PAR-1抑制剂、其手性合成方法、及其盐晶型的制备方法与用途
文献发布时间:2024-04-18 20:00:50
技术领域
本发明属于医药化工技术领域,具体涉及PAR-1抑制剂、其手性合成方法、及其盐晶型的制备方法与用途。
背景技术
血栓症,即局部血液凝块形成。其中,动脉血栓可导致如心肌梗塞、中风、急性冠状动脉综合症和外周动脉疾病等;而静脉血栓则可引发肺栓塞。动静脉血栓是引发心血管疾病的发病与死亡的首要原因,同时,它也是癌症患者死亡的首要原因之一。抗血栓药物包括抗凝血药、抗血小板聚集药和溶血栓药。然而,在这三种不同作用类型的药物中,只有抗凝血药和抗血小板聚集药物效果还可以。现有抗凝血药物的缺陷以及强有力的市场机会将驱动抗凝血药物的发展。而那些疗效至少与现有抗凝血药物相当,安全性(尤其是降低出血风险)更佳且使用方便(可供口服,尤其是可长期使用)的新型抗凝血药物是迫切需要的。
凝血酶受体(PARs)是G蛋白偶联受体(GPCRs)中的一种,目前的PAR-1类抑制类药物虽然具有较好的抗凝血活性,但在长期的临床应用中,患者出现了不同程度的出血的副作用,该药有效半衰期长达3-4天,终末清除半衰期达8天。短时间的停药对于治疗出血效果不佳,在停药后作用持续至少4周。目前,还没有合适的治疗方法来对抗这个药物的抗血小板作用。
CN105732595A和CN115043820A都公开了一种PAR-1抑制剂,其结构通式为:
虽然以上都制备了PAR-1抑制剂,但是其药物与靶点结合不紧密,药效差,药代动力学参数差,成药性差,因此仍旧存在体内代谢快,不利于成药的问题。
发明内容
本发明要解决的技术问题是克服现有技术存在的上述缺陷,提供一种PAR-1抑制剂,具有靶点明确、作用机制清晰、结构新颖、安全性高、活性高的特点,其手性合成方法得到构型翻转化合物;本发明还提供PAR-1抑制剂的盐晶型的制备方法,制备的晶型稳定,将本发明制备的PAR-1抑制剂应用在治疗血栓性疾病中,价值高。
本发明所述的PAR-1抑制剂,结构式如下所示:
。
所述的PAR-1抑制剂的制备方法,包括以下步骤:
(1)以穿心莲内酯为起始原料,经反应制得中间体Ⅱ,中间体Ⅱ的结构式如下所示:
(2)将中间体Ⅱ的羰基进行手性选择还原胺化,同时对邻位上的甲基构型进行重新构建,制得化合物Ⅲ,化合物Ⅲ的结构式如下所示:
;
(3)将化合物Ⅲ中的氨基采用氯甲酸乙酯修饰,制得化合物Ⅳ,化合物Ⅳ的结构式如下所示:
(4)将化合物Ⅳ在酸催化的作用下发生水解、酯交换,制得化合物Ⅴ,化合物Ⅴ的结构式如下所示:
(5)在二异丙基氨基锂的作用下,将化合物Ⅴ与化合物Ⅵ进行维蒂希反应,制得PAR-1抑制剂;化合物Ⅵ的结构式如下所示:
。
步骤(2)中手性选择还原胺化采用的催化剂为Ru((R)-BINAP)(OAc)
所述的PAR-1抑制剂盐晶型的制备方法,包括以下步骤:将PAR-1抑制剂溶于溶剂,加入酸溶液,搅拌均匀,降温析晶,抽滤或离心,所得固体烘干,得到PAR-1抑制剂盐晶型。
所述的酸溶液为盐酸、硫酸、磷酸、富马酸、酒石酸中的一种,优选为硫酸。
降温至-20℃-20℃析晶,优选为0℃-10℃。
