一种防己黄芪汤的体内外多成分分析方法
文献发布时间:2023-06-19 11:19:16
技术领域
本发明涉及样品中多成分的检测与分析,具体地说,涉及一种防己黄芪汤的体内外多成分分析方法。
背景技术
防己黄芪汤是我国古代经方,出自汉·张仲景《金匮要略·痉湿病脉症并治》篇与《金匮要略·水气病脉症并治》篇中,由防己、黄芪、甘草、白术组成。方中防己苦涩辛散,祛风除湿利水消肿,黄芪补气健脾补肺,尤能固表行水,二药共为君药,补气除湿利水,祛风散邪固表。白术补脾燥湿,既助黄芪补气固表,又助防己祛湿利水,为方臣药。甘草益气健脾,调和诸药,为方中佐使药。诸药合用具有益气祛风、健脾利水之功。组方药味虽简单,但配伍却精炼,是治疗风水、风湿症的经典方。防己黄芪汤在临床上常用于治疗包括慢性肾小球肾炎、心源性水肿、风湿性关节炎等属风水、风湿的疾病。
防己黄芪汤复方的物质基础研究:目前吕翠平等
综上,目前对于防己黄芪汤复方的质量控制多以单独检测生物碱类、黄酮类、皂苷或糖类的一种或几种混合物,并未见从防己黄芪汤的五大类化学成分群(生物碱类、皂苷类、黄酮类、内酯类和五环三萜类)全面分析该复方的化学成分种类及含量的报道,也未见从化合物群角度检测入血成分的系统分析方法报道,因此,本发明针对该方进行反复和创新性优化实验,通过对质谱条件和色谱条件进行不断探索和改良,从整体、全面、准确、快速、动态、操作简单兼顾的出发点,最终发现并建立了多成分同步检测的UPLC-MS/MS体内外分析方法,不仅为该方的经典名方开发、复方提取物新药开发及全方药动学研究提供了一种系统、可靠的快速检测方法,也为今后阐明该方的药效物质基础提供了可能的手段。
经调研防己黄芪汤研究文献,发现防己黄芪汤的物质基础研究存在以下两点不足:(1)防己黄芪汤为中药复方,遵循中医配伍理论,组方成分复杂,目前对其质量评价方法大多选择其中几个成分作为质量控制指标,难以反映复方整体水平;(2)对防己黄芪汤有效成分在体内动态过程的研究则不够全面,被吸收入血的成分是发挥药效的物质基础,了解这类成分在体内的动态变化,特别是配伍前后对药动学参数的影响,对于揭示该复方的物质基础、配方规律和指导临床应用具有重要意义。
发明内容
本发明的目的是针对现有技术中的不足,提供防己黄芪汤的体内外多成分分析方法。
第一方面,本发明提供了一种防己黄芪汤中18种活性成分的定量分析方法,所述18种活性成分为木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、甘草苷、异甘草苷、甘草素、5-O-甲基维斯阿米醇苷、芒柄花苷、毛蕊异黄酮、异甘草素、芒柄花素、白术内酯Ⅲ、白术内酯Ⅱ、黄芪皂苷Ⅲ、黄芪甲苷、黄芪皂苷Ⅰ和甘草次酸,所述定量分析方法为UPLC-MS/MS分析方法,质谱条件为:离子源:ESI源;扫描模式:正负离子切换,甘草苷、异甘草苷、甘草素和异甘草素采用负离子模式,木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、芒柄花苷、毛蕊异黄酮、芒柄花素、白术内酯Ⅲ、白术内酯Ⅱ、黄芪皂苷Ⅲ、黄芪甲苷、黄芪皂苷Ⅰ和甘草次酸采用正离子模式;喷雾电压:正离子选择3.5KV,负离子选择2.5KV;去溶剂温度:400℃;离子传输管温度:350℃;鞘气流速:45psi;辅气流速:15psi;色谱条件为:色谱柱为C18柱;流动相为A(甲醇)-B(0.1%甲酸水);洗脱条件为:0~1min:20%A,2~8min:20%A→100%A,8~12min:100%A,12.1~16min:20%A。
优选地,所述色谱条件还包含:柱温为40℃;流速为0.3ml·min
优选地,供试品溶液的制备方法为:精密称定防己黄芪汤原料药,加入甲醇,超声提取,冷却至室温后加甲醇补足减失重量,取上清液,于4℃,13200rpm,离心10min,取续滤液,即得。
优选地,对照品溶液的制备方法包含以下步骤:称取各对照品,以甲醇溶解。
第二方面,本发明提供了一种防己黄芪汤的质量控制方法,所述防己黄芪汤的质量控制方法包含对防己黄芪汤中18种活性成分的定量分析步骤,所述防己黄芪汤中18种活性成分的定量分析步骤如前任一所述。
