碱基质量值小于等于1,则过滤掉窗口右边的碱基。在实施例中,去除长度小于51的reads通过商业软件的模块完成,如调用fastp 0.20.0,采用--length_required 51去除。在实施例中,以窗口大小为1个碱基从read前到后进行滑窗,如果窗口内平均碱基质量值小于等于1,则过滤掉窗口右边的碱基通过商业软件的模块完成,如调用fastp 0.20.0,采用--cut_right-W1-M1去除。在实施例中,去除接头序列通过商业软件的模块完成,如adapter_fasta TruSeq3-PE.fa。在实施例中,所述比对至参考基因组生成Bam文件包括将预处理后的数据比对到hg19人类参考基因组序列并生成Bam文件。在实施例中,比对至参考基因组生成Bam文件通过商业软件的模块完成,如调用bwa-0.7.17的BWA-MEM算法将双端测序的reads比对到hg19人类参考基因组序列,使用samtools-1.3的view将比对的sam文件转化成bam文件,使用samtools-1.3的sort进行排序,index对排序后的bam文件建索引。
S2:以S1中生成的Bam文件为输入文件,构建参考基因组一致性序列并进行reads水平过滤,其中:构建参考基因组一致性序列,即将突变频率大于等于99%的碱基作为该位点的真实碱基类型;reads水平过滤包括过滤掉没有比对到参考基因组的reads对、非主要比对的reads、平台/供应商质量检查失败的reads和嵌和比对但不是主要代表性比对的reads,同时不对read1和read2重叠部分的碱基依据碱基质量值过滤。在实施例中,非主要比对的reads、平台/供应商质量检查失败的reads和嵌和比对但不是主要代表性比对的reads是商业软件过滤时的通用定义。在实施例中,reads水平过滤通过商业软件的模块完成,依次是设置flag_filter为2820,read unmapped,flag(4,8),not primary alignment,flag 256,read fails platform/vendor quality checks,flag 512,supplementaryalignment,flag 2048。在实施例中,使用min_base_quality参数设置为0,将ignore_overlaps设置为false,即不对read1和read2重叠部分的碱基依据碱基质量值做过滤,而是两个都保留下来,用于后续分析。在实施例中,reads信息通过商业软件提取。在实施例中,采用Python 3.9.12的pysam模块的pileup方法按照参考基因组的位置逐个位点从bam文件中读取比对在该位置的reads信息。
S3:从过滤后的Bam文件中提取重叠碱基及其特征信息,并区分错配错误来源,具体包括:基于S2的过滤结果,依据read name提取比对到基因组某一位置的reads对,区分reads对的来源是F1R2还是F2R1,即确定属于read1还是read2;提取read比对到该位点的碱基,与测序扩增方向的前一个碱基组成的二核苷酸(dinucleotide);碱基的测序循环数(cycle);提取测序仪报告的碱基质量值(RQS);将read1的碱基与read2的碱基不配对,且其中与参考基因组一致性序列碱基不匹配的碱基标记为测序错误碱基;将read1的碱基与read2的碱基配对,且都与参考基因组一致性序列碱基不匹配的碱基标记为非测序错误碱基。在实施例中,如read E100034389L1C010R03202285505:TCA_TAT是来源于参考基因组一致性序列正链的测序片段,所以其reads组成是F1R2,即read1比对到参考基因正链,其总长为95 bp,第80位碱基C比对到参考基因组一致性序列正链chr10的101620719的A位点,获得其测序仪给出的碱基质量值14,cycle为80,dinucleotide为TA,而对应的read2,总长95bp,其碱基T比对到参考基因组一致性序列chr10的101620719位点的负链上,其测序仪给出的测序质量值为37,cycle为22,dinucleotide为CT,由于参考基因组一致性序列chr10的101620719位点原始正链碱基是A,负链为T,因此给read1比对到该位置的C碱基标记为测序错误(sequencing error),而read2比对到该位置的T标记为正确测序碱基。