一种治缺血性心肌再灌注损伤的药物制剂及其应用
文献发布时间:2024-04-18 19:58:21
技术领域
本发明涉及一种治疗缺血性心肌再灌注损伤的药物制剂及其应用,属于药物新剂型领域。
背景技术
众所周知,急性心肌梗死(AMI)具有发病率高、致死性强,是导致全世界人口死亡的主要原因之一。对于急性心梗治疗包括:静脉药物溶栓治疗、直接经皮冠状动脉介入治疗(PPCI)、以及急诊冠状动脉搭桥术(CABG)等,其能使缺血心肌快速恢复血液灌流及氧供应。其中,PPCI是ST段抬高型心肌梗死(STEMI)再灌注治疗的首选治疗策略。尽管血运重建(PPCI、CABG以及药物溶栓等),心肌缺血后及时恢复心肌血流灌注是减轻心肌缺血损伤最有效的治疗策略,显著降低了心肌梗死患者急性期的死亡率,但仍有相当一部分病人由于心肌缺血再灌注损伤最终走向心室负性重构和心衰。
随着PPCI治疗STEMI患者的临床疗效的持续提升,虽然目前心肌梗死患者的病死率较以前明显下降,但是心肌梗死患者心力衰竭发生率却越来越高。因此,仍需要探索新的能够预防心肌再灌注损伤,及减少心梗面积且可以维持左室收缩功能和预防心力衰竭的发生的治疗方案。(罗峰,苏强.心肌缺血再灌注损伤防治的研究现状及展望[J].广东医学,2019,40(2):3)
心肌缺血再灌注损伤(MIRI)是临床心脏病学的重要问题,缺血的心肌在血管开放后导致了比血管闭塞时更严重的急性损伤,临床上会导致严重的心律失常,心肌坏死面积扩大,心脏破裂甚至死亡。因此,探讨引起再灌注损伤的机制,寻找有效的药物减轻这一损伤过程,对临床急性心肌梗死的救治有十分重要的意义。
目前,针对急性心肌梗死患者(AMI)在治疗过程中控制心肌缺血/再灌注损伤(MIRI)发生的手段主要有两种,即,手术过程血流控制和药物治疗。
手术过程血流控制,采用机械干预手段(包括缺血后处理和远隔缺血处理),在进行PPCI时选择性应用远端保护装置和血栓抽吸导管等措施。心肌缺血药物治疗经过了心肌缺血预适应-心肌缺血后适应的发展过程。从治疗时机不同分为心肌缺血预适应药物和后适应药物。预适应药物是指应用各种药物(如腺苷、内源性阿片肽等)模拟机体缺血预适应机制而发挥心肌保护作用。预适应保护心脏的主要机制是抑制钾通道、激活内源性物质(如腺苷、阿片肽类、缓激肽类等)、酶信号通路以及线粒体调整等。
然而临床中急性心肌梗死的时间通常不是预知的,患者到达医疗机构的时间是缺血发生后,因此对心肌缺血再灌注损伤的药剂治疗时机必须在至少在介入手术进行血管开通再灌注时进行使用,而不可能在心肌梗死事件前。因此,从临床治疗的角度讲,预适应药物的临床应用价值有很大的局限性。
与预适应相比,后适应(Myocardial ischemic postconditioning,MIP)药物可以在缺血再灌注时和再灌注后使用,则更具操作性和潜在的临床应用价值。理想的药物后适应处理,可以在恢复冠状动脉血流的同时,减轻心肌缺血再灌注损伤,进一步改善心肌缺血的治疗效果,明显提高患者的生活质量,已经成为目前研究的热点之一。
虽然在研究的后适应药物,可能对抗/拮抗药物再灌注损伤有一定作用,但已目前投入临床研究的药物都未能达到临床试验终点。例如,β受体阻断剂奈比洛尔、钠钙交换抑制剂阿米洛利、氧自由基清除剂依达拉奉(ClinicalTrials.gov ID:NCT00265239)、线粒体核孔MPTP调节剂制剂TRO40303(ClinicalTrials.gov ID:NCT01379261、NCT01374321),以及Stealth Biotherapeutics公司的线粒体靶向性小分子肽MTP-131(ClinicalTrials.govID:NCT01572909)等在临床2-3期结束后均未达到临床终点而终止研究。
其他机制的药物,例如HMG-CoA还原酶选择性抑制剂阿托伐他汀(ClinicalTrials.gov ID:NCT01780740)、免疫抑制剂环孢素A(ClinicalTrials.gov ID:NCT01650662)、GLP-1类似物利拉鲁肽(ClinicalTrials.gov ID:NCT02001363)等,都在早期曾被给予厚望,而其药物效果初步看来只在小样本临床实验或动物实验上有效果,在后期临床大样本队列的人群研究中尚未获得可靠的临床结果。因此,开发更加安全有效的抗心肌缺血/再灌注损伤药物仍然是全世界心血管病医生的共同研究热点。
随着对心肌缺血再灌注损伤机制的深入研究,在其诸多机制中,近年来自噬引起了研究者的关注。
自噬是细胞内一种通过溶酶体降解长寿命蛋白和受损细胞器的代谢途径,在细胞应激过程中起到了重要作用。自噬在心肌缺血再灌注损伤中的有利作用包括1)自噬通过产生ATP缓解缺血引起的能量危机;2)自噬被激活以清除功能紊乱的线粒体,促进线粒体更新,维持心肌细胞功能;3)在缺血再灌注后,自噬被激活以清除泛素化蛋白,维持细胞内蛋白质稳态。
虽然有关缺血再灌注时自噬现象的发生机制研究已取得较大的进步,mTOR、Atg、LC3及AMPK等对自噬的调节已经研究得比较清楚。但从众多的自噬调节靶点中探寻合适的自噬调节药物,还是一项具有挑战性的工作。
