一种青蒿素含量快速检测方法
文献发布时间:2023-06-19 12:00:51
技术领域
本发明涉及青蒿素含量检测方法技术领域,具体涉及一种青蒿素含量快速检测方法。
背景技术
青蒿素及其衍生物联合疗法是目前治疗疟疾的最有效的方法,由于国际市场对青蒿素晶体原料的旺盛需求,而青蒿素在相当长的时间内难以实现工业合成对植物提取替代,导致近年来青蒿种植和原料提取加工产业在我国重庆、广西、广东和湖南等地迅猛发展。然而,不同种子来源、不同生长环境,甚至同一环境下不同栽培技术措施对青蒿素含量均有较大影响,因此,开发出一种能快速抽样检测的青蒿素含量检测方法显得尤为重要。
目前,青蒿素检测方法主要有高效液相法检测和紫外分光光度法检测法。中国药典2020(二部)中采用石油醚(30~60℃)索氏提取+高效液相色谱法(HPLC)检测,该方法虽检测结果准确,但对仪器设备和检测人员素质要求高,且前处理时间较长,操作复杂繁琐的缺点,难以满足青蒿原料加工厂对检测速度的要求。
紫外分光光度法(以下简称紫外法或UV法)一般用在原料质量的快速检测中,该方法对检测准确度要求不高,但能加快检测速度,降低操作难度,大大降低原料质检成本。在采用紫外分光光度法检测时,主要以弱极性提取剂石油醚为溶剂,利用索氏提取或超声水浴提取样品中青蒿素,来降低非青蒿素背景干扰成分的溶出,提高检测的准确性,但索氏提取时长均在2h~24h之间,且需要多次提取、洗脱,然后用适当浓度乙醇来溶解浸膏,以充分溶解氢氧化钠促进青蒿素结构转化,此过程较为繁琐,且费时费力。另外,超声提取由于难以控制提取过程温度导致同一样品不同批次检测结果重复性较差。 此外,前人研究主要以空白溶液或完全未与氢氧化钠反应的样品溶液做参比,不能准确有效地消除检测中的背景干扰问题,导致检测结果与标准HPLC法检测结果相比,存在不同程度的随机误差和系统误差,虽然采用线性拟合的方法进行了修正,但未能有效消除因样品不同带来的随机误差。此外,前人也尝试以紫外法检测原料青蒿素含量时以无水乙醇为提取剂,采用C-18萃取柱洗脱杂质的方式,有效避免了非青蒿成分对λ259紫外光的吸收,实现了青蒿素的准确检测,但该方法依然存在前处理过程复杂、萃取柱昂贵、超声提取温度难以控制导致的重复性差等问题。
发明内容
本发明针对上述问题提供一种青蒿素含量快速检测方法,包括以下步骤:
S1:确定最佳吸收波长;
S2:背景干扰的确定和消除;
S3:建立标准曲线;
S4:样品的处理及测定;
所述样品的处理及测定包括以下步骤:
S41:青蒿叶50℃烘干后粉碎过筛;
S42:取粉碎后的青蒿叶并加入95%乙醇进行水浴加热提取,对提取液进行离心;
S43:取离心后的上清液0.32mL加入95%乙醇1.28mL后再加入0.2%NaOH溶液6.40mL配制青蒿素样品溶液;
S44:将所述青蒿素样品溶液50℃水浴加热30min,快速冷却后加入弱酸性水稀释10倍得到待测样品;
S45:取步骤S43中的青蒿素样品溶液0.32mL,加入95%乙醇1.28mL,再加入6.4mL弱酸性水,摇匀,取0.8mL混合液,加入6.56mL弱酸性水,检测前加入0.2%NaOH 640ul的摇匀作为参比溶液,20s内进行样品检测。
进一步地,步骤S1中确定所述最佳吸收波长的具体步骤包括:
S11:取青蒿素标准品0.1000g加95%乙醇配制成浓度为1.0mg/mL的第一青蒿素母液;
S12:取第一青蒿素母液10mL加95%乙醇配制成浓度为0.1mg/mL的第二青蒿素母液;
S13:取第二青蒿素母液0.0mL,加入95%乙醇1.6mL 、弱酸性纯水6.4mL,计为WA-1;取第二青蒿素母液0.0 mL,加入95%乙醇1.6mL、0.2%NaOH溶液6.4mL,计为WA-2;取第二青蒿素母液0.4mL,加入95%乙醇1.2mL、弱酸性水6.4mL,计为WA-3;取第二青蒿素母液0.4mL,加入95%乙醇1.2mL、0.2%NaOH 溶液6.4mL,计为WA-4;
S14:将WA-1、WA-2、WA-3、WA-4摇匀后50℃下水浴加热30min,快速冷却后以空气为参比在200~500nm进行光谱扫描确定最大吸收峰的波长位置。
更进一步地,步骤S3中所述建立标准曲线的具体步骤包括:
分别取第二青蒿素母液0.0mL、0.1mL、0.2mL、0.5mL、1.0mL、1.5mL、2.0mL至10mL具有刻度的比色管中,分别加入95%乙醇2mL、1.9mL、1.8mL、1.5mL、1.0mL、0.5mL、0.0mL,再分别加入0.2%NaOH 8mL,配成浓度分别为0.00mg/mL、0.01mg/mL、0.02mg/mL、0.05mg/mL、0.10mg/mL、0.15mg/mL、0.20mg/mL的标准溶液,50℃水浴加热30min,快速冷却,以空气为参比测吸光度值后再以0.00mg/mL标准液为参比作背景扣除,建立标准曲线。
更进一步地,步骤S2中所述背景干扰的确定和消除的具体步骤包括:
S21:取步骤S42中离心后的提取液0.4mL加入95%乙醇1.2mL,再加入弱酸性纯水6.4mL;
S22:取步骤S42中的提取液0.4mL加入95%乙醇1.2mL,再加入0.2%NaOH 6.4mL摇匀后于50℃下水浴加热30min后快速冷却;
S23:将步骤S21和步骤S22中处理后的提取液均稀释10倍,以空气为参比测样品的吸光度;
S24:分别取2个青蒿含量差异较大的青蒿样品乙醇提取液40ul,加入95%乙醇160ul,再加入弱酸性纯水9mL,最后加入0.2%NaOH 800ul,测样品的吸光度值;
对比步骤S23和S24样品测定过程中背景干扰的变化,确定最佳的参比液。
更进一步地,所述弱酸性水的pH值为6.0≤pH≤7.0。
更进一步地,所述青蒿素标准品、95%乙醇和氢氧化钠均为分析纯。
本发明的优点:
本发明的检测方法直接采用95%乙醇水浴加热浸提的方法,减少了样品前处理环节,降低前处理时长,与超声提取相比,随机误差降低;对于青蒿提取物中非青蒿素干扰成分增多从而导致背景干扰增大的问题,本发明通过利用NaOH溶液与非青蒿素干扰物质、NaOH溶液与青蒿素在常温下反应存在的时间差来最大限度消除背景干扰——加入0.2%NaOH溶液后迅速测定,将吸光度降低至一稳定值作为背景干扰值来消除背景干扰对检测结果的影响,结果表明,其检测结果与标准HPLC法检测值一致性较好,且不需要进行线性拟合修正。
传统的索氏提取法进行青蒿素提取,时长均在2 h~24 h之间,再加上浸膏洗脱、溶解、过滤等过程,以6个索氏提取器同时提取计,单一样品的前处理平均时间均大于0.5h,且受限于索氏提取器数量限制,难以实现批量检测。本发明的检测方法可以实现批量水浴加热提取1h,批量离心,批量水浴反应衍生,以6个水浴锅容纳300个样品计,每一样品前处理平均时长可以控制在2 min以内,1d可实现超300个样品的检测,检测效率大大提高。此外,相较于乙醇提取+C-18萃取柱洗脱方法中单支C-18萃取柱超过17元,综合成本超过20元,本研究方法单个样品的检测成本可控制在0.5元内,真正实现了简便高效低成本检测。
除了上面所描述的目的、特征和优点之外,本发明还有其它的目的、特征和优点。下面将参照附图,对本发明作进一步详细的说明。
附图说明
构成本申请的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
图1是本发明实施例的不同参比液和标准青蒿素反应液光谱扫描图;
图2是本发明实施例的青蒿样品溶液碱转化衍生前后光谱扫描图;
图3是本发明实施例的不同参比溶液的动力学曲线图;
图4是本发明实施例的青蒿素标准曲线图;
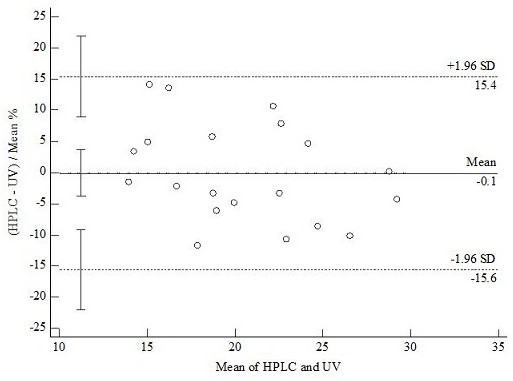
图5是本发明实施例的UV法和HPLC法Bland-Altman一致性分析图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
本申请的实施例所用仪器和试剂如下:
主要仪器设备
高效液相色谱(安捷伦,1220 Infinity II),紫外分光光度计(日本岛津,UV-1800),万分之一天平(日本岛津,AUW220),恒温水浴锅(上海亚荣生化仪器厂,HH-8),高速冷冻离心机(常州金坛良友仪器有限公司,TGL-16M)。
主要试剂
青蒿素标准品(98%,AR),95%乙醇(AR),石油醚(30~60℃,GR),乙腈(GR),氢氧化钠(AR),弱酸性纯水(6.0≤pH≤7.0)。