所述的溶剂为乙醇、甲醇、乙腈中的一种,其加入量为PAR-1抑制剂的5-100倍体积,优选为10-20倍体积。
所述的PAR-1抑制剂盐晶型的制备方法制得的PAR-1抑制剂盐晶型的用途:用于治疗血栓性疾病的药物,可用于口服。
具体的,所述的PAR-1抑制剂盐晶型的制备方法,包括以下步骤:
(1)以穿心莲内酯为起始原料,经多步反应制得中间体Ⅱ,中间体Ⅱ的结构式如下所示:
(2)将中间体Ⅱ加入反应瓶中,加入三氟乙醇(TFE)溶解,依次加入乙酸铵(NH
。
(3)将化合物Ⅲ粗品用二氯甲烷溶解,并转移至圆底烧瓶中,加氯甲酸乙酯,0℃搅拌下缓慢加入三乙胺(TEA),加料完毕,室温搅拌2h,加水搅拌5min,搅拌完成,将体系转移至分液漏斗中,静置分层,放出下层有机相,上层水相加二氯甲烷萃取,萃取完成,弃去水相,合并有机相,有机相加水洗涤,转移至锥形瓶中,无水硫酸钠干燥有机相,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到白色固体化合物,即化合物Ⅳ,化合物Ⅳ的结构式如下所示:
(4)将化合物Ⅳ加入反应瓶中,加入对甲苯磺酸,再加丙酮和水,80℃下加热搅拌4h,反应完成后,加饱和碳酸氢钠溶液搅拌5min,并转移至分液漏斗中,加乙酸乙酯摇匀,静置分层,放出上层有机相,无水硫酸钠干燥,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到白色固体化合物,即化合物Ⅴ,化合物Ⅴ的结构式如下所示:
(5)将化合物Ⅴ加入反应瓶中,加四氢呋喃溶解,-20℃下搅拌5min,缓慢滴加二异丙基氨基锂,加料完毕,反应1h,加入化合物VI,0℃下搅拌2h,反应完成,加饱和氯化铵水溶液搅拌5min,并转移至分液漏斗中,摇匀,静置分层,放出上层有机相,下层水相加丙烯酸乙酯(EA)萃取,萃取完成,弃去水相,合并有机相,无水硫酸钠干燥,抽滤,减压蒸馏,粗品用无水乙醇精制,得到白色固体化合物,即PAR-1抑制剂;反应过程如下所示:
。
(6)将PAR-1抑制剂溶于5-100倍体积溶剂,加入酸溶液,搅拌均匀,降温至-20℃-20℃析晶,抽滤或离心,所得固体烘干得到PAR-1抑制剂盐的α晶型。
与现有技术相比,本发明具有的有益效果是:
(1)本发明制备的PAR-1抑制剂盐的α晶型,具有好的稳定性,适于长期储存,有效避免药物储存以及开发过程中发生转晶,从而避免生物利用度以及药效的改变。
(2)本发明的PAR-1抑制剂盐晶型的制备方法,工艺简单,成本低廉,重复性好,所采用的溶剂无毒,对该药物的应用和开发具有重要价值。
附图说明
图1为实施例1制备的化合物Ⅳ核磁图谱。
图2为实施例1制备的化合物Ⅳ质谱图谱。
图3为实施例1制备的化合物V核磁图谱。
图4为实施例1制备的化合物V质谱图谱。
图5为实施例1制备的PAR-1化合物核磁图谱。
图6为实施例1制备的PAR-1化合物质谱图谱。
图7为实施例2制备的PAR-1抑制剂盐晶型的XRPD图。
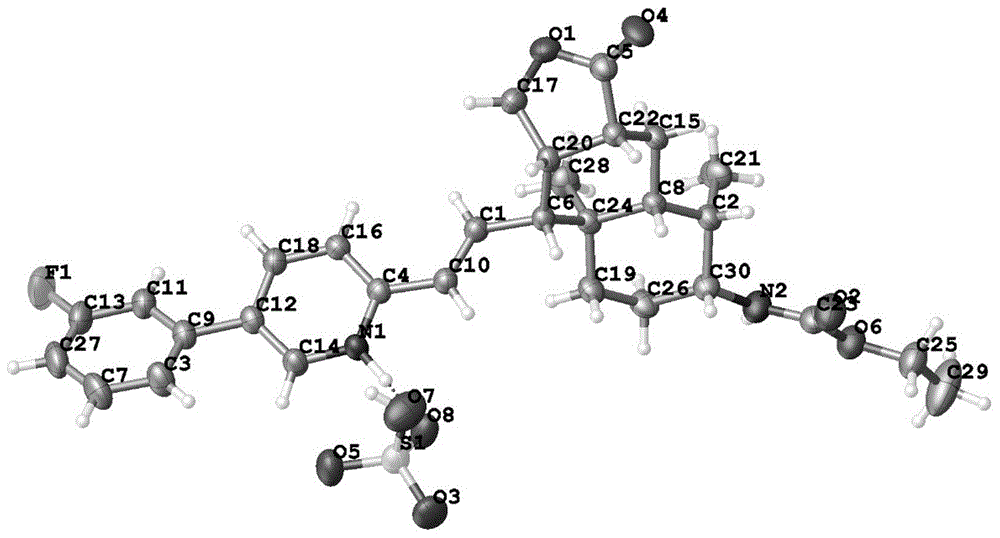