欣喜的是,抗肿瘤药物HDAC抑制剂辛二酰苯胺异羟肟酸(SAHA;伏立诺他)被发现可以防止I/R诱导的线粒体功能障碍和线粒体膜电位的损失,并减少心肌缺血前和缺血后造成的ROS产生。而且这些保护作用在敲除自噬功能相关基因Atg7和Atg5被抵消,证实了SAHA确实通过调节自噬通量来减少I/R心肌损伤;此外还发现SAHA独立于自噬途径的可调节由PGC-1α介导的线粒体功能来挽救I/R心脏损伤,发挥保护作用,也说明HDAC抑制剂作为缺血性心脏病中的自噬调节剂具有重要的临床意义。(Min Xie,Yida Tang,JosephA.Hill,HDAC inhibition as a therapeutic strategy in myocardial ischemia/reperfusion injury,J Mol Cell Cardiol.2019April;129:188–192)
更惊喜的是,不论是对模型动物用SAHA进行手术前预给药还是再灌注时给药,都可以降低模型动物的心肌再灌注损伤,可以减少心肌梗死面积,同时部分挽救了再灌注之前的收缩功能,两种给药方式在药效上没有显著性差别。进一步的研究发现用SAHA进行再灌注时给药更有利于梗死边缘区维持有益的自噬流,保护心肌细胞。
(Markus Wallner et al.HDAC inhibition improves cardiopulmonaryfunction in a feline model of diastolic dysfunction.Science TranslationalMedicine,2020,January 08;12(525))
(Xie M,Hill JA.HDAC-dependent ventricular remodeling.Trends,Cardiovasc Med.2013;23:229–235)
(Targeting Autophagy for the Therapeutic Application of HistoneDeacetylase Inhibitors in Ischemia/Reperfusion Heart Injury.Circulation March11,2014Mar 11;129(10):1088-91)
相似的实验结果,也反映在先申请的专利描述里(请号:201810076352.3,申请日:2018.01.26),SAHA配置成乳剂浓度为30mg/ml,预适应组按照50mg/kg剂量在手术前一小时一次性预适应给药,再灌注后适应组于手术前1小时和再灌注操作时,分别给予25mg/kg剂量药物,这两种给药方案都能显著的减少心肌梗死面积和心功能减退。
上述这些研究成果为SAHA成为治疗心肌缺血再灌注损伤后适应药物奠定了基础。后适应药物的理想状态是开发成注射剂,这样能够对患者进行皮下注射、静脉注射或直接PPCI手术时冠脉内给药,有利于心脏介入手术围术期的患者(可能已经处于昏迷状态),更具有临床意义。而SAHA几乎不溶于水,易溶于有机溶剂,其二甲基亚砜(DMSO)的溶解度>15mg/ml。
虽然,人们早期将SAHA作为抗肿瘤药物开发时,曾被设计为静脉注射溶液(浓度5mg/ml,pH值在11.0-11.2)。(W.K.Kelly etl,Phase I Clinical Trial of HistoneDeacetylase Inhibitor:Suberoylanilide Hydroxamic Acid AdministeredIntravenously Clinical Cancer Research,Vol.9,3578–3588,September 1,2003)该SAHA静脉注射制剂pH值11.2,对血管刺激性很大,容易造成静脉炎,不适合于心血管疾病患者静脉注射使用,更不适合于围术期的冠脉内注射,对心脏直接产生强烈的刺激。因此,曾在先申请专利(申请号:201810076352.3,申请日:2018.01.26)对SAHA制备成乳剂进行了描述。
随着我们对上述专利所述乳剂研究的深入,出乎意料的发现乳剂的药物代谢规律相对于以DMSO作为溶媒以及普通水溶性注射剂发生了较大的变化。主要的变化有:1)乳剂较DMSO溶媒注射剂及普通水溶注射剂药物半衰期明显延长;2)药物的组织分布规律与乳剂的配方以及乳剂的粒径有密切关系。
考虑到作为治疗心肌缺血再灌注损伤的药物,应当考虑到心梗急救手术期临床使用的便利性,以及安全性。因此,乳剂药物半衰期延长的特性有利于降低药物使用剂量,有利于降低药剂浓度,改小容量针剂为大容量输液,便于手术过程中建立静脉通道,减少医护人员的操作,方便临床用药和及时控制用药剂量。这可以进一步提高用药安全性,有着不言而喻的益处。此外,不同粒径和组方的乳剂的体内组织分布规律有较大差别,优化筛选具有心脏组织分布更佳的乳剂可以更进一步有效降低药物的剂量,保证药效,提高药物的安全性。
发明内容
本发明的目的在于克服现有技术的不足,提供一种治疗缺血性心肌再灌注损伤的药物制剂及其应用。
本发明药物制剂的优势在于:本药物制剂延长了SAHA的体内半衰期,延长药物作用时间,提高了生物利用度,提高了受损心脏部位的组织分布的靶向性,从而减少了实际临床剂量,提高了药物安全性。此外,本制剂为输液型乳剂,制剂药物浓度低,组织相容性好,血管刺激性小,方便于临床使用,减少医护人员的操作和掌握剂量,提高了临床使用的便利性。更有利于用于心脏手术围术期(即围绕手术的时期,包括术前、术中和术后)给药,也可以直接用于冠脉内注射,特别适用于作为心梗介入治疗围术期的治疗缺血再灌注损伤的药物的临床应用。
为实现上述目的,本发明的技术方案为:
一种治疗缺血性心肌再灌注损伤的药物制剂,所述药物制剂包括药物活性成分和辅料重量份数配比为:HDAC抑制剂药物0.5~30份、油相60~250份、乳化剂8~30份、渗透压调节剂10~30份、稳定剂0~12份、注射用水500~900份。
作为本发明药物制剂的优选:伏立诺他0.5~15份、油相80~150份、乳化剂10~18份、渗透压调节剂15~25份、稳定剂0~10份、注射用水600~800份;
所述油相辅料为:大豆油(含注射级)、蓖麻油、茶油、棉籽油、芝麻油、菜籽油、红花油,芥花油、橄榄油、椰子油、棕榈油、可可豆油等其中的一种或多种;链长在C6~C12之间的中等长链脂肪酸甘油酯,如MCT中链甘油三酯、Arlacel 80、Arlacel 86、Capmul MCM、Captex 200、Captex 355、Miglyol812、Myvacet、Myverol 18-92、油酸甘油酯、亚油酸甘油酯、聚乙二醇月桂酸甘油酯、油酸乙酯、亚油酸乙酯、辛酸/癸酸甘油三酯等其中的一种或多种;还可以是上述长链脂肪酸酯和中链脂肪酸酯的混合物中的一种或多种;
所述乳化剂辅料为:大豆卵磷脂及其修饰物(天然或合成)、蛋黄卵磷脂及其修饰物(天然或合成),PEG磷脂、氢化大豆磷脂HSPC、培化磷脂DSPE-MPEG2000、Ophase31、伯洛沙姆、聚氧乙烯氢化蓖麻油、水溶性VE(TPGS)、solutol HS-15、PEG300、PEG400、PEG1750及其单硬脂酸酯、Tween 80、Span 20、磷脂酰胆碱、二硬脂酰磷脂酰胆碱、肉豆蔻酰溶血卵磷脂、二肉豆蔻酰基卵磷脂、二月桂酰基卵磷脂、二芥酰磷脂酰胆碱、聚氧丙烯聚氧乙烯嵌段共聚物这些物质中的一种或多种;
所述渗透压调节剂辅料为:氯化钠、葡萄糖、山梨醇、木糖醇、甘露醇、甘油中的一种或多种;
所述稳定剂辅料为为:油酸、油酸钠、辛酸钠、胆固醇、胆酸、脱氧胆酸及其钠盐、维生素A、维生素C、维生素E中的一种或多种。
作为本发明药物制剂的优选,其特征在于,所述药物制剂包括药物活性成分和辅料重量份数配比为:伏立诺他0.5~15份、大豆油80~120份、蛋黄卵磷脂(E80)10~15份、甘油17~25份、胆固醇0~10份、油酸钠溶液0~10份、注射用水600~800份。
作为本发明药物制剂的优选,所述药物制剂包括药物活性成分和辅料重量份数配比为:伏立诺他0.5~15份、大豆油80~120份、MCT中链甘油三酯40~60份、磷脂酰胆碱10~15份、甘油17~25份、油酸钠溶液1~10份、注射用水600~800份。
作为本发明药物制剂的优选,所述药物制剂包括药物活性成分和辅料重量份数配比可以为:伏立诺他0.5~15份、芥花油80~120份、氢化大豆磷脂HSPC 5~9份、培化磷脂DSPE-MPEG20001~3份、甘露醇17~25份、胆固醇1~10份、注射用水600~800份。
作为本发明药物制剂的优选,所述药物制剂包括药物活性成分和辅料重量份数配比还可以为:伏立诺他0.5~15份、大豆油80~120份、PEG300单硬脂酸酯10~15份、甘油17~25份、油酸1~10份、注射用水600~800份。
作为本发明药物制剂的优选,所述药物制剂包括药物活性成分和辅料重量份数配比可以为:伏立诺他0.5~15份、蓖麻油80~120份、伯洛沙姆10~15份、葡萄糖17~25份、脱氧胆酸1~10份、注射用水600~800份。
作为本发明药物制剂的进一步优选,所述药物制剂包括药物活性成分和辅料重量份数配比可以为:伏立诺他0.5~15份、茶油80~120份、大豆磷脂10~15份、氯化钠17~25份、胆酸1~10份、注射用水600~800份。
作为本发明药物制剂的进一步优选,所述药物制剂包括药物活性成分和辅料重量份数配比为:伏立诺他0.5~15份、橄榄油80~120份、蛋黄卵磷脂10~15份、山梨醇17~25份、辛酸钠1~10份、注射用水600~800份。
作为本发明药物制剂的进一步优选,所述药物制剂包括药物活性成分和辅料重量份数配比为:伏立诺他3份、大豆油100份、蛋黄卵磷脂E80 12份、甘油22.5份、胆固醇4份、注射用水750份。
作为本发明药物制剂的进一步优选,所述药物制剂包括药物活性成分和辅料重量份数配比为:伏立诺他3份、大豆油50份、MCT中链甘油三酯卵磷脂50份、磷脂酰胆碱PL-100M12份、甘油22.5份、0.3%油酸钠溶液10份、注射用水750份。
作为本发明药物制剂的进一步优选,所述药物制剂包括药物活性成分和辅料重量份数配比为:伏立诺他3份、芥花油100份、HSPC(氢化大豆磷脂酰胆碱)6份、DSPE-mPEG20002份、甘露醇25份、胆固醇4份、注射用水750份。
作为本发明药物制剂的进一步优选,所述药物制剂包括药物活性成分和辅料重量份数配比为:伏立诺他3份、大豆油100份、PEG300单硬脂酸12份、甘油25份、油酸1份、注射用水750份。
作为本发明药物制剂的优选,所述药物制剂为静脉乳制剂,冠脉内注射液。
所述药物制剂的剂型为注射剂,其还可以为静脉注射乳剂、冠脉内注射液。优选为静脉注射乳剂,其注射乳剂的pH为6.3-7.9。
本发明药物制剂所选的药物可以为HDAC抑制剂药物,具体药物为伏立诺他,其化学结构如下:
SAHA(伏立诺他)
油相辅料选择:
油相在本发明所涉及的乳剂中质量分数(W/W)为0-50%,在本发明中要求能以较少的油相溶解处方量的药物,并在低温储藏条件下也不会有药物析出,同时在乳化剂的作用下,能与水相生成稳定的乳剂。在本发明中所应用的油相是含长链脂肪酸酯的天然植物油或经过结构改造和水解后的植物油或脂肪酸酯,如大豆油(含注射级)、蓖麻油、茶油、棉籽油、芝麻油、菜籽油、红花油,芥花油、橄榄油、椰子油、棕榈油、可可豆油等其中的一种或多种;或者是链长在C6~C12之间的中等长链脂肪酸甘油酯,如MCT中链甘油三酯、Arlacel80、Arlacel 86、Capmul MCM、Captex 200、Captex 355、Miglyol 812、Myvacet、Myverol18-92、油酸甘油酯、亚油酸甘油酯、聚乙二醇月桂酸甘油酯、油酸乙酯、亚油酸乙酯、辛酸/癸酸甘油三酯等其中的一种或多种;还可以是上述长链脂肪酸酯和中链脂肪酸酯的混合物。
优选的油相为大豆油、芥花油、MCT中链甘油三酯、大豆油:MCT中链甘油三酯(1:1)的混合物。
乳化剂的选择:
本发明采用的乳化剂是非离子表面活性剂和离子型表面活性剂的一种或一种以上的混合物,可使用磷脂酰胆碱PC含量大于70%的磷脂,包含大豆卵磷脂及其修饰物(天然或合成)、蛋黄卵磷脂及其修饰物(天然或合成),PEG磷脂、培化磷脂DSPE-MPEG2000、Ophase31、伯洛沙姆108、伯洛沙姆188、伯洛沙姆407、聚氧乙烯氢化蓖麻油、水溶性VE(TPGS)、solutol HS-15、PEG300、PEG400、PEG1750及其单硬脂酸酯、Tween 80、Span 20这些物质中的一种或两种和两种以上的混合物。在制备注射剂时,要选择溶血性小的,精制的乳化剂。
其中本发明优选的乳化剂的具体类型有:大豆磷脂PC80,氢化大豆磷脂HSPC、蛋黄卵磷脂E80、培化磷脂DSPE-MPEG2000、磷脂酰胆碱PL-100M、PC98T、二硬脂酰磷脂酰胆碱DSPC、肉豆蔻酰溶血卵磷脂M-lysoPC、二肉豆蔻酰基卵磷脂DMPC、二月桂酰基卵磷脂DLPC、二芥酰磷脂酰胆碱DEPC、聚氧丙烯聚氧乙烯嵌段共聚物Poloxamer,最优选蛋黄卵磷脂E80、培化磷脂DSPE-MPEG2000、磷脂酰胆碱PL-100M。
渗透压调节剂的选择:
优选的,所述渗透压调节剂包括氯化钠、葡萄糖、山梨醇、木糖醇、甘露醇、甘油中的一种或者由这些物质中的两种或两种以上组成的混合物最优先的渗透压调节剂为甘油和甘露醇。
稳定剂的选择:
优选的,所述稳定剂为油酸、油酸钠、辛酸钠、胆固醇、胆酸、脱氧胆酸及其钠盐、维生素A、维生素C、维生素E等或以上物质任意比例混合物,调节pH和降低乳化剂在高温灭菌时的水解速度,同时能够使乳剂的胃里带有负电荷,通过静电斥力来提高乳剂物理稳定性的物质。
优选的,所述稳定剂为油酸钠、胆固醇的一种或两种;
更优选的,所述稳定剂的油酸钠为浓度为0.1~0.6%油酸钠溶液;所述稳定剂的胆固醇使用量是上述乳化剂中磷脂用量的20~50%;所述稳定剂是上述稳定剂的一种或两种。
本发明药物制剂的制备工艺为:
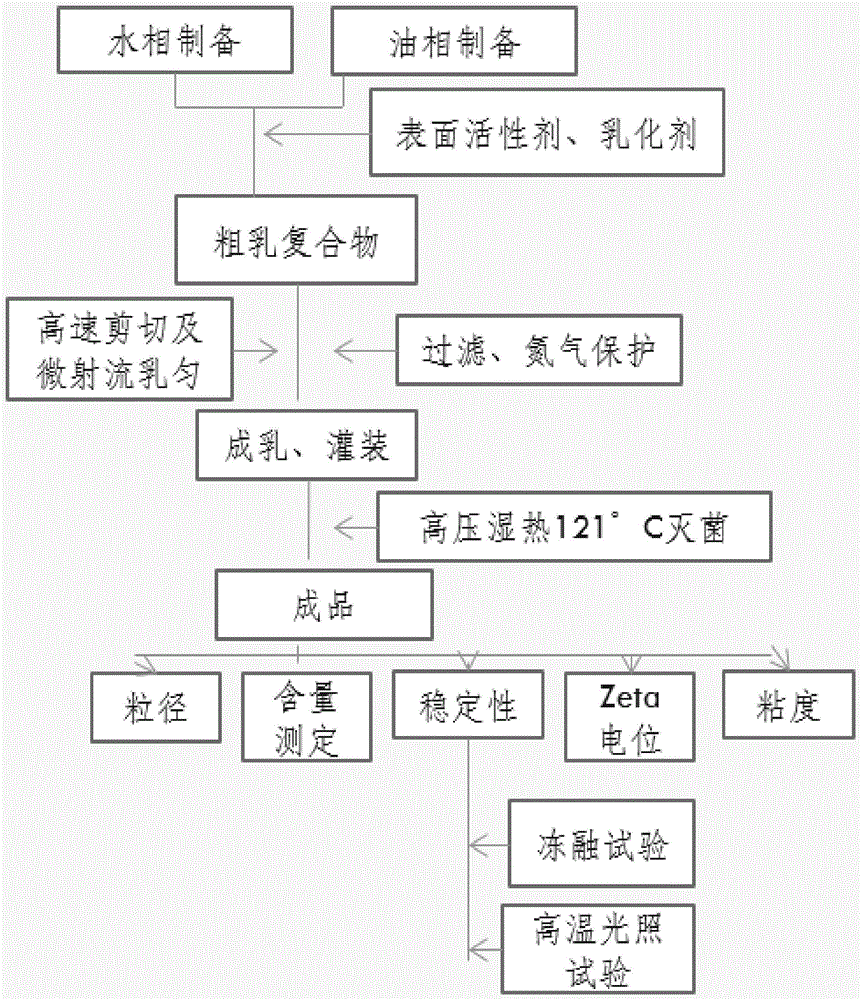
SAHA伏立诺他静脉乳剂的制备方法分初乳制备、均质、灭菌和质控等步骤。初乳的制备是利用超声法或高速剪切法(Y25型实验室高剪切分散乳化机,上海翼悾机电有限公司)。均质是采用高压均质机(ATS实验室通用型高压均质机AH-MINI 1)或微射流技术(NanoGenizer20K,实验型高压微射流均质机)。灭菌采用湿热灭菌法。质量控制主要采取粒径测定。粒径的控制范围为50~300nm,优选80-250nm。
本发明药物制剂制备操作工艺过程:
先将SAHA(伏立诺他)与油相、乳化剂混合后在超声振荡器上加热溶解(温度85-105℃),制备油相,另取纯水与稳定剂、渗透调节剂等混合,在磁力搅拌器下进行充分搅拌,制备水相;将油相在高速剪切机的搅拌下徐徐加入水相,控制温度60-80℃,在液体高速剪切机中(传动速度5000-8000RPM)剪切5-15min,高速剪切重复2-3次,控制温度不超过60-80℃,同时进行通氮保护,制备成初乳。
初乳在高压均质机或高压微射流均质机中匀化6-8次,控制温度60-80℃,同时进行通氮保护,乳剂冷却到室温后收集乳剂,测量乳剂粒径在100-250nm范围,调整pH值在6.3-7.9之间。
乳剂分装,充氮罐封压盖后,121℃蒸汽湿热灭菌,10-15min,既得。
用该方法制备得到的各个配方的SAHA乳剂的粒径为在150-250nm范围内,其他指标符合注射剂的要求。
本发明药物制剂的有益效果:
1、本发明经过大量实验研究,意外的发现可以通过调整组方和配比有效延长药物半衰期。相比于在先申请专利201810076352.3,组方配比中特别针对于乳化剂、稳定剂、渗透压调节剂等进行了筛选和优化,特别筛选出油酸钠或胆固醇等作为乳剂稳定剂,磷脂酰胆碱PC含量大于70%的磷脂作为乳化剂,增加与药物的亲和力,提高乳剂的稳定性,减缓药物从乳剂颗粒的释放速度,从而延长药物体内半衰期,延长药物作用时间。
2、由于乳剂组方的调整,延长了药物半衰期,使得可以降低给药剂量。进一步的实验结果表明,使用调整后的乳剂组方,相比于在先申请专利201810076352.3的小鼠给药剂量50mg/kg,最低起效剂量降低到6mg/kg,且在非预处理给药的情况下,再灌注时单剂量给药,依然有效的减轻心肌梗死面积,减少心肌细胞的凋亡。
