检测基因突变及表达量的方法及装置
文献发布时间:2023-06-19 09:58:59
技术领域
本发明涉及生物学技术领域,具体而言,涉及一种检测基因突变及表达量的方法及装置。
背景技术
基因突变是指基因组DNA分子发生的突然的、可遗传的变异现象(genemutation)。从分子水平上看,基因突变是指基因在结构上发生碱基对组成或排列顺序的改变。基因虽然十分稳定,能在细胞分裂时精确地复制自己,但这种稳定性是相对的。在一定的条件下基因也可以从原来的存在形式突然改变成另一种新的存在形式,就是在一个位点上,突然出现了一个新基因,代替了原有基因,这个基因叫做突变基因。于是后代的表现中也就突然地出现祖先从未有的新性状。
基因突变是生物进化的重要因素之一,所以研究基因突变除了本身的理论意义以外还有广泛的生物学意义。有的基因突变是由于染色体发生结构变异形成。在自然条件或人为因素的影响下,染色体发生的结构变异主要有:缺失、重复、倒位和易位,其中,基因融合也是染色体发生结构变异的一种。
随着测序技术的发展,成本的降低,在人类健康领域,人全基因组测序必将成为今后的主流趋势,精准医疗将是测序的最终目的。准确注释人类基因组的变异是实现精准医疗的必要手段。
目前常规方法一般利用全基因组测序WGS或DNA panel进行SNV、CNV和融合的检测。但是在DNA水平检测突变,不能反映突变在转录水平的真实表现。
发明内容
本发明旨在提供一种检测基因突变及表达量的方法及装置,检测基因突变及表达量。
本发明基于RNA靶向测序(targeted RNA sequencing)的基因突变(包括基因融合)及表达量检测方法,能够高效富集肿瘤相关基因所表达的RNA转录本,并完整检测这些基因表达的转录本上的包含融合、单碱基与多碱基替换(SNV/MNV)、插入缺失突变(indel)等多种突变类型,同时分析这些肿瘤驱动基因在肿瘤组织中的表达量。
现有技术中一般利用全基因组测序WGS或DNA panel进行SNV、CNV和融合的检测。传统方法在DNA水平检测突变,不能反映突变在转录水平的真实表现,利用RNA进行突变检测,功能相关性更强。例如两个SNV突变频率都是1%,但因为表达量不同,突变的临床影响会有差异。本发明不仅能够检测RNAseq常规的基因表达量、基因融合,还能够检测DNApanel的SNV和CNV,并且能够检测各种突变的表达量。实现一次检测,覆盖所有突变类型和相对表达量。
本发明系统进行RNA panel靶向目标基因,相对RNAseq检测全转录组,测序费用更低,并且能显著富集目标区域,特别是对于低表达的基因或突变,检测灵敏度更高。并且RNA靶向测序panel设计只需要覆盖外显子区域,相比DNA panel设计需要覆盖外显子和内含子,更节省探针和测序成本,更适用于临床试剂盒开发。
为了实现上述目的,根据本发明的一个方面,提供了一种检测基因突变及表达量的方法。该方法包括以下步骤:S1,提取待检测样本RNA,将待检测样本RNA打断,进行反转录,得到cDNA;S2,采用cDNA通过末端修复、接头连接和文库富集步骤构建基因文库;S3,利用捕获探针与目标区域特异性杂交从基因文库中捕获并富集目标基因;S4,利用高通量测序仪测序,获得RNA靶向测序数据;S5,分析RNA靶向测序数据中基因突变及表达量的变化;S5具体包括:S51,基因表达量分析:使用RPKM方法定量评估检测样本中目标基因的表达量;S52,基因过表达分析:调取基线样本群体,分析目标基因的RPKM值分布,确定目标基因表达量高低的阈值,根据待检测样本的目