第三方面,本发明提供了一种同时测定血浆中7种成分含量的方法,所述7种成分为木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、甘草苷、黄芪甲苷和甘草次酸,所述方法为UPLC-MS/MS分析方法,质谱条件为:离子源:ESI源;扫描模式:正负离子切换,甘草苷采用负离子模式,木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、黄芪甲苷和甘草次酸采用正离子模式;喷雾电压:正离子选择3.5KV,负离子选择2.5KV;去溶剂温度:400℃;离子传输管温度:350℃;鞘气:45psi;辅气:15psi;色谱条件为:色谱柱:C18柱,流动相:A(甲醇)-B(0.1%甲酸水);洗脱条件为:0~1min:20%A,2~8min:20%A→100%A,8~12min:100%A,12.1~16min:20%A。
优选地,色谱条件还包含:柱温:40℃;流速:0.3ml/min;进样器温度:4℃;进样量:10μL。
优选地,供试品溶液的制备方法为:取血,置于含有肝素钠的采血管中,10000rpm离心15min,取上层血浆,加入混标溶液和内标溶液,涡旋混合,再加入甲醇漩涡混合,4℃下13200rpm离心15min,取上清液,37℃下氮气吹干,20%甲醇复溶,涡旋,13200rpm离心15min,去上清液,即得供试品溶液。
优选地,对照品溶液的制备方法为:称取各对照品,以甲醇溶解;内标为地西泮。
第四方面,本发明提供了一种研究防己黄芪汤在体内的药代动力学的测定方法,所述研究防己黄芪汤在体内的药代动力学的测定方法包含同时测定血浆中7种成分含量的步骤,所述同时测定血浆中7种成分含量的步骤如前任一所述。
本文中,所述防己黄芪汤由防己12重量份、黄芪15重量份、甘草6重量份、白术9重量份四味中药组成,但对本领域技术人员而言,对各原料药重量比例的调节属于常规的操作,即以上所述方法同样适用于原料药重量比例有所调整的防己黄芪汤的检测和分析。
本发明优点在于:
1、本发明为建立防己黄芪汤复方中五大类化学成分能够被同步测定的快速、准确、重现及耐用性好的体内外分析方法,在方法开发和方法验证中,为保证多成分同步检测,本发明对该方法的色谱条件、质谱条件进行了不断地探索和优化实验。通过多反应监测(Multi Reaction Monitor,MRM)扫描模式和正负离子切换模式解决18种成分同步质谱检测。对色谱条件的方法学进行专属性、最低定量限、线性、日内、间精密度、加样回收率、样品重复性和稳定性进行了全面考察,创新性建立了全面、同步、快速检测复方中化合物成分群的UPLC-MS/MS体内外分析方法。
2、基于发明人丰富的研究经验,确定了合适的质谱和色谱条件,建立了灵敏度高、分析质量可靠、分析效率高且能对防己黄芪汤复方中多成分同时测定的UPLC-MS/MS分析方法,能用于防己黄芪汤中的生物碱类(木兰花碱、防己诺林碱和粉防己碱)、皂苷类(毛蕊异黄酮葡萄糖苷、甘草苷、异甘草苷、5-O-甲基维斯阿米醇苷、芒柄花苷、黄芪皂苷Ⅲ、黄芪甲苷和黄芪皂苷Ⅰ)、黄酮类(毛蕊异黄酮、甘草素、异甘草素和芒柄花素)、内酯类(白术内酯Ⅲ和白术内酯Ⅱ)和五环三萜类(甘草次酸)共18种成分的定量分析研究,有助于防己黄芪汤的质量控制,反映复方整体化学成分的种类和含量水平。
3、基于发明人丰富的研究经验,确定了质谱和色谱条件,建立了专属性好、精密度高、基质效应小、回收率高、稳定性强的同时测定血浆中木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、甘草苷、黄芪甲苷、甘草次酸7种成分的UPLC-MS/MS分析方法,为防己黄芪汤在体内的药代动力学研究奠定了工作基础。在该方法中,虽然采用单一溶剂甲醇进行除蛋白操作,但是由于检测条件恰当,仍然获得了优异的检测性能,同时还减少了对环境的污染,节约了实验的时间和经济成本。
附图说明
图1:防己黄芪汤18种活性成分的化学结构图。
图2-4:防己黄芪汤中18种成分的色谱图。图2:空白溶液;图3:混合标准品溶液;图4:样品溶液。(1)木兰花碱,(2)防己诺林碱,(3)粉防己碱,(4)毛蕊异黄酮葡萄糖苷,(5)甘草苷,(6)异甘草苷,(7)甘草素,(8)5-O-甲基维斯阿米醇苷,(9)芒柄花苷,(10)毛蕊异黄酮,(11)异甘草素,(12)芒柄花素,(13)白术内酯Ⅲ,(14)白术内酯Ⅱ,(15)黄芪皂苷Ⅲ,(16)黄芪甲苷,(17)黄芪皂苷Ⅰ,(18)甘草次酸。
图5:防己黄芪汤在SD大鼠血浆中的7种化合物及内标的化学结构图。
