一类具有近红外吸收及发射性能的金属有机配合物及其制备方法
文献发布时间:2023-06-19 09:57:26
技术领域
本发明属于有机合成领域,具体涉及一类具有近红外吸收及发射性能的金属有机配合物及其制备方法。
背景技术
近红外吸收、发光材料在能源、通信、生物成像、传感、催化、和光电等一些领域具有重要的应用,特别是近红外成像技术在军事领域和民用用途中都有很大需求。典型的有机近红外材料大致分为金属有机配合物,离子染料,π共轭发色团和电荷转移发色团等。和传统无机材料相比,金属有机配合物具有材料来源丰富,光学特性可调节范围大等优点。寻找和设计新型的分子结构,以及对配体和配合物加以修饰,让配合物具有优良的光学性能,仍是研究近红外材料的热点。
发明内容
本发明的目的之一在于提供一类新型具有近红外吸收及发射性能的金属有机配合物,所述金属有机配合物具有近红外吸收及发射性能,在近红外区域有较大的红移,表现出了优良的光学特性。
本发明的目的之二在于提供一种上述具有近红外吸收及发射性能的金属有机配合物的制备方法。通常,金属有机配合物的合成方法有溶剂热法、水热法、界面扩散法、凝胶扩散法、微波法和紫外光照射法,与之相比,本发明所述方法简单快捷,条件温和,产率较高,可操作性强。
本发明实现目的之一采用以下技术方案:
一类具有近红外吸收及发射性能的金属有机配合物,所述金属有机配合物由含醛基的配体前驱体与噻吩或呋喃或吡咯或苯环或硒吩经缩合反应制得配体、配体再与Pd(DMSO)
所述含醛基的配体前驱体的结构式为下式L1~L20中任一种,
其中,
R
A为具有可提供配位原子的芳香结构体,其结构为以下任一种,其中E代表氨基(-NH
当所述含醛基的配体前驱体结构式为L1或L2或L5或L6或L7时,所述配体结构式包含下式S1~S12中的任一种,
其中,
X
R
R
R
A为具有可提供配位原子的芳香结构体,其结构为以下任一种,其中E代表氨基(-NH
当所述含醛基的配体前驱体结构式为L1或L2或L5或L6或L7时,所述配合物单体的结构式包含下式Y1~Y12任一种,
其中,
X
R
R
R
M为Sc、Ti、Fe、Co、Cu、Pd、Pt、Ir、Ru、Rh、Ni、Au具有配位功能的金属元素中的任一种;
A为具有可提供配位原子的芳香结构体,其结构为以下任一种,其中E代表氨基(-NH
当所述含醛基的配体前驱体结构式为L1或L2或L5或L6或L7时,所述金属有机配合物的结构式包含下式(Ⅰ)~式(Ⅻ)中任一种:
其中,
X
R
R
R
M为Sc、Ti、Fe、Co、Cu、Pd、Pt、Ir、Ru、Rh、Ni、Au具有配位功能的金属元素中的任一种;
A为具有可提供配位原子的芳香结构体,其结构为以下任一种,其中E代表氨基(-NH
本发明目的之二通过以下技术方案实现:
一种上述一类具有近红外吸收及发射性能的金属有机配合物的制备方法,所述方法主要包括以下步骤:
(1)以合成的含醛基的配体前驱体为原料,与噻吩或呋喃或吡咯或苯环或硒吩一同加入到冰乙酸溶剂中,加入催化剂,通过缩合反应得到含噻吩或呋喃或吡咯或苯环或硒吩单元的配体;
(2)将上述步骤(1)得到的配体与Pd(DMSO)
(3)将上述步骤(2)得到的配合物单体溶解于溶剂D中,并搅拌12小时-14小时,将上述搅拌后的溶解了配合物单体的溶剂D进行过滤,所得滤渣用甲醇洗涤,再于真空干燥箱中烘干,即得到配合物二聚体,所述配合物二聚体即为具有近红外吸收及发射性能的金属有机配合物。