标基因的RPKM值,判断待检测样本的目标基因是否为过表达;S53,基因融合分析:过滤掉属于同一基因家族的融合基因、属于同一旁系同源组的融合基因、来源于同一基因模型的融合基因,根据阈值过滤未满足条件的融合基因,获得检测样本中融合基因;S54,融合突变相对表达量分析:根据看家基因的表达定量结果和S53中获得的基因融合分析的结果进行表达量校正标准化,得到融合基因的相对融合表达量;S55,单核苷酸变异分析:通过基因比对确定变异单核苷酸;S56,单核苷酸变异表达量分析:根据单核苷酸变异分析的结果和看家基因的表达定量结果和序列比对的统计结果,进行单核苷酸变异的表达定量分析,得到单核苷酸变异的表达量。
进一步地,S5还包括:过滤掉低质量的测序数据和含有接头序列的reads并进行质控后,得到符合标准的数据再进行分析RNA靶向测序数据中基因突变及表达量的变化,其中,质控步骤包括:将过滤掉低质量的测序数据和含有接头序列的reads后得到的测序数据比对到参考基因组,得到序列比对结果,对比对结果进行质量控制评估,符合如下三项指标后进行后续分析:1)序列回帖比对率,阈值,>=80%;2)目标区域数据量,阈值,>=2M;3)表达的看家基因个数>=4。
进一步地,S53中,阈值如下表:
。
进一步地,S54中,融合表达量校正标准化采用的标准化公式如下:
其中,SeedReads+RescueReads表示跨融合断点的reads,HKA表示看家基因A,HKB表示看家基因B,HKC表示看家基因C,count表示测序序列与参考基因组比对上的序列数目,length表示测序序列与参考基因组比对上的序列长度。
进一步地,S4中采用双端或单端模式进行测序。
进一步地,S56中,单核苷酸变异的表达量计算公式为:
其中,Gene Average Depth表示基因的平均深度;
ALT count表示突变的深度;
HK_expression_Coeffient表示根据样本中看家基因的表达量与标准品中看家基因的表达量计算表达量变化系数。
根据本发明的另一个方面,提供一种检测基因突变及表达量的装置。该装置包括:RNA提取模块,设置为提取待检测样本RNA,将待检测样本RNA打断,进行反转录,得到cDNA;基因文库构建模块,设置为采用cDNA通过末端修复、接头连接和文库富集步骤构建基因文库;目标基因富集模块,设置为利用捕获探针与目标区域特异性杂交从基因文库中捕获并富集目标基因;测序模块,设置为利用高通量测序仪测序,获得RNA靶向测序数据;分析模块,设置为分析RNA靶向测序数据中基因突变及表达量的变化;分析模块具体包括:基因表达量分析子模块,设置为使用RPKM方法定量评估检测样本中目标基因的表达量;基因过表达分析子模块:设置为调取基线样本群体,分析目标基因的RPKM值分布,确定目标基因表达量高低的阈值,根据待检测样本的目标基因的RPKM值,判断待检测样本的目标基因是否为过表达;基因融合分析子模块:设置为过滤属于同一基因家族的融合基因、属于同一旁系同源组的融合基因、来源于同一基因模型的融合基因,根据阈值过滤掉未满足条件的融合基因,获得检测样本中融合基因;融合突变相对表达量分析子模块:设置为根据看家基因的表达定量结果和基因融合分析子模块中获得的基因融合分析的结果进行表达量校正标准化,得到融合基因的相对表达量;单核苷酸变异分析子模块:设置为通过基因比对确定变异单核苷酸;单核苷酸变异表达量分析子模块:设置为根据单核苷酸变异分析的结果和看家基因的表达定量结果和序列比对的统计结果,进行单核苷酸变异的表达定量分析,得到单核苷酸变异的表达量。