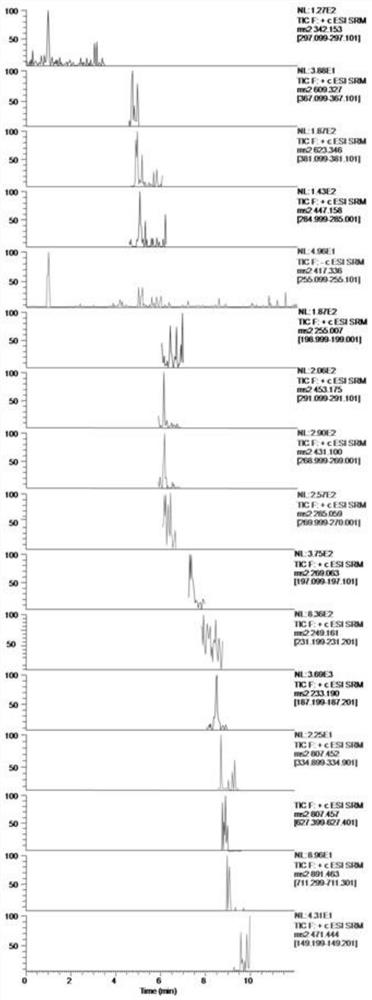
图6:7种入血成分的色谱图。A:空白血浆加内标;B:空白血浆加混合标准品;C:血浆样品。(1)木兰花碱;(2)防己诺林碱;(3)粉防己碱;(4)毛蕊异黄酮葡萄糖苷;(5)甘草苷;(6)黄芪甲苷;(7)甘草次酸;(8)内标。
具体实施方式
下面结合附图对本发明提供的具体实施方式作详细说明。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。其中,所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1防己黄芪汤中18种活性成分的定量分析
1仪器、材料与试剂
1.1仪器
Dionex UPLC 3000超高效液相-TSQ quantum Access MAX三重四级杆质谱联用系统(Thermo,美国);
Waters Acquity UPLC BEH C18 column(2.1*100mm 1.7μm,Waters,美国);
FA2004N型电子天平(上海精密科学仪器有限公司);
CP225D型电子天平(Sartorius AG,上海精密科学仪器有限公司);
Milli-Q型超纯水仪(Millipore公司,美国);
YC-18X型九阳电磁炉(山东九阳小家电有限公司);
SK250LHC型超声清洗器(上海科导超声仪器有限公司);
微量移液器0.1-10μL、10-100μL、20-200μL、200-1000μL(Eppendorf,德国);
Centrifuge 5427R冷冻离心机(Eppendorf,德国);
DZF-6050型真空干燥箱(上海精宏实验设备有限公司);
SHB-IIIA型循环水式多用真空泵(上海豫康科教仪器设备有限公司)。
1.2材料
各化学对照品、对照药材信息见表1、表2。
表1化学对照品信息
表2药材信息
1.3试剂
甲醇(色谱纯,Merck公司,德国)、甲酸(色谱纯,CNW公司,德国)、去离子水,其它试剂均为分析纯,符合分析测定要求。
2实验方法
2.1防己黄芪汤的处方分析
2.1.1复方组方及方解
《金匮要略》中的防己黄芪汤由防己(12g)、黄芪(15g)、甘草(6g)、白术(9g)四味中药组成。该方是治疗风水、风湿属表虚证的常用方剂,方中防己苦涩辛散,祛风除湿利水消肿,黄芪补气健脾补肺,尤能固表行水,二药共为君药,补气除湿利水,祛风散邪固表。白术补脾燥湿,既助黄芪补气固表,又助防己祛湿利水,为方臣药。甘草益气健脾,调和诸药,为方中佐使药。诸药合用具有益气祛风、健脾利水之功。
2.1.2汤剂制备
遵循《金匮要略》中复方以水煎煮的制备方法,按照复方中防己、黄芪、甘草、白术四味中药比例称取四种饮片适量,加10倍量水,浸泡0.5h,煎煮2次,每次1小时,煎液滤过,滤液合并凝缩至适量。65℃真空干燥后保存至干燥器中,阴性样品同法制备。
2.2多成分定量分析的UPLC-MS/MS方法的建立
2.2.1对照品溶液的制备
分别称量各对照品适量,用甲醇溶解制成对照品储备液于-20℃冰箱保存。使用时分别吸取相应18种对照品储备液体积适量,至10mL容量瓶内,用纯水和甲醇稀释至刻度,制成相应浓度的70%甲醇水混合标准品溶液。用70%甲醇水溶液将该混合标准品溶液稀释,制成一系列相应浓度的标准品工作液,4℃存储备用,质控(QC)样品制备方法同混标制备方法。
2.2.2质谱条件的优化
质谱条件的优化包括指标性成分正负离子检测模式和质谱参数的优化。本实验对本复方中18种成分(化学结构见图1)的喷雾电压、去溶剂温度、离子传输管温度、碰撞电压、鞘气、辅气等质谱参数进行优化。
为满足复方中多成分可同时检测的要求,首先对每个成分的离子检测模式进行优化,结果显示甘草苷、异甘草苷、甘草素、异甘草素在负离子模式下检测峰响应值明显优于正离子模式,木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、5-O-甲基维斯阿米醇苷、芒柄花苷、毛蕊异黄酮、芒柄花素、白术内酯Ⅲ、白术内酯Ⅱ、黄芪皂苷Ⅲ、黄芪甲苷、黄芪皂苷Ⅰ和甘草次酸在正离子模式下检测响应值明显优于负离子模式,为使所测化合物均具有较好的响应,本实验采用正负离子切换模式对复方中的18种成分进行同时检测。