3、本发明药物制剂提高了受损心脏部位的代谢组织分布;经过药物代谢组织分布的测试,本静脉注射用乳剂有易于分布到心脏受损部位的靶向特性。实验表明,调整后的乳剂组方,以相对摄取率(re)为指标,Heart(re)=4.89>1,有良好的心脏组织分布的靶向性。
4、本发明药物制剂通过优化乳剂的配方辅料,筛选出油酸钠作为pH调节剂,筛选出甘油作为乳剂渗透调节剂,控制pH值和渗透压在适宜的范围,减少了血管刺激性。
5、本发明的SAHA注射乳剂,经过6个月的加速稳定性试验观察,其外观形状为白色乳状液,乳滴平均粒径在100-200nm之间,并未发生分层现象,其乳液稀释后的Zeta电位在-12~-32之间,也说明其药物乳液所带的负电荷均匀稳定。其药物制剂的包封率均在89.6%~96.2%,其药物乳液pH值在6.3~7.9,上述实验结果表明,表明现在的伏立诺他乳剂工艺稳定,可重现性强。
附图说明
此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
图1-本发明药物制剂的制备工艺流程图;
图2-本发明药物制剂实施例1、2、3、4、5组方的静脉注射药时曲线;
图3-本发明药物制剂实施例1组方不同剂量对小鼠心肌梗死面积/心肌缺血面积百分比(%)组间比较;
图4-本发明药物制剂实施例1组方不同剂量对小鼠心肌梗死面积/心肌缺血面积百分比(%)组间比较。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
实施例1
1.1本发明药物制剂的制备处方及工艺方法
本发明对处方1、2、3、4以及处方5在先专利(201810076352.3)的乳剂配方、制备过程、乳剂表征、稳定性研究进行系统研究。
1.1.1、本发明药物制剂的处方剂量
表1本发明药物制剂涉及5种处方配比剂量
1.1.2、本发明药物制剂的工艺过程
处方1:称取大豆油10g、蛋黄卵磷脂E80 1.2g,磁力搅拌器充分混合后,加热至75-80℃,加入0.3gSAHA、胆固醇0.4g,混合后在超声振荡器上维持温度在75-80℃,待溶液澄清后可得油相;将油相在高速剪切机的搅拌下徐徐加入预先加热的水相(水75g,甘油2.25g),控制温度60-80℃,在液体高速剪切机中(传动速度5000-8000RPM)剪切5-15min,高速剪切重复2-3次,同时进行通氮保护,制备成初乳。初乳在微射流高压均质机中匀化6-8次(压力1000-1200bar),控制温度60-80℃,同时进行通氮保护,乳剂冷却到室温后收集乳剂,测量乳剂粒径在100-250nm范围,调整pH值在6.3-7.9之间。
乳剂分装,充氮罐封压盖后,121℃蒸汽湿热灭菌15min,灭菌后所有样品均匀,未见分层,既得。
处方2:称取大豆油:MCT(1:1)混合物10g,磷脂酰胆碱PL-100M 1.2g,磁力搅拌器充分混合后,加热至75-80℃,加入0.3gSAHA,混合后在超声振荡器维持温度在75-80℃,待溶液澄清后,可得油相;将油相在高速剪切机的搅拌下徐徐加入水相(水75g,甘油2.25g,0.3%油酸钠溶液1.0g),控制温度60-80℃,在液体高速剪切机中(传动速度5000-8000RPM)剪切5-15min,高速剪切重复2-3次,同时进行通氮保护,制备成初乳。初乳在高压均质机中匀化6-8次(压力600-700bar),控制温度60-80℃,同时进行通氮保护,乳剂冷却到室温后收集乳剂,测量乳剂粒径在100-250nm范围,调整pH值在6.3-7.9之间。
乳剂分装,充氮罐封压盖后,121℃蒸汽湿热灭菌15min,灭菌后所有样品均匀,未见分层,既得。
处方3:称取芥花油10g、氢化大豆磷脂HSPC 0.6g、培化磷脂DSPE-MPEG2000 0.2g,磁力搅拌器充分混合后,加热至75-80℃,加入0.3gSAHA、胆固醇0.4g,混合后在超声振荡器维持温度在75-80℃,待溶液澄清后,可得油相;将油相在高速剪切机的搅拌下徐徐加入水相(水75g,甘露醇2.5g),控制温度60-80℃,在液体高速剪切机中(传动速度5000-8000RPM)剪切5-15min,高速剪切重复2-3次,同时进行通氮保护,制备成初乳。初乳在微射流高压均质机中匀化6-8次(压力1000-1200bar),控制温度60-80℃,同时进行通氮保护,乳剂冷却到室温后收集乳剂,测量乳剂粒径在100-250nm范围,调整pH值在6.3-7.9之间。
乳剂分装,充氮罐封压盖后,121℃蒸汽湿热灭菌15min,灭菌后所有样品均匀,未见分层,既得。
处方4:称取大豆油10g、PEG300单硬脂酸酯1.2g、油酸0.1g,磁力搅拌器充分混合后,加热至75-80℃,加入0.3g SAHA,混合后在超声振荡器维持温度在75-80℃,待溶液澄清后,可得油相;将油相在高速剪切机的搅拌下徐徐加入水相(水75g,甘油2.5g),控制温度60-80℃,在液体高速剪切机中(传动速度5000-8000RPM)剪切5-15min,高速剪切重复2-3次,同时进行通氮保护,制备成初乳。初乳在高压均质机中匀化6-8次(压力600-700bar),控制温度60-80℃,同时进行通氮保护,乳剂冷却到室温后收集乳剂,测量乳剂粒径在100-250nm范围,调整pH值在6.3-7.9之间。
乳剂分装,充氮罐封压盖后,121℃蒸汽湿热灭菌15min,灭菌后所有样品均匀,未见分层,既得。