进一步地,分析模块还包括过滤子模块:设置为过滤掉低质量的测序数据和含有接头序列的reads并进行质控后,得到符合标准的数据再进行分析RNA靶向测序数据中基因突变及表达量的变化,其中,质控包括:将过滤掉低质量的测序数据和含有接头序列的reads后得到的测序数据比对到参考基因组,得到序列比对结果,对比对结果进行质量控制评估,符合如下三项指标后进行后续分析:1)序列回帖比对率,阈值,>=80%;2)目标区域数据量,阈值,>=2M;3)表达的看家基因个数>=4。
进一步地,基因融合分析子模块中,阈值如下表:
。
进一步地,融合突变表达量分析子模块中,表达量校正标准化采用的标准化公式如下:
其中,SeedReads+RescueReads表示跨融合断点的reads,HKA表示看家基因A,HKB表示看家基因B,HKC表示看家基因C,count表示测序序列与参考基因组比对上的序列数目,length表示测序序列与参考基因组比对上的序列长度。
进一步地,测序模块中采用双端或单端模式进行测序。
进一步地,单核苷酸变异表达量分析子模块中,单核苷酸变异的表达量计算公式为:
其中,Gene Average Depth表示基因的平均深度;
ALT count表示突变的深度;
HK_expression_Coeffient表示根据样本中看家基因的表达量与标准品中看家基因的表达量计算表达量变化系数。
应用本发明的技术方案,以待检测样本总RNA或mRNA为检测对象,采用RNA靶向测序(targeted RNA sequencing)的方法,能够高效富集肿瘤相关基因表达的RNA转录本,并完整检测这些基因表达的转录本上的包含融合、单碱基与多碱基替换(SNV/MNV)、插入缺失突变(indel)等多种突变类型,同时分析这些肿瘤基因在肿瘤组织中的表达量。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
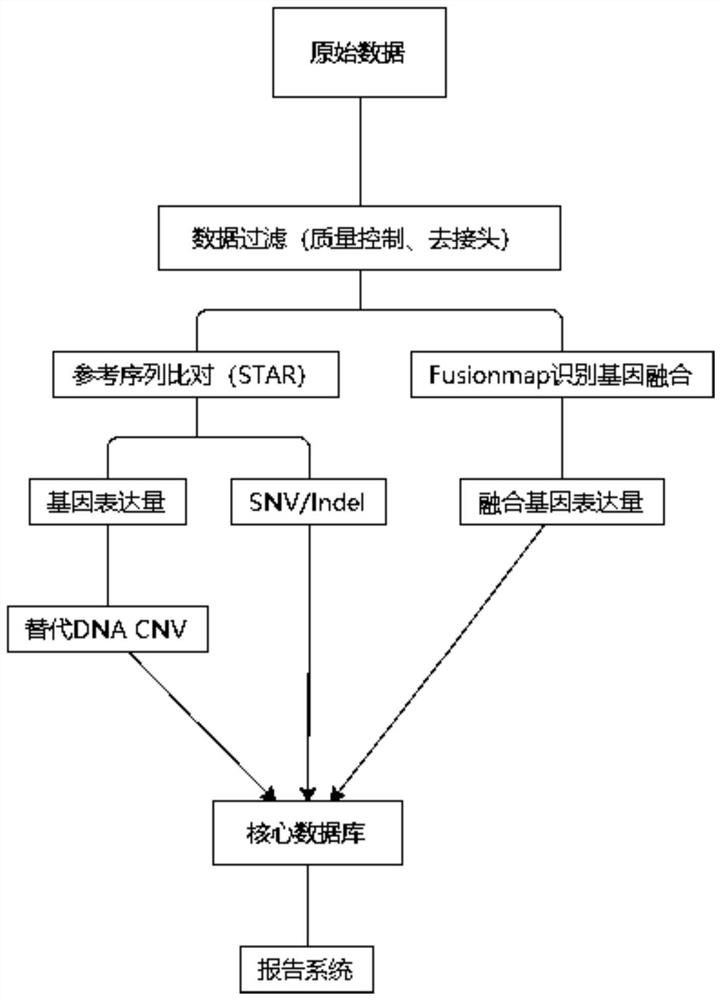
图1示出了根据本发明实施方式的检测基因突变及表达量的方法的流程示意图;
图2示出了实施例中RNA Panel和RNAseq测序基因表达量的相关性示意图;
图3示出了实施例中重要的癌症驱动基因RNA Panel和RNAseq测序基因表达量的相关性示意图。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
基于液相探针捕获的RNA靶向测序相较于传统RNA-seq,能够以超高深度的测序水平覆盖主要肿瘤驱动基因所表达的转录本以及融合、激活突变、耐药突变,并且保留所有转录本相对于看家基因的相对表达量信息。并且由于仅覆盖少数肿瘤目标基因,测序数据量少、成本低,更适用于临床检测试剂盒开发。
相较于DNA,RNA更靠近下游功能蛋白,更适于阐释细胞功能通路的活性状态。但既往很少用RNA检测体细胞突变SNV/Indel,也不会使用RNA表达量替代DNA的拷贝数分析,主要是因为存在一些影响检测准确度的因素,这些因素主要包括:1)单链;2)反转错误;3)RNA质量引起噪音;4)受表达量影响,非表达的突变无法检测;5)转录水平的突变导致不一致等,而针对这些技术问题,本发明进行了技术改进,主要包括:1)通过锚定SNV/Indel的基因列表和优化RNA SNV突变的过滤标准,提高了激活突变与耐药突变SNV/indel的准确度;2)突变等位基因转录本与野生型等位基因的相对表达量;3)融合突变与耐药点突变的顺式分析以及相对表达量关联分析;4)建立肿瘤驱动基因拷贝数增加与表达量的对应关系,可以通过RNA表达量替代DNA的拷贝数分析。