质谱的喷雾电压和离子传输管温度根据仪器使用说明书推荐的值进行设定,去溶剂温度根据喷雾状态设定在400℃,鞘气和辅气的流速能够影响化合物的离子化程度,根据化合物的响应值,鞘气和辅气最终分别设置为45psi、15psi。化合物的碰撞电压和Tub Lens在优化子离子时由系统确定。
质谱主要参数设置值见表3,各成分的离子模式、母离子、子离子、碰撞电压、TubLens和出峰时间见表4。
表3质谱喷雾电压、去溶剂温度、鞘气、辅气的设定值
表4 18种化合物的离子模式、母离子、子离子、碰撞电压、Tub Lens及出峰时间
2.2.3液相条件的优化
色谱柱选择Acquity BEH C18(2.1mm×100mm,1.7μm;Waters,Milford,MA)色谱柱。流动相首先考察了乙腈-水、甲醇-水、乙腈-甲酸水溶液、甲醇-甲酸水溶液等系统,结果发现甲醇-甲酸水溶液系统较好,并继续优化了0.05%,0.1%和0.2%等不同比例甲酸浓度对系统分离的影响,最终确定甲醇-0.1%甲酸水溶液系统可满足本复方的分析要求。为使18种成分均具有较好的分离度和峰型,对流动相梯度、柱温、进样器温度进行了优化,最终确定的液相条件为:色谱柱为Acquity BEH C18(2.1mm×100mm,1.7μm;Waters,Milford,MA);柱温为40℃;流动相为A(甲醇)-B(0.1%甲酸水);洗脱条件为:0~1min:20%A,2~8min:20%A→100%A,8~12min:100%A,12.1~16min:20%A;流速为0.3ml·min-1;进样器温度为10℃;进样体积为2μL。
2.2.4方法学考察
2.2.4.1专属性试验
按照本章2.3项下操作制备防己黄芪汤的供试样品,70%甲醇水溶液作为空白溶液。取空白溶液、混合标准品溶液、防己黄芪汤的供试样品各2μl,按照建立的质谱条件和液相条件进行测定,观察在各成分相应的出峰时间点是否存在干扰。
2.2.4.2标准曲线的制备
按照本章2.2.1项下配制7种不同浓度的混合标准品溶液,每个浓度三份,按照建立的质谱条件和液相条件进行测定,记录各成分的峰面积(A)。以各成分的浓度作为X值,峰面积作为Y值,对纵横坐标值进行线性回归,所得的回归方程,即为标准曲线,同时计算各成分的线性相关系数。标准曲线中所设的最低浓度至最高浓度,即为该成分的线性范围,将线性范围的最低浓度继续稀释后进样,将信噪比S/N=10时的进样浓度记为最低定量限(LOQ)。
2.2.4.3日内、日间精密度试验
日内精密度:将制备的低、中、高浓度的QC样品24h内连续进样5次,每个QC浓度平行三份,根据随行标曲得出每个QC样品的检测浓度,计算5次检测浓度的RSD值,用于评价日内精密度。
日间精密度:将制备的低、中、高浓度的QC样品每天进样一次,每个QC浓度平行三份,连续三天,根据随行标曲得出每个QC样品的检测浓度,计算三天内各成分检测浓度的RSD值,用于评价日间精密度。
2.2.4.4加样回收率试验
按照2.2.1项下制备低、中、高三个剂量的混合标准品。精密称取9份防己黄芪汤样品适量,分为三组,加入适量的低、中、高三个剂量的混合标准品,按照供试品处理方法制备供试样品,按照建立的质谱条件和液相条件进行测定,得各成分的峰面积A,根据随行标曲计算各成分含量。加入的标准品浓度记为C
2.2.4.5重复性试验
取同一批次防己黄芪汤粉末6份,按照供试品处理方法制备供试样品,按照建立的质谱条件和液相条件进行测定,得各成分的峰面积A,根据随行标曲计算各成分含量的RSD值,评价样品制备分析过程的重复性。
2.2.4.6稳定性试验
取同一批次防己黄芪汤粉末6份,按照供试品处理方法制备供试样品,分别在0h、12h、24h、36h、48h按照建立的质谱条件和液相条件进行测定,记录各成分的峰面积A,根据随行标曲计算各成分含量,计算各成分不同时间点之间检测浓度的RSD值,用于评价样品48h内的稳定性。
2.3防己黄芪汤供试品的制备
为了有效提取防己黄芪汤中的药效物质成分,本实验对影响提取率的关键因素也进行了考察,分别对提取溶剂(50、75和100%,v/v)、溶剂体积(1:250,1:500和1:750,w/v)和提取时间(20、45和90min)进行了单因素考察,结果显示0.1g样品粉末加50mL甲醇超声提取45min为最佳提取条件。
供试品制备方法:根据2.1.2项下操作分别制备不同批次防己黄芪汤供试品及阴性样品。精密称定相应批次粉末约0.1g,加入甲醇50ml,称定重量,超声提取45min,冷却至室温后加70%甲醇补足减失重量,取1ml上清液于4℃,13200rpm,离心10min,续滤液2μL进样,分析检测。