优选地,所述步骤(3)中,将上述步骤(2)得到的配合物单体溶解于溶剂D后,再加入适量诱发剂使溶液呈碱性,然后再进行搅拌12小时-14小时,将搅拌后的加入诱发剂的溶解了配合物单体的溶剂D进行过滤,所得滤渣用甲醇洗涤,再烘干,即得到配合物二聚体,所述配合物二聚体即为具有近红外吸收及发射性能的金属有机配合物。
所述诱发剂为三乙胺或乙二胺。
所述步骤(1)中,所述催化剂为三氟化硼乙醚络合物、氢氟酸、三氟乙酸中的任一种。
所述步骤(3)中,所述溶剂D为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷酮、吡啶、苯胺中的任一种。
为更好地理解本发明,举例说明如下:
当选用的含醛基的配体前驱体结构式为L14,R
配体结构式:
配合物单体结构式:
配合物二聚体结构式:
与现有技术相比,本发明具有以下有益效果:
(1)本发明一类具有近红外吸收及发射性能的金属有机配合物的制备方法,以合成的一类联吡啶或邻二氮菲或吡啶席夫碱等为骨架,包含杂环单元的配体,与金属Pd/Pt等具有催化活性的金属生成配合物单体,方法简单、普适度高,可操作性强。
(2)在碱性条件下,诱导配合物单体生成配合物二聚体,该方法简单,条件温和,产率高,所得配合物二聚体较单体在近红外区域有较大的红移,从而表现出了优良的光学特性,可作为吸波材料应用于近红外成像领域、隐形墨水和防伪印刷、近红外条码隐形识别、等离子显示面板、染料敏化太阳能电池、近红外发光二极管等方面。
附图说明
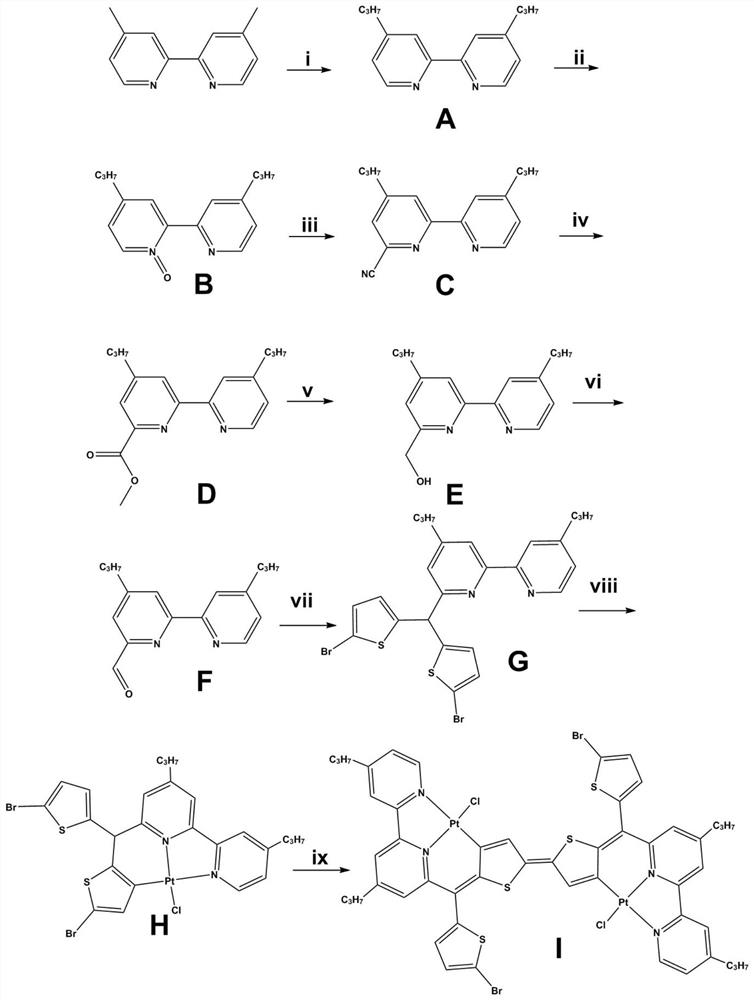
图1为本发明实施例(一)含噻吩单元2,2’-二联吡啶骨架的具有近红外吸收及发射性能的金属有机配合物合成流程示意图;
图2为本发明实施例(一)中化合物A的核磁氢谱图;
图3为本发明实施例(一)中化合物B的核磁氢谱图;
图4为本发明实施例(一)中化合物D的核磁氢谱图;
图5为本发明实施例(一)中化合物E的核磁氢谱图;
图6为本发明实施例(一)中化合物F的核磁氢谱图;
图7为本发明实施例(一)中化合物G的核磁氢谱图;
图8为本发明实施例(一)中化合物H的核磁氢谱图;
图9为本发明实施例(一)中化合物H和化合物I在二氯甲烷溶液中的紫外吸收和荧光发射光谱图;
图10为本发明实施例(二)以2,6-吡啶二羧酸作为原始原料合成具有近红外吸收及发射性能的含席夫碱结构的金属有机配合物的流程图;
图11为本发明实施例(二)中化合物J的核磁氢谱图;
图12为本发明实施例(二)中化合物K的核磁氢谱图;
图13为本发明实施例(二)中化合物L的核磁氢谱图;
图14为本发明实施例(二)中化合物M的核磁氢谱图;
图15为本发明实施例(二)中化合物N的核磁氢谱图;
图16为本发明实施例(二)中化合物O的核磁氢谱图。
具体实施方式
通过以下详细说明结合附图可以进一步理解本发明的特点和优点。所提供的实施例仅是对本发明方法的说明,而不以任何方式限制本发明揭示的其余内容。
下述实施例中,具体操作未涉及温度的,均是指室温下进行。
由于所选用原始化合物底物不同,在合成含醛基的配体前驱体的设计方案上有所不同。