readE100034389L1C003R02404263679:AGC_ATC是来源于参考基因组一致性序列负链的测序片段,所以其reads组成是F2R1,即read2比对到参考基因正链,其总长为95 bp,第68位碱基A比对到参考基因组一致性序列正链chr10的104268962 G位点,获得其测序仪给出的碱基质量值38,cycle为68,dinucleotide为TG,而对应的read1,总长95 bp,其碱基T比对到参考基因组一致性序列chr10的104268962负链上的C位点,其测序仪给出的碱基质量值为36,cycle为5,dinucleotide为AC,由于参考基因组一致性序列chr10的104268962位点原始正链碱基是G,负链为C,因此给read1比对到该位置的的T和read2比对到该位置的的A碱基标记为非测序错误(non sequencing error)。
S4:测序平台系统性偏好评估及非测序导致的错配模式,包括如下步骤:基于S3的结果,分别以read位置、dinucleotide和cycle作为特征,描述测序仪的测序错误的模式及非测序错误的模式,即统计不同dinucleotide,不同的read来源的各个cycle中碱基的测序错误和非测序错误的比例和分布,结果如图1-图4所示,从图中可以看出,测序和非测序错误的错误率在不同测序特征(dinucleotide,cycle)中存在明显差异,如测序错误来源的A(A
S5:碱基质量值准确性评估,所述碱基质量值准确性为经验质量值与测序仪报告碱基质量值的差,具体包括如下步骤:基于S4的分析结果,统计分析NOVA-seq 6000测序平台给定碱基质量值与经验质量值之间的关系,统计分析发现不同测序特征(read朝向,dinucleotide,cycle)错误率存在差异,为了衡量在不同测序特征(read朝向,dinucleotide,cycle)下测序平台碱基质量值准确性,将提取的重叠碱基划分到不同的分组形成特征bins,即将测序特征相同的碱基划分至一个小组生成一个特征bin,统计在测序仪报告的碱基质量值下,不同read朝向的各个cycle和dinucleotide下的总碱基数目和测序错误的碱基数目,计算碱基的经验质量值以衡量测序平台碱基质量值的准确性,经验质量值的计算公式如式(1)所示。NovaSeq 6000测序平台在不同dinucleotide和cycle下碱基质量值的准确性分布图如图5所示,从图中可以看出,不同测序特征(dinucleotide,cycle)的碱基质量值准确性存在明显差异。
其中,empricial quality代表bin的经验质量值(empricial quality score,EQS),mismatches代表bin的内测序错误碱基数目,bases代表bin内所有碱基数目;
S6:采用python的sm.nonparametric模块的lowess方法,即局部加权回归模型(locally weighted regression,lowess)对仪器报告的碱基质量值(report qualityscore,RQS)和经验质量值(empricial quality score,EQS)进行多项式拟合建模,参数frac设置为0.1。基于该模型对仪器报告的碱基质量值进行矫正,获得矫正后的碱基质量值。矫正前后碱基质量值与经验碱基质量值的关系分布如图6所示,从图上可以看出,与矫正前仪器报告的碱基质量值相比,矫正后的质量值与经验质量值更接近;
S7:矫正后不同测序批次碱基质量值准确性评估。
(1)参考步骤S5评估矫正后碱基质量值的准确性,不同dinucleotide和cycle矫正后的碱基质量值的准确性分布如图7所示,可以看出,经过碱基质量值矫正之后不同的dinucleotide,cycle的碱基质量值的偏差都得到了很好的矫正。