2.4防己黄芪汤中18种成分的含量测定
精密称取防己黄芪汤粉末0.1g,5批次,每个批次平行6份,共36份样品按照2.3项下的方法制备供试品,采用2.2项下建立的分析方法对复方中的18种成分进行定量检测。
3实验结果
3.1方法学考察
3.1.1专属性
空白溶液、混合标准品溶液、防己黄芪汤的供试样品的色谱图见图2-4,各成分的相应出峰时间点无干扰,表明测定18种成分的分析方法具有良好的专属性。
3.1.2线性
18种成分的线性方程、相关系数、线性范围、定量限的结果见表5。18种成分相关系数r均满足r>0.995,具有较好的线性关系、合理的线性范围。
表5 18种成分的线性方程、相关系数、线性范围、定量限和检测限
3.1.3日内、日间精密度
18种成分相应的日内、日间精密度结果见表6。结果显示18种成分的RSD值均小于5%,表明仪器精密度良好,符合分析要求。
表6 18种成分的日内、日间精密度结果
3.1.4加样回收率
18种成分的加样回收率的结果见表7。低、中、高三个剂量的平均回收率在92.91%~102.86%之间,各浓度的平均回收率均在90%~110%范围内,满足分析要求。
3.1.5样品重复性
18种成分的样品重复性试验结果见表7,各成分的浓度RSD值均小于5%,表明重复性良好。
3.1.6样品稳定性
18种成分的样品在48h内的稳定性结果见表7,结果显示各成分的浓度的RSD值均小于5%,表明18种成分在48h内具有良好的稳定性。
表7 18种成分的加样回收率、重复性和稳定性结果
3.2防己黄芪汤样品中18种活性成分含量测定结果
含量测定结果表明不同批次的防己黄芪汤均含有以下18种成分:木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、甘草苷、异甘草苷、甘草素、5-O-甲基维斯阿米醇苷、芒柄花苷、毛蕊异黄酮、异甘草素、芒柄花素、白术内酯Ⅲ、白术内酯Ⅱ、黄芪皂苷Ⅲ、黄芪甲苷、黄芪皂苷Ⅰ、甘草次酸,各成分的含量结果见表8。
表8不同批次防己黄芪汤中18种成分含量测定结果
4小结与讨论
本发明的质谱条件采用多反应监测(Multi Reaction Monitor,MRM)扫描模式和正负离子切换模式,基于该领域丰富的研究经验确定了合适的检测条件并不断优化。为确保所建立的UPLC-MS/MS分析方法能够满足复方的定量研究,本发明对方法的专属性、最低定量限、线性、日内、间精密度、加样回收率、样品重复性和稳定性进行了考察。结果表明18种指标成分专属性良好,具有较好的线性关系(r>0.995),日内、日间精密度的RSD<5%,加样回收率在92.91%~102.86%范围内,重复性的RSD<5%,且在48h内稳定。以上结果表明:本发明建立的多成分测定方法灵敏度高、分析效率高、分析时间短及检测范围较宽,满足分析要求,可用于防己黄芪汤的质量控制研究。
防己黄芪汤多成分定量结果表明:不同批次的防己黄芪汤样品主要含有18种成分,包括生物碱类(粉防己碱、防己诺林碱和木兰花碱)、皂苷类(黄芪甲苷、黄芪皂苷Ⅲ、黄芪皂苷Ⅰ)、黄酮类(甘草素、毛蕊异黄酮、毛蕊异黄酮葡萄糖苷、甘草苷、异甘草苷、5-O-甲基维斯阿米醇苷、芒柄花苷、异甘草素和芒柄花素)、内酯类(白术内酯Ⅲ和白术内酯Ⅱ)和五环三萜类(甘草次酸)成分。
实施例2大鼠血浆中多成分同时测定的UPLC-MS/MS分析方法的建立
1仪器、试剂、材料与动物
1.1仪器
Dionex UPLC 3000超高效液相-TSQ quantum Access MAX三重四级杆质谱联用系统(Thermo,美国);
Waters Acquity UPLC BEH C18 column(2.1*100mm,1.7μm,Waters,美国);
FA2004N型电子天平(上海精密科学仪器有限公司);
Milli-Q超纯水器(Millipore公司,美国);
QT-1涡旋混合器(上海琪特分析仪器有限公司);
Centrifuge 5427R冷冻离心机(Eppendorf,德国);
CP225D型电子天平(Sartorius AG,上海精密科学仪器有限公司);
SK250LHC型超声清洗器(上海科导超声仪器有限公司);
微量移液器0.1-10μL、10-100μL、20-200μL、200-1000μL(Eppendorf,德国);