本发明实施例(一)采用以2,2’-二联吡啶为原料,加入间氯过氧苯甲酸(m-CPBA),得到2,2’-二联吡啶-1-氧化物,然后加入氰化三甲基硅烷和苯甲酰氯,得到无色油状物6-氰基-2,2’-二联吡啶;所得产物在碱性条件下水解,然后在酸性环境下酯化;将上一步所得产物加入硼氢化钠经还原,提纯得到6-甲醇-2,2’-二联吡啶,6-甲醇-2,2’-二联吡啶经二氧化硒氧化,得到所述含醛基的配体前驱体6-醛基-2,2’-二联吡啶;再以含醛基的双配体前驱体6-醛基-2,2’-二联吡啶为原料,与2-溴噻吩共同加入到冰乙酸溶剂中,加入三氟化硼乙醚络合物,得到含噻吩单元的配体;将得到的含噻吩单元的配体与Pt(DMSO)
本实施例(二)以2,6-吡啶二羧酸为原料,在浓硫酸的环境下,加入乙醇反应得到2,6-二甲酸乙酯吡啶,在甲醇溶液中经硼氢化钠还原得到2,6-二羟基甲基吡啶,再以1,4-二氧六环作溶剂,经二氧化硒氧化,得到2,6-二醛基吡啶。将上一步所得产物与2-氯噻吩共同加入到冰乙酸溶剂中搅拌均匀,加入三氟化硼乙醚络合物,得到含醛基和噻吩单元产物6-(双(5-氯噻吩-2-基)甲基)吡啶甲醛;将所得产物在无水乙醇溶剂中,加入四苯乙烯氨基得到含席夫碱的配体,在将所得配体溶解到二氯甲烷中,滴加到Pd(DMSO)
实施例(一):以4,4’-二甲基-2,2’-二联吡啶作为原始原料详细说明各化合物合成步骤。
(1)化合物A的合成
250mL三口瓶加入2.76g(15mmol)4,4’-二甲基-2,2’-二联吡啶,通氮除氧,加入100mL四氢呋喃,降低温度到-78℃,滴加30mmol二异丙基氨基锂,搅拌2小时,滴加90mmol溴乙烷。滴加完毕缓慢升温至室温,搅拌12小时。加入氯化铵溶液猝灭反应,加入混合有机相(石油醚:乙酸乙酯=2:1)萃取,合并有机相,用无水硫酸钠干燥,减压除去溶剂,以流动相(石油醚:乙酸乙酯=10:1)过硅胶柱,得到无色油状物,即化合物A,产率95%。
(2)化合物B的合成
将所得A化合物溶于15mL二氯甲烷中,将间氯过氧苯甲酸(m-CPBA)(2.03g,85%,10mmol)溶于20mL二氯甲烷并于5℃控温下滴加到上述溶液中,滴加完毕室温搅拌12小时,于0℃下加入Na
(3)化合物C的合成
氮气氛围下,将所得B物质5.6mmol和氰化三甲基硅烷3.16mL(23.7mmol)加入20mL无水四氢呋喃中,于0℃下加入苯甲酰氯(1.29mL,11.2mmol)。滴加完毕,室温搅拌过夜,小心加入Na
(4)化合物D的合成
将上部所得C的混合物和0.33g(6.1mmol)甲醇钠溶于15mL干燥甲醇中,室温搅拌过夜,加入冰乙酸(0.367g,6.1mmol)和适量碳酸氢钠,搅拌15min,减压除去有机相,剩余物用乙酸乙酯和碳酸氢钠溶液(8mL+2mL)溶解,合并有机相并旋干溶剂,剩余物加入甲醇水溶液,用5%浓硫酸调节pH≈1,搅拌2小时,用氢氧化钠调节溶液pH≈9,再次用乙酸乙酯萃取,合并有机相,以流动相(石油醚:乙酸乙酯=10:1)过硅胶柱,得到无色油状物,即化合物D,产率91%。
(5)化合物E的合成
将上一步所得产物D(3.25mmol)加入到甲醇/四氢呋喃(20/5mL)溶液中,分批加入硼氢化钠(0.756g,20mmol),室温搅拌2小时,加入饱和碳酸氢钾溶液搅拌30min,用乙酸乙酯萃取,合并有机相并减压除去,所的白色固体无需进一步提纯,即化合物E,产率87%。
(6)化合物F的合成
取二氧化硒(0.145g,1.3mmol),加入1,4-二氧六环溶剂6mL,升温到65℃并搅拌30min,将所得化合物E(0.7g,2.6mmol)溶于9mL 1,4-二氧六环中并缓慢滴加到上述混浊液中,升温110℃回流2小时,降至室温,过滤掉灰色沉淀,并用乙酸乙酯冲洗滤渣,合并有机相,减压除去溶剂,以流动相(石油醚:乙酸乙酯=4:1)过硅胶柱,得到白色固体,即化合物F(含醛基的配体前驱体),产率91%。