图8为实施例2制备的PAR-1抑制剂盐晶型的单晶结构图。
图9为实施例2制备的PAR-1抑制剂盐进行样本测试时的PAR-1抑制剂在SD大鼠体内平均血药浓度与时间的关系图。
具体实施方式
下面结合具体实施例对本发明作进一步说明。
以下实施例和对比例所采用的原料为市售产品。
实施例1
所述的PAR-1抑制剂的制备方法,包括以下步骤:
(1)以穿心莲内酯为起始原料,经反应制得中间体Ⅱ,其具体制备步骤参照2022年9月13日公开的中国专利CN115043820A中的实施例1中说明书【0095-0114】段。
(2)将10g中间体Ⅱ加入反应瓶中,加入50mL三氟乙醇(TFE)溶解,依次加入4.78g乙酸铵(NH
(3)将化合物Ⅲ粗品用100mL二氯甲烷溶解,并转移至圆底烧瓶中,加16.8g氯甲酸乙酯,0℃搅拌下缓慢加入31.4g三乙胺(TEA),加料完毕,室温搅拌2h,加100mL水搅拌5min,搅拌完成将体系转移至分液漏斗中,静置分层,放出下层有机相,上层水相加100mL二氯甲烷萃取,萃取完成,弃去水相,合并有机相,有机相加100mL水洗涤,转移至锥形瓶中,无水硫酸钠干燥有机相,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到7.9g白色固体化合物,即化合物Ⅳ,产率为64.44%,化合物Ⅳ的核磁图如图1所示,质谱图如图2所示。
(4)将7.00g化合物Ⅳ加入反应瓶,加入4.0g对甲苯磺酸,再加20mL丙酮和2mL水,80℃下加热搅拌4h,反应完成后,加20mL饱和碳酸氢钠溶液搅拌5min,并转移至分液漏斗中,加20mL乙酸乙酯摇匀,静置分层,放出上层有机相,无水硫酸钠干燥,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到4.85g白色固体化合物,即化合物Ⅴ,产率为81.65%,化合物Ⅴ的核磁图如图3所示,质谱图如图4所示。
(5)将4.00g化合物Ⅴ加入50mL反应瓶,加10mL四氢呋喃溶解,-20℃下搅拌5min,缓慢滴加2.4mL二异丙基氨基锂,加料完毕,反应1h,加入4.60g化合物VI,0℃下搅拌2h,反应完成,加15mL饱和氯化铵水溶液搅拌5min,并转移至分液漏斗中,摇匀,静置分层,放出上层有机相,下层水相加15mL丙烯酸乙酯(EA)萃取,萃取完成,弃去水相,合并有机相,无水硫酸钠干燥,抽滤,减压蒸馏,粗品用无水乙醇精制,得到5.02g白色固体化合物,即PAR-1抑制剂,产率为83.67%。PAR-1化合物的核磁图如图5所示,质谱图如图6所示。
实施例2
所述的PAR-1抑制剂盐晶型的制备方法,包括以下步骤:
将4.00g实施例1制备的PAR-1抑制剂溶于40mL乙醇中,称1.00g浓硫酸用20mL乙醇溶解,再加入PAR-1抑制剂的乙醇溶液中,搅拌均匀,降温至5℃析晶,搅拌析晶5h,抽滤,所得固体烘干,得到PAR-1抑制剂硫酸盐的α晶型。