SHB-IIIA型循环水式多用真空泵(上海豫康科教仪器设备有限公司)。
1.2试剂
甲醇(色谱纯,Merck公司,德国)、甲酸(色谱纯,CNW公司,德国)、去离子水(Millipore公司,美国),其它试剂均为分析纯,符合分析测定要求。
1.3材料
表9对照品来源信息
1.4实验动物
Sprague-Dawley(SD)雄性大鼠(270±20g),清洁级,购于北京维通利华实验动物技术有限公司,饲养于上海中医药大学动物实验中心,动物生产许可证号:SCXK(京)2012-0001,动物使用许可证号:SYXK(沪)2014-0008。实验前,大鼠在室温22±2℃,相对湿度45-60%,每天光照12h的条件下适应性饲养一周。动物实验操作方法遵循上海中医药大学实验动物伦理委员会相关规定。
2实验方法
2.1空白血浆样品的采集
SD大鼠禁食过夜(12h)后,灌胃给予0.3%CMC-Na,2小时后,腹主动脉取血,置于含有肝素钠的5ml采血管中,10000rpm离心15min,取上层血浆保存于-20℃冰箱中,备用。
2.2对照品溶液和QC混标溶液的制备
精密称取各对照品和内标(地西泮)粉末适量,于棕色10ml容量瓶中,甲醇充分溶解,-20℃条件下保存,作为储备液。使用前,取各对照品储备液适量体积于同一容量瓶中,甲醇稀释至刻度线,配制成混标溶液。取内标储备液适量体积,甲醇稀释至200ng/ml,备用。
取一定量的混标溶液(混标母液),用甲醇稀释成不同浓度低、中、中高、高QC混标溶液,各成分的低、中、中高、高QC终浓度分别为木兰花碱:0.75ng/ml,3.75ng/ml,7.5ng/ml,30ng/ml;防己诺林碱:3ng/ml,15ng/ml,30ng/ml,120ng/ml;粉防己碱:7.5ng/ml,37.5ng/ml,75ng/ml,300ng/ml;毛蕊异黄酮葡萄糖苷:0.75ng/ml,3.75ng/ml,7.5ng/ml,30ng/ml;黄芪甲苷:3ng/ml,15ng/ml,30ng/ml,120ng/ml;甘草苷:1.87ng/ml,9.35ng/ml,18.7ng/ml,74.8ng/ml;甘草次酸:45ng/ml,225ng/ml,450ng/ml,1800ng/ml,其化学结构图见图5。
2.3血浆样品的前处理条件
本实验考察了不同倍量的有机试剂(乙腈/甲醇)及SPE-C18固相萃取法除蛋白方法,结果显示5倍量的有机试剂和SPE-C18固相小柱除蛋白效果均较好,且可靠稳定。但考虑到要减少毒性气体污染和节约实验成本,最终确定的血浆样品前处理方法为:取空白血浆200μL,加入100μL混标溶液和50μL内标溶液(200ng/ml),涡旋混合1min,加入850μL甲醇漩涡混合2min,4℃下13200rpm离心15min,上清液转移至1.5mL离心管,37℃下氮气吹干,100μL 20%甲醇复溶,涡旋3min,13200rpm离心15min,10μL上清液进样。
2.4色谱和质谱条件
色谱条件:色谱柱Acquity BEH C18(2.1mm×100mm,1.7μm;Waters,Milford,MA)色谱柱;流动相:甲醇-0.1%甲酸水溶液体系,流动相梯度同实施例1;柱温:40℃;流速:0.3ml/min;进样器温度:4℃;进样量:10μL。
质谱条件:离子源:ESI源;扫描模式:正负离子切换;喷雾电压:正离子选择3.5KV,负离子选择2.5KV;去溶剂温度:400℃;离子传输管温度:350℃;鞘气:45psi;辅气:15psi;各化合物的母离子→子离子:木兰花碱342.15→297.10,碰撞能量18V;防己诺林碱609.33→367.10,碰撞能量38V;粉防己碱623.35→381.10,碰撞能量43V;毛蕊异黄酮葡萄糖苷447.16→285.00,碰撞能量19V;甘草苷417.29→255.10,碰撞能量20V;黄芪甲苷807.46→627.40,碰撞能量49V;甘草次酸471.44→149.20,碰撞能量36V;IS(地西泮)284.99→193.00,碰撞能量27V。
2.5方法学考察
2.5.1专属性考察
低浓度的QC样品、45min的血浆样品及空白血浆按2.3项下处理后,采用2.4项下的检测条件进行检测,观察各成分相应保留时间是否存在干扰。
2.5.2线性、定量限及检测限考察