另外,现有技术中DNA panel在融合检测中有漏检问题(原因:DNA水平复杂结构变异导致的RNA水平融合,或DNA panel探针没有覆盖断点等),因此融合检测需要RNA方法的补充。由于实体肿瘤靶向药物的有功能的突变(actionable mutations)主要以SNV/indel/CNV为主,因此临床样本NGS初筛以DNA方法为主,辅助以RNA或者FISH/IHC等复核方法,造成流程复杂,样本需求量高,成本高等问题。在本发明一典型的实施例中,本发明以一个高通量测序(NGS)捕获panel内涵盖肿瘤主要TKI靶向药物的所有突变类型,大大简化操作流程、节省样本、成本减少情况下提高测序深度、融合突变与激活点突变准确度提高、并且能够获得驱动基因的表达量以及突变等位基因的特异性表达量等DNA panel不能提供的信息,为肿瘤靶向药物的选择提供辅助参考。
本发明基于RNA靶向测序(targeted RNA sequencing)的基因突变(包括基因融合)及表达量检测方法,能够高效富集肿瘤相关基因所表达的RNA转录本,并完整检测这些基因表达的转录本上的包含融合、单碱基与多碱基替换(SNV/MNV)、插入缺失突变(indel)等多种突变类型,同时分析这些肿瘤驱动基因在肿瘤组织中的表达量。
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
在本发明实施方式中,RNA靶向测序数据的获取方法可以包括以下步骤:从FFPE样本中提取总RNA,无需去除核糖体RNA,将样本总RNA打断,并反转录为cDNA;通过包括末端修复、接头连接和文库富集步骤构建基因文库;捕获探针利用能够与目标区域特异性杂交的核酸探针从所构建的cDNA文库中捕获并富集目标基因;利用高通量测序仪以双端模式进行测序,由此获取RNA靶向测序数据。
根据本发明一种典型的实施方式,提供一种检测基因突变及表达量的方法。参见图1,该方法包括以下步骤:S1,提取待检测样本RNA,将待检测样本RNA打断,进行反转录,得到cDNA;S2,采用cDNA通过末端修复、接头连接和文库富集步骤构建基因文库;S3,利用捕获探针与目标区域特异性杂交从基因文库中捕获并富集目标基因;S4,利用高通量测序仪测序,获得RNA靶向测序数据;S5,分析RNA靶向测序数据中基因突变及表达量的变化;S5具体包括:S51,基因表达量分析:使用RPKM方法定量评估检测样本中目标基因的表达量;S52,基因过表达分析:调取基线样本群体,分析目标基因的RPKM值分布,确定目标基因表达量高低的阈值,根据待检测样本的目标基因的RPKM值,判断待检测样本的目标基因是否为过表达;S53,基因融合分析:过滤属于同一基因家族的融合基因、属于同一旁系同源组的融合基因、来源于同一基因模型的融合基因,根据阈值过滤掉未满足条件的融合基因,获得检测样本中融合基因;S54,融合突变相对表达量分析:根据看家基因的表达定量结果和S53中获得的基因融合分析的结果进行表达量校正标准化,得到融合基因的相对表达量;S55,单核苷酸变异分析:通过基因比对确定变异单核苷酸;S56,单核苷酸变异表达量分析:根据单核苷酸变异分析的结果和看家基因的表达定量结果和序列比对的统计结果,进行单核苷酸变异的表达定量分析,得到单核苷酸变异的表达量。