将以上实施例2制备的PAR-1抑制剂盐的α晶型进行XRPD(X射线粉末衍射)分析,如图7所示,采用的是Bruker D8 FOCUS X射线粉末衍射仪,由图7所示,在2θ值为4.5°±0.2°、9.0°±0.2°、14.5°±0.2°、14.8°±0.2°、15.4°±0.2°、15.6°±0.2°、18.2°±0.2°、18.4°±0.2°、19.8°±0.2°处具有特征峰。
并对实施例2的单晶结构分析,其单晶结构图如图8所示,采用的仪器品牌:德国布鲁克,型号:D8 VENTURE,检测方法:X-ray工作电压50kV,工作电流50mA,最大2θ值68.395°,检测温度248K,用Bruker APEX-II CCD面探测仪收集衍射强度数据,CuKα辐射,总衍射点为5948个,独立衍射点为5644个(Rint=0.0510),数据完整度为99.1%。晶体呈无色透明块状,属于正交晶系,空间群为P2
实施例3
所述的PAR-1抑制剂的制备方法,包括以下步骤:
(1)以穿心莲内酯为起始原料,经反应制得中间体Ⅱ,其具体制备步骤参照2022年9月13日公开的中国专利CN115043820A中的实施例1中说明书【0095-0114】段。
(2)将10g中间体Ⅱ加入反应瓶中,加入50mL三氟乙醇(TFE)溶解,依次加入4.78g乙酸铵(NH
(3)将化合物Ⅲ粗品用100mL二氯甲烷溶解,并转移至圆底烧瓶中,加16.8g氯甲酸乙酯,0℃搅拌下缓慢加入31.4g三乙胺(TEA),加料完毕,室温搅拌2h,加100mL水搅拌5min,搅拌完成将体系转移至分液漏斗中,静置分层,放出下层有机相,上层水相加100mL二氯甲烷萃取,萃取完成,弃去水相,合并有机相,有机相加100mL水洗涤,转移至锥形瓶中,无水硫酸钠干燥有机相,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到5.3g白色固体化合物,即化合物Ⅳ,产率为43.23%。
(4)将7.00g化合物Ⅳ加入反应瓶,加入4.0g对甲苯磺酸,再加20mL丙酮和2mL水,80℃下加热搅拌4h,反应完成后,加20mL饱和碳酸氢钠溶液搅拌5min,并转移至分液漏斗中,加20mL乙酸乙酯摇匀,静置分层,放出上层有机相,无水硫酸钠干燥,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到4.85g白色固体化合物,即化合物Ⅴ,产率为81.02%。
(5)将4.00g化合物Ⅴ加入50mL反应瓶,加10mL四氢呋喃溶解,-20℃下搅拌5min,缓慢滴加2.4mL二异丙基氨基锂,加料完毕,反应1h,加入4.60g化合物VI,0℃下搅拌2h,反应完成,加15mL饱和氯化铵水溶液搅拌5min,并转移至分液漏斗中,摇匀,静置分层,放出上层有机相,下层水相加15mL丙烯酸乙酯(EA)萃取,萃取完成,弃去水相,合并有机相,无水硫酸钠干燥,抽滤,减压蒸馏,粗品用无水乙醇精制,得到5.02g白色固体化合物,即PAR-1抑制剂,产率为82.85%。
实施例4
所述的PAR-1抑制剂的制备方法,包括以下步骤:
(1)以穿心莲内酯为起始原料,经反应制得中间体Ⅱ,其具体制备步骤参照2022年9月13日公开的中国专利CN115043820A中的实施例1中说明书【0095-0114】段。