实施例1
一种青蒿素含量快速检测方法,包括以下步骤:
S1:确定最佳吸收波长;
母液配制:准确称取青蒿素标准品0.1000g,用95%乙醇溶解并定容至100mL容量瓶,制成浓度为1.0mg/mL的第一青蒿素母液;用移液管准确移取第一青蒿素母液10mL至100mL容量瓶,采用95%乙醇定容,得0.1mg/mL的第二青蒿素母液;
光谱扫描:取第二青蒿素母液0.0mL,加入1.6 mL95%乙醇至10mL离心管中,再加入弱酸性水6.4mL,计为WA-1;取第二青蒿素母液0.0mL,加入1.6mL95%乙醇至10mL离心管中,加入0.2%NaOH溶液6.4mL,计为WA-2;取第二青蒿素母液0.4mL,加入95%乙醇1.2mL至10mL离心管中,再加入弱酸性水6.4mL,计为WA-3;取第二青蒿素母液0.4mL,加入95%乙醇1.2mL至10mL离心管中,再加入0.2%NaOH 6.4 mL,计为WA-4;上述样品摇匀后50℃下水浴加热30min,取出冷水浴快速冷却,以空气为参比在200~500nm进行光谱扫描。通过对比WA-1、WA-2、WA-3、WA-4溶液光谱扫描图,即可确定最大吸收峰的波长位置。
为确定青蒿素溶液经0.2%NaOH衍生后的最大吸收波长,对可能参与反应的不同成分反应液在紫外-可见光范围内(200~500nm)进行背景光谱扫描,如图1所示。通过对比WA-1和WA-3,发现青蒿素在弱酸性水中无法进行碱转化衍生,仅在200nm左右存在末端吸收,且背景吸收较小;对比WA-2和WA-4,发现青蒿素经0.2%NaOH转化后在290~292nm处出现明显吸收峰,而220nm处的强吸收峰应为NaOH溶液的末端吸收。综合来看,反应液中可能构成背景干扰的CH
S2:背景干扰的确定和消除;
步骤一:取乙醇提取样品溶液0.4mL至10mL离心管中,加入95%乙醇1.2mL,再加入弱酸性水6.4mL。另取乙醇提取液0.4mL至10 mL离心管中,加入95%乙醇1.2mL,再加入0.2%NaOH6.4mL至10mL离心管中,将二者摇匀后置于50℃水浴加热30min,取出冷水浴快速冷却,然后分别稀释10倍,以空气为参比进行光谱扫描。
步骤二:分别吸取2个青蒿含量差异较大的青蒿样品乙醇提取液40ul,加入95%乙醇160ul,再加入弱酸性纯水9mL,最后加入0.2%NaOH 800ul至10mL离心管中,室温下观察二者在291nm处吸光度值的变化。
通过对比步骤一和步骤二中溶液背景干扰的变化,确定最佳的参比液。
分析青蒿叶95%乙醇提取液经碱转化衍生前后的光谱扫描图,发现未经衍生的青蒿提取原液在320~340nm处有较强吸收峰,在290~292nm处吸收较弱;经碱转化衍生后,青蒿提取液在320~340nm处吸收峰消失,在360~370nm处隆起一新的弱吸收峰,且在290~292nm处隆起一强吸收峰,该强吸收峰为青蒿素衍生物吸收峰。由此可见,以290~292nm为检测波长进行青蒿提取液中青蒿素含量检测具有可行性。
从图2可以看出,青蒿叶95%乙醇提取液中背景成分较为复杂,若简单以相应量的95%乙醇+0.2%NaOH(图1WA-2)为参比,或相应量青蒿提取液+95%乙醇+弱酸性纯水为参比(图3中未转化CK1、CK2),均不能有效的消除提取液中非青蒿素成分的背景值。为此,本发明以反应量的青蒿95%乙醇提取液+95%乙醇+弱酸性纯水为参比,以及相应量青蒿95%乙醇提取液+95%乙醇,最后加入0.2%NaOH迅速摇匀后为参比在291nm处进行动力学观察,如图3所示,CK1和CK2代表青蒿素含量有较大差异的样品所配制的参比液。分析发现,未经碱转化衍生的青蒿提取液吸光度值从放入样品开始便不再变化,且吸光度值较碱转化衍生的参比液低。将反应液配好迅速加入0.2%NaOH进行动力学观察,发现其吸光度在加入后5~10s内有略微下降,后保持5~10s的稳定,而后因青蒿素碱化衍生反应开始而逐步增大。同时,不同样品所配制的参比在碱转化衍生前后的差值差异较大。
由此可以证明,固定参比(与反应液相应量的95%乙醇+0.2%NaOH)与未经碱转化衍生的参比(与反应液相应量的青蒿提取液+95%乙醇+弱酸性纯水)均不能作为正确参比进行背景扣除,而采用与反应液相应量的青蒿提取液+95%乙醇,在迅速加入0.2%NaOH后检测,待吸光度在最低值稳定后作为参比值即本发明制备的特异性碱转化参比较为可行。