具体的,在本发明一实施方式中,S5还包括:过滤掉低质量的测序数据和含有接头序列的reads并进行质控后,得到符合标准的数据再进行分析RNA靶向测序数据中基因突变及表达量的变化,其中,质控步骤包括:将过滤掉低质量的测序数据和含有接头序列的reads后得到的测序数据比对到参考基因组,得到序列比对结果,对比对结果进行质量控制评估,符合如下三项指标后进行后续分析:1)序列回帖比对率,阈值,>=80%;2)目标区域数据量,阈值,>=2M;3)看家基因表达个数>=4。
优选地,S53中,融合阈值如下表1:
表1融合突变阈值标准
优选地,S54中,表达量校正标准化采用的标准化公式如下:
其中,SeedReads+RescueReads表示跨融合断点的reads,HKA表示看家基因A,HKB表示看家基因B,HKC表示看家基因C,count表示测序序列与参考基因组比对上的序列数目,length表示测序序列与参考基因组比对上的序列长度。
优选的,S56中,单核苷酸变异的表达量计算公式为:
其中,Gene Average Depth表示基因的平均深度;
ALT count表示突变的深度;
HK_expression_Coeffient表示根据样本中看家基因的表达量与标准品中看家基因的表达量计算表达量变化系数。
为了更方便的实施本发明的上述方法,根据本发明一种典型的实施方式,提供一种检测基因突变及表达量的装置。该装置包括RNA提取模块、基因文库构建模块、目标基因富集模块、测序模块和分析模块,其中,RNA提取模块设置为提取待检测样本总RNA或mRNA,将待检测样本RNA打断,进行反转录,得到cDNA;基因文库构建模块设置为采用cDNA通过末端修复、接头连接和文库富集步骤构建基因文库;目标基因富集模块设置为利用捕获探针与目标区域特异性杂交从基因文库中捕获并富集目标基因;测序模块设置为利用高通量测序仪测序,获得RNA靶向测序数据;分析模块设置为分析RNA靶向测序数据中基因突变及表达量的变化;其中,分析模块具体包括基因表达量分析子模块、基因过表达分析子模块、基因融合分析子模块、融合突变表达量分析子模块、单核苷酸变异分析子模块和单核苷酸变异突变表达量分析子模块,基因表达量分析子模块设置为使用RPKM方法定量评估检测样本中目标基因的表达量;基因过表达分析子模块设置为调取基线样本群体,分析目标基因的RPKM值分布,确定目标基因表达量高低的阈值,根据待检测样本的目标基因的RPKM值,判断待检测样本的目标基因是否为过表达;基因融合分析子模块设置为过滤掉属于同一基因家族的融合基因、属于同一旁系同源组的融合基因、来源于同一基因模型的融合基因,根据阈值过滤未满足条件的融合基因,获得检测样本中融合基因;融合突变相对表达量分析子模块设置为根据看家基因的表达定量结果和基因融合分析子模块获得的基因融合分析的结果进行表达量校正标准化,得到融合基因的相对表达量;单核苷酸变异分析子模块设置为通过基因比对确定变异单核苷酸;单核苷酸变异表达量分析子模块设置为根据单核苷酸变异分析的结果和看家基因的表达定量结果和序列比对的统计结果,进行单核苷酸变异的表达定量分析,得到单核苷酸变异的表达量。
具体的,在本发明一实施方式中,分析模块还包括过滤子模块:设置为过滤掉低质量的测序数据和含有接头序列的reads并进行质控后,得到符合标准的数据再进行分析RNA靶向测序数据中基因突变及表达量的变化,其中,质控包括:将过滤掉低质量的测序数据和含有接头序列的reads后得到的测序数据比对到参考基因组,得到序列比对结果,对比对结果进行质量控制评估,符合如下三项指标后进行后续分析:1)序列回帖比对率,阈值,>=80%;2)目标区域数据量,阈值,>=2M;3)看家基因表达个数>=4。