处方5(在先专利申请201810076352.3):称取SAHA 3g、加入吐温80和甘油,加热到80℃,加水约100ml,至原料药溶解,加入大豆磷脂,剪切(6000rpm)混合均匀,制成水相;称取油酸和注射用大豆油,混合均匀,制成油相;油水两相在70℃条件下,高速剪切混合(6000rpm)30分钟制成初乳。将初乳在800bar压力下进行均质化,循环6遍,装入安瓿,封口,115℃下灭菌30分钟即得。
1.1.3本发明药物制剂乳剂表征上述乳剂制备后,检测相应的乳剂并进行粒径、Zeta电位包封率、pH值等质量检测,检测结果见下表2。
表2本发明药物制剂质量检测指标结果
1.1.4、本发明药物制剂稳定性研究
对上述处方静脉乳剂组方,进行药物制剂稳定性考察(考察指标:平均粒径、Zeta电位、包封率含量和pH值变化)进行了6个月稳定性考察,在室温条件下,分别在0、1、3、6个月对样品进行了外观、平均粒径、Zeta电位、包封率含量和pH值变化进行考察。其中平均粒径和Zeta电位采用Zetasizer Lab纳米粒度电位仪测定,pH值用pH计进行测量,包封率(含量)采用超速离心法将油相、乳化层和水相分开,通过测定水中SAHA的含量计算包封率,伏立诺他的含量测定方法采用高效液相色谱法,色谱条件为C18柱,(5μm,50mm×4.6mm),流动相为乙腈-0.1%磷酸水(30:70)(用三乙胺调pH至3.0);检测波长为241nm,流速为1.0ml/min,以上药物制剂稳定性测定结果见表3。
表3本发明药物制剂质量检测各项稳定性指标
实施例2
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他4g、大豆油100g、蛋黄卵磷脂13g、甘油20g、胆固醇6g、注射用水700g;制成本发明药物静脉乳剂。
实施例3
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他5g、大豆油120g、蛋黄卵磷脂10g、甘油25g、胆固醇10g、注射用水800g;制成本发明药物静脉乳剂。
实施例4
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他2g、大豆油80g、蛋黄卵磷脂15g、甘油17g、胆固醇1g、注射用水600g;制成本发明药物静脉乳剂。
实施例5
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他4g、大豆油50g、MCT中链甘油三酯50g、磷脂酰胆碱12g、甘油21g、油酸钠溶液5g、注射用水700g;制成本发明药物静脉乳剂。
实施例6
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他2g、大豆油60g、MCT中链甘油三酯60g、磷脂酰胆碱10g、甘油25g、油酸钠溶液10g、注射用水800g;制成本发明药物静脉乳剂。
实施例7
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他5g、大豆油40g、MCT中链甘油三酯40g、磷脂酰胆碱15g、甘油17g、油酸钠溶液1g、注射用水600g;制成本发明药物静脉乳剂。
实施例8
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他4g、芥花油100g、氢化大豆磷脂6g、培化磷脂2g、甘露醇21g、胆固醇6g、注射用水700g;制成本发明药物静脉乳剂。
实施例9
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他2g、芥花油80g、氢化大豆磷脂7g、培化磷脂1g、甘露醇25g、胆固醇10g、注射用水800g;制成本发明药物静脉乳剂。
实施例10
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他5g、芥花油120g、氢化大豆磷脂5g、培化磷脂3g、甘露醇17g、胆固醇1g、注射用水600g;制成本发明药物静脉乳剂。
实施例11
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他4g、大豆油90g、PEG300单硬脂酸酯12g、甘油19g、油酸6g、注射用水750g;制成本发明药物静脉乳剂。
实施例12
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他5g、大豆油80g、PEG300单硬脂酸酯15g、甘油25g、油酸10g、注射用水800g;制成本发明药物静脉乳剂。
实施例13
按照实施例1制备方法的制备本发明药物制剂,其处方的剂量配比为:伏立诺他2g、大豆油120g、PEG300单硬脂酸酯10g、甘油17g、油酸1g、注射用水600g;制成本发明药物静脉乳剂。
实施例14本发明药物制剂的药代动力学研究
14.1本发明药物制剂的半衰期和药时曲线测定
14.1.1试验方法
用SD大鼠分别给予实施例1的处方1、2、3、4、5静脉给药,测量血药浓度。选取体重200g左右SD大鼠15只,随机分5组,每组3只,分别按照10mg/kg剂量分别尾静脉注射处方1、2、3、4、5(3mg/ml)分别于0、1、5、15、30、60、120、240、360、720min,于眼底静脉丛取血约0.5ml,肝素抗凝,精密吸取100微升血浆与200μL乙腈混合(含0.15%甲酸,300ng/mL苄基苯甲酰胺做内标)。将样品旋转15s,孵育在室温下旋转10分钟,并以13200rpm的转速在标准微量离心机,所得血浆于-20℃冷冻待用。然后,取上清液10μl液相色谱进样分析。