(2)利用式(2)计算均方根误差(RMSE)用于衡量样本层面矫正前后碱基质量值的准确度,均方根误差是预测值与真实值偏差的平方与观测次数n比值的平方根,衡量的是预测值与真实值之间的偏差,并且对数据中的异常值较为敏感:
NOVA-seq 6000测序平台的两个批次13个样本(run1:4;run2:9)矫正前后碱基质量值与经验质量值之间的RMSE分布如图8所示,经过矫正碱基质量值能够更准确的描述碱基错误率。
(3)以Q30为阈值比较矫正前后测序错误率的分布。对NOVA-seq 6000四个样本,使用矫正前后碱基质量值阈值30,对重叠区域的存在碱基进行过滤,基于过滤结果重新计算在不同dinucleotide类型和不同cycle下的测序错误率,如图9-图10所示,碱基质量值矫正能够有效降低不同dinucleotide类型和不同cycle的造成的测序偏差,有效提高同一阈值下检测精度的均一性。
(4)以Q30为阈值比较矫正前后非测序错误率的分布。对NOVA-seq 6000四个样本,使用矫正前后碱基质量值阈值30,基于过滤结果重新计算在不同Dinucleotide类型和不同cycle下的非测序错误率,如图11-图12所示,矫正前后非测序错误率未发生显著变化也与预期相符。
实施例2
本实施例提供MGISEQ-2000测序平台的碱基质量值矫正方法,具体步骤参考实施例1。
测序平台系统性偏好评估及非测序导致的错配模式,不同dinucleotide,不同的read链来源的各个cycle中碱基的测序错误和非测序导致的错配的比例和分布,结果如图13-图16所示,从图中可以看出,测序和非测序错误的错误率在不同测序特征(dinucleotide,cycle)中存在明显差异,如测序错误来源的G(A
MGISEQ-2000测序平台在不同dinucleotide和cycle下碱基质量值的准确性分布图如图17所示,从图中可以看出,可以看出,MGISEQ-2000不同测序特征(dinucleotide,cycle)的碱基质量值准确性存在明显差异。reads末端碱基中部分碱基质量值的准确性比经验质量值高,低估了该特征下的测序错误率。
碱基质量值矫正模型构建,使用局部加权平均算法对MGISEQ-2000测序平台不同特征bin下测序仪报告的碱基质量值与经验碱基质量值进行拟合,并对碱基质量值进行矫正,矫正前后碱基质量值与经验碱基质量值的关系分布如图18所示,从图上可以看出,与矫正前仪器给出的碱基质量值相比,矫正后的质量值与经验质量值更接近。
MGISEQ-2000测序平台在不同dinucleotide和cycle下矫正后的碱基质量值的准确性分布如图19所示,可以看出,测序平台给出的碱基质量值与经验质量值相比均存在偏差,并且在不同的dinucleotide,cycle下也存在差异,经过碱基质量值矫正之后其偏差都得到了很好的矫正。
MGISEQ-2000测序平台的两个批次共9个样本(run1:4;run2:5)矫正前后碱基质量值与经验质量值之间的RMSE分布如图8所示,经过矫正碱基质量值能够更准确的描述碱基错误率。
以Q30为阈值比较矫正前后测序错误率的分布。对MGISEQ-2000四个样本,使用矫正前后碱基质量值阈值30,对重叠区域的存在碱基进行过滤,基于过滤结果重新计算在不同Dinucleotide类型和不同cycle下的测序错误率,如图20-图21所示,碱基质量值矫正能够有效降低不同dinucleotide类型和不同cycle的造成的测序偏差,有效提高同一阈值下检测精度的均一性。
以Q30为阈值比较矫正前后非测序错误率的分布。对NOVA-seq 6000四个样本,使用矫正前后碱基质量值阈值30,对重叠区域的存在碱基进行过滤,基于过滤结果重新计算在不同Dinucleotide类型和不同cycle下的非测序错误率,如图22-图23所示,矫正前后非测序错误率未发生显著变化也与预期相符。
实施例3
本实施例提供MGISEQ-T7测序平台的碱基质量值矫正方法,具体步骤参考实施例1。
测序平台系统性偏好评估及非测序导致的错配模式,不同dinucleotide,不同的read链来源的各个cycle中碱基的测序错误和非测序导致的错配的比例和分布,结果如图24-图27所示,从图中可以看出,测序和非测序错误的错误率在不同测序特征(dinucleotide,cycle)中存在明显差异,如测序错误来源的G(T