优选地,基因融合分析子模块中,阈值如表1。
优选地,融合突变表达量分析子模块中,表达量校正标准化采用的标准化公式如下:
其中,SeedReads+RescueReads表示跨融合断点的reads,HKA表示看家基因A,HKB表示看家基因B,HKC表示看家基因C,count表示测序序列与参考基因组比对上的序列数目,length表示测序序列与参考基因组比对上的序列长度。
优选地,单核苷酸变异突变表达量分析子模块中,单核苷酸变异的表达量计算公式为:
其中,Gene Average Depth表示基因的平均深度;
ALT count表示突变的深度;
HK_expression_Coeffient表示根据样本中看家基因的表达量与标准品中看家基因的表达量计算表达量变化系数。
现有技术中一般利用全基因组测序WGS或DNA panel进行SNV、CNV和融合的检测。传统方法在DNA水平检测突变,不能反映突变在转录水平的真实表现,利用RNA进行突变检测,功能相关性更强。例如两个SNV突变频率都是1%,但因为表达量不同,突变的临床影响会有差异。本发明不仅能够检测RNAseq常规的基因表达量、基因融合,还能够检测DNApanel的SNV和CNV,并且能够检测各种突变的表达量。实现一次检测,覆盖所有突变类型和相对表达量。
本发明系统进行RNA panel靶向目标基因,相对RNAseq检测全转录组,测序费用更低,并且能显著富集目标区域,特别是对于低表达的基因或突变,检测灵敏度更高。并且RNA靶向测序panel设计只需要覆盖外显子区域,相比DNA panel设计需要覆盖外显子和内含子,更节省探针和测序成本,更适用于临床试剂盒开发。
下面将结合实施例进一步说明本发明的有益效果。
实施例
一、实验:
1.RNA提取:
使用肺癌患者石蜡包埋的病理切片,采用Qiagen的RNeasy FFPE Kit(Cat No./ID:73504)进行总RNA提取。使用Qubit RNA HS对RNA的含量进行测定,使用Labchip检测对RNA进行质控。
2.杂交前核苷酸文库制备:
使用ABclonal公司的mRNA-seq Lib Prep Module for illumina进行核苷酸文库构建:包括cDNA反转录、片段化、末端修复、接头连接、文库富集等步骤。所构建文库使用Agencourt AMpure XP磁珠纯化后,使用Qubit 3.0以及Agilent 2100毛细管电泳用于浓度检测和质控。
3.探针捕获杂交:
根据选取的36个靶基因(ALK、ESR1、FGFR1、NRG1、RET、ERG、BRAF、ETV1、FGFR2、NTRK1、ROS1、EWSR1、CD74、ETV4、FGFR3、NTRK2、SLC34A2、MET、EGFR、ETV5、FGFR4、NTRK3、SLC45A3、PPARG、EML4、ETV6、KIF5B、PDGFRA、TPM3、PDGFRB、SFT2D3、CNTF、EPM2A、NOL10、HEATR4和RPGRIP1),根据其转录本序列设计non-overlapping的平铺探针序列,探针5’端用生物素标记。将2ug制备好的杂交前文库与5uL Human Cot DNA(IDT),2uL xGen UniversalBlockers-TS Mix混合,使用真空离心浓缩仪蒸干(60℃,约20min-1hr)后,再复溶于杂交液中,室温孵育10min后,移至PCR仪中65℃杂交16h。将捕获过夜的杂交产物与链霉亲和素磁珠混合,在PCR仪中孵育45min后,用清洗液对磁珠进行清洗。将洗脱产物进行下一步PCR扩增实验,后续用Agencourt AMPure XP磁珠纯化,使用Qubit 3.0以及Agilent 2100毛细管电泳进行浓度测定和质控。