14.1.2血药浓度测定方法
本发明试验采用液相色谱-串联质谱法(HPLC-MS/MS)进行血药浓度测定。将100微升血浆与200μL乙腈混合(含0.15%甲酸,300ng/mL苄基苯甲酰胺做内标)。将样品旋转15s,孵育在室温下旋转10分钟,并以13 200rpm的转速在标准微量离心机。然后,取上清液10μl液相色谱进样分析。SAHA伏立诺他(分子离子峰265.1/232.021)和内标(分子离子峰212.2/91.1)的保留时间分别为4.1和4.5min。
流动相:缓冲液A:水+0.1%甲酸;缓冲液B:甲醇+0.1%甲酸;流量,1.5mL/min;色谱柱:安捷伦C18 XDB柱,(5μm,50mm×4.6mm);液相条件:0-1分钟95%A,1-1.5分钟梯度至95%B;1.5至2.5分钟100%B,2.5至2.6分钟梯度至95%A;2.6至3.5分钟95%A。
采用电喷雾离子源,正离子多反应离子监测模式检测。根据内标计算伏立诺他的峰面积。测定不同时间点血浆中药物浓度,扣除血浆本底药物浓度,即为所得血药浓度。得到处方1、2、3、4、5的静脉注射药时曲线,可参见附图2。
14.1.3本发明药物制剂药物动力学参数
采用DAS2.2.2.1药物动力学软件对血药浓度-时间数据进行房室模型拟合,结果表明SAHA制剂大鼠尾静脉注射给药的药物动力学过程均符合双隔室模型,结果见表。由表4结果可知,处方1、2、3、4、5的AUC随着组方的不同而有较大的差距。其中处方1有效的延长了半衰期,且AUC面积提示有良好的生物利用度。
表4不同处方的药物动力学研究结果
14.1.4本发明药物制剂的药物组织分布
1.1.4.1试验方法
取禁食12h小鼠135只随机分成5组,分别尾静脉注射处方1、2、3、4、5,给药剂量10mg/kg,每个时间点3只鼠,并于给药后1、5、15、30、60、120、240、360、720min眼眶取血于肝素抗凝管中,13200r/min离心分离血浆,迅速处死动物,取出心、肝、脑、脾、肺、肾,用冰生理盐水冲洗干净,滤纸沾干,称取0.5g。所得组织于-20℃冷冻保存待用。
14.1.4.2血浆样品的处理:
取小鼠血浆0.1ml于1.5ml离心管中,加入甲醇0.2ml,正己烷0.4ml,4℃下10000r/min离心10min,取上清液20μL进样测定,避光操作。
14.1.4.3组织样品的处理及检测:
精密称取0.1g组织进行匀浆,匀浆介质位0.2ml生理盐水,将匀浆液置于1.5ml离心管中,加入甲醇0.2ml,正己烷0.4ml,涡旋3min,4℃下10000r/min离心10min,移取正己烷于1.5ml离心管中,重复操作2次,合并正己烷层,离心浓缩挥干正己烷,200μL流动相溶解,涡旋3min,10000r/min离心10min,取上清液20μL进样测定,避光操作。
流动相:缓冲液A:水+0.1%甲酸;缓冲液B:甲醇+0.1%甲酸;流量,1.5mL/min;色谱柱:安捷伦C18 XDB柱,(5μm,50mm×4.6mm);液相条件:0-1分钟95%A,1-1.5分钟梯度至95%B;1.5至2.5分钟100%B,2.5至2.6分钟梯度至95%A;2.6至3.5分钟95%A。采用电喷雾离子源,正离子多反应离子监测模式检测。根据内标计算伏立诺他的峰面积。
测定不同时间点组织中的药物浓度,扣除组织中本底药物浓度,即为所得组织中的血药浓度,得到不同组织中药物经时变化计算不同组织中SAHA的药时曲线下面积AUC值,结果见表5。
表5不同组织中SAHA的AUC值
14.1.4.4靶向性初步评价
以相对摄取率(r
表6不同组织中SAHA的靶向性
相对摄取率结果显示,在考察时间内,处方1在心、脾的含量是在先专利处方5组的5.75和3.6倍,处方2在心、脾的含量是在先专利处方5的2.75、2.32倍,处方3和处方4在肺的含量分别是处方5的2.4和2.17倍,处方4的乳剂粒径最小,在脑中的含量是处方5的3.17倍,这也说明经过优化的本发明静脉乳剂组方的调整,以及通过对粒径的控制可以有效的调整药物的组织分布。
实施例15本发明药物制剂的体外溶血实验
取家兔血5ml,置抗凝离心管中,放入装载小玻璃珠的锥形瓶中振摇10min,除去纤维蛋白原,离心取上清液,将红细胞层用0.9%氯化钠注射液冲洗4次,每次10ml,离心取上清液,直到上清液无红色为止,然后吸取一定量红细胞,用0.9%氯化钠注射液按体积比稀释制备2%红细胞混悬液,4℃冰箱冷藏备用。
取7支试管,编号为1-7,溶血试验设计见表6,分别在各管内加入适量2%红细胞混悬液和0.9%氯化钠注射液(7号管内加蒸馏水做阳性对照)置37℃恒温水浴中30min后又分别加入不同量的处方1-5(6号管做阴性空白对照),摇匀放入37℃恒温水浴中,分别于0.5、1、2、3、4h时观察试管内红细胞溶血情况。溶血试验设计加样表。
试验结果观察判断标准:分级:试管情况;溶血:溶液澄明红色,管底无细胞残留或有少量红细胞残留;无溶血:红细胞全部下沉,上层液体无色澄明;凝聚:溶液中有棕红色或红棕色絮状沉淀,振摇之后不能分散。