(7)化合物G的合成
将所得化合物F(1mmol)和2-溴噻吩(10mmol)加入5mL冰乙酸中,搅拌10min,加入三氟化硼乙醚络合物1ml,升温至50℃,点板检测至无化合物F点,加入20mL水猝灭反应,分3次加入1:1的石油醚和乙酸乙酯混合溶液100mL萃取,合并有机相,经饱和碳酸钠溶液和饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压除去溶剂,以流动相(石油醚:乙酸乙酯=10:1)过硅胶柱,得到黄色油状物,即化合物G(含噻吩单元的配体),产率93%。
(8)化合物H的合成
取Pt(DMSO)
(9)化合物I的合成
取配合物H(0.081g,0.1mmol)加入到20mL N,N-二甲基甲酰胺溶液中,加0.5mL三乙胺,搅拌12小时-14小时,过滤,所得滤渣用甲醇洗涤,于真空干燥箱中烘干得到褐色粉末,即化合物I(配合物二聚体)。
实施例(二):以2,6-吡啶二羧酸作为原始原料详细说明各化合物合成步骤。
(1)化合物J的合成
500mL三口瓶加入无水乙醇300mL,2,6-吡啶二羧酸33.4g,搅拌均匀后再加入浓硫酸10mL,90℃回流5小时,减压旋走乙醇,残渣中加入200mL水,用碳酸钠调节pH为中性,加入混合有机相(石油醚:乙酸乙酯=2:1)150mL萃取,合并有机相,用无水硫酸钠干燥,减压除去溶剂,经硅胶柱净化,得白色晶体2,6-二羟基甲基吡啶,即化合物J,产率95%。
(2)化合物K的合成
500mL单口瓶加入无水甲醇300mL,加入2,6-二甲酸乙酯吡啶17.8g,搅拌均匀,分批次加入硼氢化钠3.6g,搅拌3小时,分批次加入无水氯化钙10.7g,室温搅拌4小时,减压去除溶剂,加入100mL饱和碳酸钾溶液,搅拌4小时,去掉溶剂水,减压烘干,快速过硅胶柱得9.88g白色固体,即化合物K,产率94.5%。
(3)化合物L的合成
取2,6-二羟基甲基吡啶2.8g,溶于250mL三口瓶中,加入1.4-二氧六环80mL,搅拌溶解,加入二氧化硒粉末2.66g,105℃回流2小时,待瓶内冷至室温,过滤掉固体,减压旋走液体,以混合有机相(石油醚:乙酸乙酯=8:1)过硅胶柱,得灰白色晶体,即化合物L,产率80%。
(4)化合物M的合成
取2,6-二醛基吡啶0.54g,与2-氯噻吩0.95g共同加入到10mL冰乙酸溶剂中搅拌均匀,加入三氟化硼乙醚络合物5mL,得到红色粘稠物0.33g,即化合物M(含醛基的配体前驱体);产率23.3%。
(5)化合物N的合成
将上步所得产物71mg溶在无水乙醇溶剂中,加入四苯乙烯氨基76mg,加热50℃2小时,得到含席夫碱的配体,即化合物N,直接旋干溶剂,此步不做纯化直接进行下一步反应。
(6)化合物O的合成
取66mg Pd(DMSO)
(7)化合物P的合成
将得到的配合物单体82.5mg溶解于20mL N,N-二甲基甲酰胺溶液中,加0.5mL三乙胺,搅拌12小时-14小时,过滤,所得滤渣用甲醇洗涤,于真空干燥箱中烘干得到配合物二聚体,即化合物P。
- 一类具有近红外吸收及发射性能的金属有机配合物及其制备方法
- 一类具有近红外吸收及发射性能的金属有机配合物及其制备方法