(2)将10g中间体Ⅱ加入反应瓶中,加入50mL三氟乙醇(TFE)溶解,依次加入4.78g乙酸铵(NH
(3)将化合物Ⅲ粗品用100mL二氯甲烷溶解,并转移至圆底烧瓶中,加16.8g氯甲酸乙酯,0℃搅拌下缓慢加入31.4g三乙胺(TEA),加料完毕,室温搅拌2h,加100mL水搅拌5min,搅拌完成将体系转移至分液漏斗中,静置分层,放出下层有机相,上层水相加100mL二氯甲烷萃取,萃取完成,弃去水相,合并有机相,有机相加100mL水洗涤,转移至锥形瓶中,无水硫酸钠干燥有机相,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到4.2g白色固体化合物,即化合物Ⅳ,产率为34.26%。
(4)将7.00g化合物Ⅳ加入反应瓶,加入4.0g对甲苯磺酸,再加20mL丙酮和2mL水,80℃下加热搅拌4h,反应完成后,加20mL饱和碳酸氢钠溶液搅拌5min,并转移至分液漏斗中,加20mL乙酸乙酯摇匀,静置分层,放出上层有机相,无水硫酸钠干燥,干燥结束,抽滤,减压蒸馏,粗品用柱层析分离纯化,得到4.85g白色固体化合物,即化合物Ⅴ,产率为80.01%。
(5)将4.00g化合物Ⅴ加入50mL反应瓶,加10mL四氢呋喃溶解,-20℃下搅拌5min,缓慢滴加2.4mL二异丙基氨基锂,加料完毕,反应1h,加入4.60g化合物VI,0℃下搅拌2h,反应完成,加15mL饱和氯化铵水溶液搅拌5min,并转移至分液漏斗中,摇匀,静置分层,放出上层有机相,下层水相加15mL丙烯酸乙酯(EA)萃取,萃取完成,弃去水相,合并有机相,无水硫酸钠干燥,抽滤,减压蒸馏,粗品用无水乙醇精制,得到5.02g白色固体化合物,即PAR-1抑制剂,产率为81.62%。
实施例5
所述的PAR-1抑制剂盐晶型的制备方法,包括以下步骤:
将1.00g实施例1制备的PAR-1抑制剂溶于100mL乙腈中,称0.50g硫酸用20mL乙腈溶解,再加入PAR-1抑制剂的乙腈溶液中,搅拌均匀,降温至20℃析晶,搅拌析晶5h,抽滤,所得固体烘干,得到PAR-1抑制剂硫酸盐的α晶型。
实施例6
所述的PAR-1抑制剂盐晶型的制备方法,包括以下步骤:
将1.00g实施例1制备的PAR-1抑制剂溶于50mL甲醇中,称0.50g硫酸用20mL甲醇溶解,再加入PAR-1抑制剂的甲醇溶液中,搅拌均匀,降温至-20℃析晶,搅拌析晶5h,抽滤,所得固体烘干,得到PAR-1抑制剂硫酸盐的α晶型。
将以上实施例1制备的PAR-1抑制剂进行生物利用度测定。
(1)生物样本采集
8只SD大鼠(♂)随机分成2组,每组4只。所有动物禁食12h后,称重,给药PAR-1抑制剂,各组于药前及给药后0.083h、0.25h、0.5h、1h、2h、4h、6h、8h、12h和24h经大鼠颈静脉穿刺采血,每个样品采集约0.2mL全血/只/时间点,取血后置冰盒肝素化离心管中,并于1h之内离心分离血浆(离心条件:2-8℃,8000rpm,10min),收集的血浆分析前存放于-80℃冰箱内。
(2)样本测试