4.高通量测序:使用Illumina Nextseq、Novaseq等,以双端模式进行测序。
二、测序数据分析:
根据RNA panel捕获reads进行上机测序,得到原始测序下机序列,使用Trimmomatic-0.36对序列进行如下处理得到高质量的测序序列
a)除低质量的测序序列
b)去掉含有接头序列的reads
将高质量的测序序列(标准采用本领域通用标准)使用STAR比对到参考基因组,得到序列比对结果,并对比对结果进行质量控制评估,符合如下表2指标进行下一步分析(包括:基因表达量分析、基因融合分析、融合突变相对表达量分析、SNV分析、SNV突变表达量分析)。
表2 RNA panel下机质控标准
1.基因表达量分析
根据序列比对结果和参考基因组的注释文件,使用RPKM方法定量评估基因表达量,RPKM公式如下:
Total exon reads:比对到基因所有外显子的序列数目,使用FeatureCounts软件根据基因注释文件和比对结果进行评估。
Mapped reads(millions):比对到基因组上所有序列的数目,根据比对结果的统计结果得到。
Exon length(KB):基因的外显子长度,根据基因组的注释文件计算得到。
2.基因融合分析
将高质量的测序序列使用FusionMap用于识别基因融合,得到初步的基因融合结果,根据基因融合结果按照以下规则进行过滤:
1)基因融合结果中Filter标识为空,表示意思如下:
a)过滤掉属于同一基因家族的融合基因;
b)过滤掉属于同一旁系同源组(由Ensembl v74定义而来)的融合基因;
c)过滤掉来源于同一基因模型的融合基因。
2)根据制定阈值过滤掉未满足条件的融合基因,阈值标准如下表3:
表3融合突变阈值标准
3.融合突变表达量分析
根据识别到基因融合结果和看家基因的表达定量结果进行校正标准化,得到融合基因的融合表达量结果,标准化公式如下:
其中,SeedReads+RescueReads表示跨融合断点的reads,HKA表示看家基因A,HKB表示看家基因B,HKC表示看家基因C,count表示测序序列与参考基因组比对上的序列数目,length表示测序序列与参考基因组比对上的序列长度。例如,HKA
4.SNV分析
分析流程:
1)测序数据分析比对,得到bam数据文件;
2)使用VarDict caller抓取出与参考基因组(hg19)比对后的突变位点和插入缺失区域,结果文件为VCF格式;
3)对VCF文件使用ANNOVAR注释,并对部分注释不准确位点再使用transvar注释,得到全部结果文件;此处使用transvar矫正注释结果,结果更加准确全面;
4)合并两次结果;对合并文件进行正负链矫正并统计reads数和freq;
此处对链偏好性的矫正,重新矫正结果注释;
5)使用证据位点数据库过滤注释和转录本支持选择;
基因突变及基因数据库模块:
a)整理出不同肿瘤,疾病高发的基因,建立一个明确的靶向位点及化疗药物相关性的热点基因列表;
b)公共数据库,包括EXAC/千人/gnomAD/HGMD/OMIM/cosmic;
转录本选择:判断是否是用药位点转录本/Clinvar中致病性位点/Transvar结果中是否有该转录本/是否有位于内含子非splice/经典转录本/是否在外显子区;