表7不同处方静脉注射乳剂溶血试验设计(ml)
表8本发明药物制剂的观察结果
实施例16本发明药物制剂对动物的血管刺激性的影响
血管刺激性试验取健康新西兰白兔21只,雌雄各半,随机分成7组,每组3只。按无菌操作方法于耳缘静脉滴注(10ml/kg,1ml/min,1次/Day)给药,第一组右耳滴注溶媒生理盐水溶液,左耳滴注生理盐水溶液;第二组左右耳都滴注溶媒5%葡萄糖溶液;第三组至第七组分别滴注处方1-5,体积均为10ml/kg,1次/日,连续5日,于每日给药前及末次给药后72h,肉眼观察注射部位有无红肿,充血等刺激现象。处死动物,以耳缘静脉注射点向心方向2cm,连续取3段血管连同组织,进行病理学检查,观察静脉血管组织有无炎症反应,甚至变性坏死等显著性刺激反应。另一侧对照血管同法处理。
经过5日给药前后观察,以及末次给药72h后对各段静脉血管及组织进进行病理观察组织学检测,结果见表9。各个给药组3只家兔耳缘静脉注射部位均未见红肿、充血等刺激现象。病理组织学检查结果,生理盐水、5%葡萄糖左耳对照组,耳缘静脉血管内皮细胞排列正常,未见层次增多、排列紊乱的改变,血管腔内无附壁血栓,无炎细胞侵润。管壁内、中、外三层结构清晰,可见内弹力板,未见纤维组织增生。管壁无增厚、坏死及炎症等形态学改变,为正常组织结构;溶媒和各个处方给药组血管组织结构正常,与左耳对照组血管比较无异常改变。
表9兔耳缘静脉病理损伤程度比较
上述试验结果说明,本发明的各处方有良好的血管耐受性,对血管刺激小,安全性良好,为作为冠脉内注射使用的注射液提供了进一步的安全保障。实施例17本发明药物制剂对模型动物的心梗面积/缺血面积的影响
17.1建立小鼠心肌缺血再灌注模型
模型组(I/R)小鼠接受2%异氟烷吸入麻醉后,术者在其左侧前胸壁上切开一个1.2cm左右的皮肤切口,并斜着暴露左胸第四肋间隙,轻柔而迅速地撑开胸膜腔和心包腔,挤出心脏。使用6号带针缝合线于心脏前壁的左冠状动脉前降支处结扎活结,肉眼观察左室心肌组织颜色转为灰白,确认结扎成功。其后,迅速将心脏放置于胸腔内,手动排空腔内空气,关闭胸腔,缝合皮肤伤口,并留置结扎心脏左冠状动脉的前降支的活结的线头于胸外。将小鼠置于空气流通的环境中,监控其复苏情况。缺血45分钟后,通过轻柔平缓地拖拽胸外留置的线头松解结扎前降支的滑结,心肌组织获得血流再灌注(灌注时间为24h)。假手术组(sham)按照模型组(I/R)上述相同步骤挤出心脏,6号带针缝合线穿过心脏前壁的左冠状动脉前降支处但不打结,按照相同方式排除胸腔空气,关闭胸腔,缝合伤口。
17.2小鼠于心肌缺血再灌注时用处方1给药,具体分组及给药见表9。
表10本发明药物制剂处方1不同剂量分组及给药剂量
通过在线分组工具Quickcalcs(http://www.graphpad.com/quickcalcs/)对动物进行随机分组,将动物分为I/R,I/R+4.5mg/kg,I/R+6mg/kg,I/R+9mg/kgI/R+13.5mg/kg组别。处方1或溶剂通过尾静脉注射给予I/R小鼠,于心肌再灌注操作时按照体重一次给予所有药物。
17.3 Evans Blue&TTC双染色法检测小鼠心脏梗死/缺血面积
心肌缺血再灌注组的小鼠在再灌注24h后再结扎心脏血管,股动脉或主动脉注射1%的Evans Blue 200μl,显示缺血心肌;假手术组不结扎心脏血管,股动脉或主动脉注射1%的Evans Blue 200μl。取其心脏,PBS中洗去血液至PBS不变色,擦干水分,-40℃冰冻60分钟,放入模具,垂直于心脏前后长轴等距离切片(5片冠状切片),置入2%TTC磷酸缓冲液PH 7.4中,37℃孵育20分钟。图像采集系统扫描后通过Image J计算统计心肌缺血范围和梗死范围。
试验结果统计结果见表10。
表11不同给药剂量的药物制剂对小鼠心肌梗死面积(%)组间比较
表11不同给药剂量的药物制剂对心肌梗死面积/心肌缺血面积百分比%组间比较
/>
处方1(4.5mg/kg,6mg/kg,9mg/kg,13.5mg/kg)对C57BL/6雄性小鼠急性心肌缺血/再灌注损伤指标的Tukey检验的单因素方差分析。表示为mean±SD,每组n=10。*p<0.05,**p<0.01vs.I/R group。
结论:本发明药物制剂处方1剂量9mg/kg组较模型组有效降低了心肌梗死面积以及心肌梗死/缺血面积百分比(p<0.05);此外,剂量13.5mg/kg组比模型组有效降低了心肌梗死面积。因此,通过乳剂组方优化筛选出的心脏组织分布最佳的本发明药物制剂处方1在较低剂量9mg/kg,且仅在再灌注时单剂量给药,也可以显著性降低心肌梗死面积达40%,降低心肌梗死/缺血面积百分比达35%。而在先专利申请乳剂则需要在手术前给再灌注前后分别给药25mg/kg,或在手术前一小时一次性给药50mg/kg来达到显著性降低心肌梗死面积。因此,本发明的静脉乳剂可以有效降低临床药物剂量,为该药物安全性提供进一步的保障。
本发明并不局限于上述的具体实施方案,上述的具体实施方案仅仅是示意性的、指导性的,而不是限制性的。本领域的普通技术人员在本说明书的启示下,但凡在本发明的精神和实质范围内,所作的任何改变、等同替换和改进,均在本发明的保护范围之内。
- 口服富勒烯材料在制备预防和/或治疗心肌缺血再灌注损伤或缺血性心脏病药物中的应用
- 口服富勒烯材料在制备预防和/或治疗心肌缺血再灌注损伤或缺血性心脏病药物中的应用