色谱条件:色谱柱:Accucore C18(2.1×50mm,2.6µm),流动相:0.1wt.%甲酸水溶液(A):0.2wt.%甲酸乙腈(B),梯度洗脱,柱温:40℃,进样体积:5µL,如表1所示。
表1梯度洗脱条件
质谱条件:AB SCIEX 5500三重四级杆串联质谱系统,离子源为ESI源,CUR:35psi,CAD:8psi,IS:5500psi,TEM:550℃,Gas1:55psi,Gas2:50psi,MRM模式。检测条件如表2所示,
表2检测条件
样本处理:取40µL血浆样品置于1.5mL离心管中,加入160µL乙腈溶液(TBTM100ng/mL),涡旋2min,13000rpm(8℃)离心6min,取上清液进样5µL检测。
样本分析:取空白SD大鼠血浆,依次加入PAR-1抑制剂标准系列溶液,配制标准样品。以待测物浓度为横坐标,待测物与内标物的峰面积比值为纵坐标,用加权最小二乘法(权重为1/x
(3)统计分析
根据血药浓度数据,采用Phoenix WinNonlin 8.1软件非房室模型,计算PAR-1抑制剂t
(4)血药浓度-时间数据
SD大鼠灌胃和静脉给药PAR-1抑制剂后,不同时间血浆中PAR-1抑制剂浓度的测定结果如表3所示,PAR-1抑制剂在SD大鼠体内平均血药浓度与时间的关系图,如图9所示,其中A表示给药浓度为1mg/kg时PAR-1抑制剂在SD大鼠体内平均血药浓度与时间的关系曲线,B表示药浓度为10mg/kg时PAR-1抑制剂在SD大鼠体内平均血药浓度与时间的关系曲线。
表3PAR-1抑制剂在SD大鼠体内平均血药浓度与时间的关系数据
(5)药代动力学参数及生物利用度
SD大鼠灌胃和静脉给药PAR-1抑制剂后,采用Phoenix WinNonlin 8.1软件非房室模型计算药代动力学参数,平均药代动力学参数见表4,其中给药浓度(10mg/kg)一列是SD大鼠灌胃给药PAR-1抑制剂后的数据,给药浓度(1mg/kg)一列是SD大鼠静脉给药PAR-1抑制剂后的数据。
表4PAR-1抑制剂在SD大鼠体内PK参数
(6)结论
由表4可以看出,大鼠静脉给药PAR-1抑制剂1mg/kg后,血浆中PAR-1抑制剂的Cmax为829.58±48.70ng/mL,AUC
由表4可以看出,大鼠灌胃给药PAR-1抑制剂10mg/kg后,血浆中PAR-1抑制剂的Cmax为1127.57±133.59ng/mL,AUC
市售同靶点药物沃拉帕沙,大鼠口服生物利用度33%(Discovery of a Novel,Orally Active Himbacine-Based Thrombin Receptor Antagonist (SCH 530348) withPotent Antiplatelet Activity. J. Med. Chem. 2008, 51, 3061–3064.),而本发明制备的PAR-1抑制剂的生物利用度56.12%,具有明显优势。
- 具有可调节输出的光电检测器装置和用于调节光电检测器装置的输出的方法
- 用于宽带信号的光电检测器
- 具有可调节输出的光电检测器装置和用于调节光电检测器装置的输出的方法