取数份200μL SD大鼠的空白血浆,分别加入一系列不同浓度的混合标准品溶液100μL,按2.3项下处理后,采用2.4项下的检测条件进行检测,记录各活性成分及内标的峰面积(A)。纵坐标的值为各成分的峰面积与内标峰面积的比值,横坐标的值为各成分的浓度,采用1/x
2.5.3日间、日内精密度考察
制备不同浓度的低、中、中高、高QC样品,每个浓度平行五份,一天连续测定三次,计算各成分的相对准确度(RE)和精密度(RSD)用于评价日内精密度。制备不同浓度的低、中、中高、高QC样品,每个浓度平行五份,连续测定三天,计算各成分的相对准确度(RR)和精密度(RSD)用于评价日间精密度。相对准确度(RR)=(C
2.5.4回收率与基质效应考察
取生理盐水200μL,加入100μL不同浓度的低、中、中高、高QC混标溶液和50μL内标溶液(200ng/ml),涡旋混合1min,加入850μL甲醇漩涡混合2min,13200rpm离心15min,上清液转移至1.5mL离心管,37℃下氮气吹干,100μL 20%甲醇复溶,涡旋3min,13200rpm离心15min,10μL上清液进样,测定峰面积为A。
取空白血浆200μL,加入50μL内标溶液(200ng/ml),涡旋混合1min,加入850μL甲醇漩涡混合2min,13200rpm离心15min,上清液转移至1.5mL离心管,再加入100μL不同浓度的低、中、中高、高QC混标溶液,混匀,37℃下氮气吹干,100μL 20%甲醇复溶,涡旋3min,13200rpm离心15min,10μL上清液进样,测定峰面积为B。
取空白血浆200μL,加入100μL不同浓度的低、中、中高、高QC混标溶液和50μL内标溶液(200ng/ml),涡旋混合1min,加入850μL甲醇漩涡混合2min,13200rpm离心15min,上清液转移至1.5mL离心管,37℃下氮气吹干,100μL 20%甲醇复溶,涡旋3min,13200rpm离心15min,10μL上清液进样,测定峰面积为C。
峰面积C/B之比为各成分的回收率,峰面积C/A之比为成分的基质效应。
2.5.5稳定性考察
取100μL低、中、中高、高QC混标溶液于1.5mL离心管,每个浓度平行五份,37℃下氮气吹干,加入200μL空白血浆和50μL内标溶液(200ng/ml),后续处理同2.3项,室温放置12小时,用于评价样品处理过程中的稳定性。
取100μL低、中、中高、高QC混标溶液于1.5mL离心管,37℃下氮气吹干,加入200μL空白血浆,混匀后置于-20℃冰箱15天后,按2.3项下处理,用于评价样品的长期稳定性。
取100μL低、中、中高、高QC混标溶液于1.5mL离心管,37℃下氮气吹干,加入200μL空白血浆,混匀后置于-20℃冰箱5天后,于室温放置12h,反复三次后,按2.3项下处理,用于评价样品的反复冻融稳定性。
取低、中、中高、高QC样品,每个浓度平行五份,按2.3项下处理后,4℃放置24小时,按建立的方法进行检测,用于评价检测条件下的稳定性。
3实验结果
3.1专属性
7种成分及IS的选择反应检测扫描(selective reaction monitoring,SRM)模式的色谱图见图6。结果表明木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、甘草苷、黄芪甲苷、甘草次酸、IS(地西泮)在相应的保留时间无干扰,具有良好的专属性。
3.2线性及定量限
7种成分的线性方程、线性范围和定量限的结果见表10。各成分的线性相关系数r>0.995,线性良好,满足分析要求。定量限的准确度(RR≥80%)及精密度(RSD≤20%),满足分析要求。
表10 SD大鼠血浆中7种成分的线性方程、线性范围和定量限
3.3日间精密度和日内精密度
根据随行标曲计算不同水平的QC样品浓度,每个浓度5个平行样。计算每个成分的日间和日内准确度RR和精密度RSD值,结果见表11。结果表明7种成分的日间、日内准确度RR在84.35~105.50%范围内,日间、内精密度RSD在0.36~10.87%范围内,符合分析要求(RR≥85%,RSD≤15%)。
表11 SD大鼠血浆中7种成分的日间精密度、日内精密度、回收率和基质效应
3.4回收率与基质效应
验证方法的回收率和基质效应时,制备低、中、中高、高四个不同水平的QC样品,每个浓度制备5个平行样品,结果以“Mean±SD”形式表示,检测结果见表12。结果表明7种活性成分的回收率在86.68%~113.04%范围内,基质效应在52.33%~112.50%范围内。