S3:建立标准曲线;
分别移第二青蒿素母液0.0mL、0.1mL、0.2mL、0.5mL、1.0mL、1.5mL、2.0mL至10mL刻度比色管,分别加入95%乙醇2mL、1.9mL、1.8mL、1.5mL、1.0mL、0.5mL、0.0mL,再分别加入0.2%NaOH8mL,配成浓度分别为0.00 mg/mL、0.01mg/mL、0.02mg/mL、0.05mg/mL、0.10mg/mL、0.15mg/mL、0.20mg/mL的标准溶液,盖塞密封后50℃水浴加热30min,取出冷水浴快速冷却,以空气为参比在291nm处比色,得到相应的吸光度值后再以0.00mg/mL标准液为参比作背景扣除,建立标准曲线。
为保证完全扣除比色皿及可能构成背景干扰的溶剂和反应底物的背景值,本发明以0.00 mg/mL标准液为参比作背景扣除,得到图4所示标准曲线图。由图4可知,吸光度-浓度回归方程为C(mg/mL)=0.156*Abs+0.009,R
S4:样品的处理及测定;
所述样品的处理及测定包括以下步骤:
将青蒿叶在烘箱中50℃下烘8~10h,高速粉碎后过60目筛,装袋置于阴凉处密封保存;
含水率的测定:取3g烘干后的样品,采用快速水分检测仪测定含水率,重复3次。
试样制备:精确称取青蒿叶样品1.0000g;加入95%乙醇20mL至50mL三角瓶中盖紧密封摇匀,50℃水浴加热60min进行提取,重复3次。取出样品震荡摇匀,转入10mL离心管中,5000r/min离心3min。取上清液0.32mL,加入95%乙醇1.28mL,再加入0.2%NaOH 6.40mL至离心管中,配制成青蒿素样品溶液,50℃水浴加热30min,取出冷水浴快速冷却,用弱酸性水稀释10倍即得待测样品。
参比选择:准确吸取青蒿素样品溶液0.32mL,加入95%乙醇1.28mL,再加入6.4mL弱酸性水,摇匀,取0.8 mL混合液,加入6.56mL弱酸性水,测样前滴加0.2%NaOH 640ul摇匀作为参比溶液,20s内在291nm处测待测样品的吸光度值。
实施例2
验证UV法准确性
为验证紫外-可见分光光度计检测结果的准确性,本发明分别挑选了青蒿素含量高、低2组共20个样品,分别用高效液相色谱法HPLC(n=3)和紫外-可见分光光度法UV(n=5)进行检测和结果比对,高效液相色谱法参照中国药典2020(第二部)原料中青蒿素含量检测方法,紫外检测按实施例1中的检测方法进行检测。
为验证上述两种方法的可替代性,对两种方法进行Bland-Altman一致性分析。
结果如表1所示,UV法和HPLC法检测结果在低含量区间(10~20 mg/g)和高含量区间(20~30 mg/g)均较为接近,Bland-Altman一致性分析结果见图5,可以看出,两种方法一致性好(P(H0:mean=0))=0.5939,具有可替代性。
表1 UV法和HPLC法检测样品青蒿素含量比较(单位:mg/g)
UV法精密度检验:
为了验证本发明检测方法的精密度,选择已知青蒿素含量差异较大的样品10个,按实施例1中方法制备样品溶液,连续测量5次,考察日内精密度。结果见表2:
表2 UV法的精密度检测
UV法稳定性检验
为了验证本发明的检测方法的稳定性,选择已知青蒿素含量差异较大的样品10个,按实施例1中方法制备样品溶液,分别于样品制备后0h、2h、4h、6h和10h检测,考察样品中青蒿素衍生物的稳定性。
结果如表3所示,各样品青蒿素衍生物在10h内基本稳定,RSD为0.05%~0. 16%;24h后检测含量有所降低,说明衍生物开始分解或转化,故样品溶液制备完成后,应当在10h内完成检测。
表3 UV法的稳定性检测
重复性检验
称取同一批号5份样品(n=5),按实施例1中制备样品溶液,碱转化衍生后测定结果如表1所示,各样品检测结果的RSD为0.91%~3.88%(分析表略),SEM为0.06~0.68,方法重复性较好。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种青蒿素含量快速检测方法
- 一种青蒿素紫外可见光快速检测方法