MGISEQ-T7测序平台在不同dinucleotide和cycle下碱基质量值的准确性分布图如图28所示,从图中可以看出,read2的沿着cycle方向的末端,仪器给定的碱基质量远高于经验质量值,即低估了错误率。
MGISEQ-T7测序数据在不同dinucleotide和cycle下矫正后的碱基质量值的准确性分布如图30,由图可以看出经过碱基质量值矫正之后其偏差得到了很好的矫正。
MGISEQ-T7测序平台的两个批次共18个样本(run1:7;run2:11)矫正前后碱基质量值与经验质量值之间的RMSE分布如图10所示,经过矫正碱基质量值能够更准确的描述碱基错误率。
以Q30为阈值比较矫正前后测序错误率的分布。对MGISEQ-T7四个样本,使用矫正前后碱基质量值阈值30,对重叠区域的存在碱基进行过滤,基于过滤结果重新计算在不同dinucleotide类型和不同cycle下的测序错误率,如图31-图32所示,碱基质量值矫正能够有效降低不同dinucleotide类型和不同cycle 的造成的测序偏差,有效提高同一阈值下检测精度的均一性。
以Q30为阈值比较矫正前后非测序错误率的分布。对NOVA-seq 6000四个样本,使用矫正前后碱基质量值阈值30,对重叠区域的存在碱基进行过滤,基于过滤结果重新计算在不同Dinucleotide类型和不同cycle下的非测序错误率,如图33-图34所示,矫正前后非测序错误率未发生显著变化也与预期相符。
实施例4
本实施例提供一种针对测序平台特征的碱基质量值矫正装置,包括如下模块:
去接头模块:用于去除原始双端测序数据中接头序列;
比对模块:用于将输入的去接头的测序数据比对到参考基因组,生成比对到参考基因组的Bam文件;
过滤模块:用于对输入的生成比对到参考基因组的Bam文件进行构建参考基因组一致性序列并进行reads水平过滤,所述进行构建参考基因组一致性序列,即将突变频率大于等于99%的碱基作为该位点的真实碱基类型;所述reads水平过滤包括过滤掉只有一条read比对到参考基因组的reads对、没有比对到参考基因组的reads对、非主要比对的reads、平台/供应商质量检查失败的reads和嵌和比对但不是主要代表性比对的reads,但不对read1和read2重叠部分的碱基依据碱基质量值过滤,即保留read1和read2重叠部分的碱基;
重叠碱基提取和区分错误来源模块:用于从过滤后的Bam文件中提取出read1和read2所有重叠的碱基,按照在相同的参考基因组一致性序列上的坐标位置,read1的碱基与read2的碱基是否互补配对,与参考基因组一致性序列碱基是否匹配区分错误来源,将read1的碱基与read2的碱基不配对,且其中与参考基因组一致性序列碱基不匹配的碱基标记为来源于测序错误的碱基;
错误率统计模块:用于提取重叠碱基的特征,包括read朝向、测序循环数、与测序扩增方向的前一个碱基组成的二核苷酸及仪器报告的碱基质量值,并按照特征将碱基分成不同bins,统计各bin中的碱基错误率;基于bin内测序错误碱基照式(1)计算经验质量值;
其中,empricial quality代表bin的经验质量值,mismatches代表bin的内测序错误碱基数目,bases代表bin内所有碱基数目;
矫正模型建模模块:用于根据错误率统计模块计算的经验质量值,使用局部加权回归对不同特征下仪器报告的碱基质量值和经验质量值进行拟合建模,局部加权回归模型的构建是采用python的sm.nonparametric模块的lowess方法建立,参数frac设置为0.1;
矫正模块:用于根据矫正模型建模模块建立的模型对仪器报告的碱基质量值进行矫正,并输出矫正后的碱基质量值。
- 跨平台访问方法、装置、电子设备及存储介质
- 在线特征确定方法、装置、电子设备及存储介质
- 随机选取特征的方法、装置、电子设备及存储介质
- 特征生成方法、装置、电子设备及计算机可读存储介质
- 对象特征参数确定方法、装置、电子设备及可读存储介质
- 基于测序数据的碱基突变检测方法、装置及存储介质
- 碱基识别方法和装置、电子设备及存储介质