3.5稳定性
本实验使用低、中、中高、高四个不同水平的QC样品(每个浓度制备5个平行样品)来验证方法的稳定性。为了确保方法的适用性,验证了取材、储存、处理过程中不同条件的稳定性,其结果见表12,结果表明7种成分的准确度在84.70%~115.65%范围内,满足分析要求。
表12 SD大鼠血浆中7种成分的稳定性研究
4小结与讨论
在满足分离效果和节约时间前提下,选择Acquity BEH C18(2.1mm×100mm,1.7μm)色谱柱,流动相选择甲醇(A)和0.1%甲酸水溶液(B),进行梯度洗脱,结果显示7种成分及IS之间的分离效果较好,在16min内能够分析完成。本方法质谱条件选择多反应监测(MultiReaction Monitor,MRM)扫描模式和正负离子切换模式,能够实现多个化合物同时测定,节省分析测定时间。为确保分析方法能够满足生物样本分析要求,本实施例从专属性、线性、定量限、精密度、回收率、基质效应和稳定性对分析方法进行方法学验证。结果表明7种成分的专属性较好;线性的相关系数r>0.995;日间、内精密度RSD在0.36%–10.87%范围内;回收率均在86.68%~113.04%范围内;基质效应在52.33%~112.50%范围内,其中粉防己碱的基质效应在52.33%~64.76%范围内,但粉防己碱的回收率在103.49%~111.68%之间,回收率较高,说明血浆中物质只对其在质谱中的响应有抑制,因四个不同浓度的基质效应RSD<15%,说明这种抑制作用可通过建立随行标曲计算浓度予以抵消,可以满足分析要求;稳定性的准确度在84.70%~115.65%范围内,该方法的专属性、线性、定量限、精密度、回收率、基质效应和稳定性满足生物样本分析要求。
本实施例建立了同时测定大鼠血浆中木兰花碱、防己诺林碱、粉防己碱、毛蕊异黄酮葡萄糖苷、甘草苷、黄芪甲苷、甘草次酸等7种成分的UPLC-MS/MS分析方法,为后续防己黄芪汤在大鼠体内的药代动力学研究提供了准确的分析手段。
参考文献:
[1]吕翠平,郭亚健,张莹.防己黄芪汤水煎液中二种防己生物碱煎出量测定[J].中国实验方剂学杂志,2006,12(12):30-31.
[2]刘书芬,梁倩倩,陈岩,等.HPLC法同时测定防己黄芪汤中5种成分[J].中成药,2014,36(11):2312-2315.
[3]X.Wang,X.Liu,X.Xu,et al.Screening and identification of multipleconstituents and their metabolites of Fangji Huangqi Tang in rats by ultra-high performance liquid chromatography coupled with quadrupole time-of-flighttandem mass spectrometry basing on coupling data processingtechniques.Journal of chromatography B Analytical technologies in thebiomedical and life sciences,985(2015):14-28.
[4]X.Wang,X.Liu,H.Cai,et al.Ultra high performance liquidchromatography with tandemmass spectrometry method for the determination oftetrandrine and fangchinoline in rat plasma after oral administration ofFangji Huangqi Tang and Stephania tetrandra S.Moore extracts.Journal ofseparation science,(2015),38(8):1286-1293.
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
- 一种防己黄芪汤的体内外多成分分析方法
- 面向多电厂多成分电量分散交易的集中式安全分析方法、装置及系统