7)根据验证得到阈值标准对合并结果进行过滤,得到最终结果;
针对不同的基因和热点进行了独立验证和大量样本平行验证,对结果进行可视化判断矫正,计算出最优性能后逆推出一套质控阈值标准;
过滤标准:
a)过滤测序深度小于10的突变位点;
b)过滤掉黑名单中的突变,保留白名单中的突变;
c)过滤掉forward和reverse中没有reads支持的突变;
d)过滤掉freq和support reads不符合要求的突变。
5.SNV突变表达量分析
根据SNV结果,以看家基因的表达定量结果和序列比对的统计结果,进行SNV的表达定量分析,得到SNV的表达量。
Gene Average Depth:基因的平均深度
HK_expression_Coeffient:根据样本中看家基因的表达量与标准品中看家基因的表达量计算表达量变化系数;
三、结果部分:
1.RNA panel检测基因融合的准确性
通过配对DNA样本靶向测序,对RNA样本融合基因检测进行一致性验证,性能见下表4。在57例DNA融合阴性样本中,52例RNA融合检出阴性,5例RNA融合检出阳性。因此,DNA与RNA融合检出结果的阴性一致性为52/57=91.23%。RNA检出融合的5例样本均用IGV确认断点真实性,检出条数均高于过滤标准,其中3例利用一代测序确认融合真实存在,说明DNA存在融合漏检可能。在16例临检DNA融合阳性样本中,16例RNA均检出阳性,且检出融合形式与DNA一致,RNA存在融合可变剪切检出。RNA检测与DNA检测的阳性一致率为16/16=100%,阴性一致率为52/57=91.23%。
表4RNA panel检测融合性能
2.RNA panel检测SNV的准确性
考察RNA panel所覆盖的oncogene激活突变以及融合继发耐药一二级突变位点(共11个基因,226个snv位点),在DNA靶向测序与配对RNA样本snv检出结果的一致性。共计40例非小细胞肺癌临床样本,29例DNA与RNA均未检出,11例共检出样本,突变主要集中在EGFR基因上。RNA与DNA检出考察范围snv结果的阳性一致率与阴性一致率均为100%。结果见表5。
表5 RNA panel检测SNV性能
3.RNA panel检测基因表达量的准确性
30例FFPE构建RNA文库,然后分别进行RNAseq测序和使用RNA panel捕获后测序,分析RNAseq和RNA panel检测基因表达量的一致性,结果显示两种方法对于panel包含的所有基因,表达量检测的一致性R值>0.8。结果见图2RNAseq和RNA panel基因表达量的相关性结果。
对于panel中重要的癌症驱动基因,比如ALK、MET、NTRK、EGFR等,RNAseq和RNApanel基因表达量的R值>0.9。结果见图3。
4.通过RNA表达量替代DNA的拷贝数分析
165例FFPE样本使用RNA panel捕获后测序,统计EGFR基因的表达量rpkm值分布,确定EGFR表达量的阈值,将EGFR表达量top 10%且有剩余切片的样本,进行免疫组化(IHC)实验和DNA靶向测序。实验结果表明,EGFR基因的表达量和免疫组化即蛋白水平的结果,比DNA CNV结果和免疫组化结果一致性更好。结果见表6。
表6 RNA panel检测CNV性能
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 检测基因突变及表达量的方法及装置
- 检测基因融合突变的方法、装置、存储介质、处理器及转录组数据表达量标准化的方法