一种美法仑杂质D的纯化方法
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及药物纯化技术领域,特别涉及一种美法仑杂质D的纯化方法。
背景技术
美法仑(Melphalan)是氮芥类化疗药物,别名:米尔法兰、L-溶肉瘤素。是苯丙氨酸氮芥的左旋体,是一种烷化剂,由于其结构的物异性,使其可进入肿瘤细胞内而发生作用,导致细胞死亡,从而起到抗肿瘤作用。与其他烷化剂相同,与DNA及RNA发生交叉联结,导致肿瘤细胞死亡,为细胞周期非特异性药物。谷胱甘肽水平提高,药物运转减慢,DNA修复增强,可导致耐药性增加。可用与治疗多种肿瘤疾病,包括多发性骨髓瘤、恶性淋巴瘤、乳腺癌、卵巢癌、慢性白血病以及甲状腺癌等。还可动脉灌注用于治疗恶性黑色素瘤、骨肉瘤及软组织肉瘤等肢体恶性肿瘤。
而药物中含有的杂质是影响药物纯度的主要因素,如药物中含有超过限量的杂质,就有可能使理化常数变动,外观性状产生变异,并影响药物的稳定性;杂质增多也必然使药物的含量偏低或活性降低,毒副作用显著增加。因此,杂质对照品的纯度对药物的质量控制起到关键性的作用。所以对于美法仑的原料和制剂的质量控制,必须有质量合格的杂质对照品。
目前,国内外研究多集中在临床应用和衍生物的合成上,对美法仑的杂质研究较少。本发明涉及的(S)-2-氨基-3-(4-((2-氯乙基)(2-羟乙基)氨基)苯基)丙酸即美法仑杂质D作为美法仑的一个重要杂质对照品之一,目前无法检索到美法仑杂质D的相关纯化方法。而常规有机合成制得的美法仑杂质D粗品可能会有较高的初始纯度,但是该粗品含有大量的无机盐混杂,该样品由于含量虚高会产生较大的校正因子偏差,同时受到盐潮解吸收水汽的影响,该样品极易降解(式1)。
如现合成的样品现制现测时检测纯度为97.8%。放置约15min后纯度下降至97.1%并观察到保留时间为4.2min的杂质峰面积增加约1%。而进一步放置3h后纯度明显下降,检测纯度值为91%,6h后纯度下降至88%,4.2min的降解产物则对应增加(如附图2)。甚至进一步延长实验时间至72小时后,溶液中的美法仑EP杂质D几乎完全分解,4.2min左右的降解杂质成为主产物。
随着美法仑杂质D放置时间的延长,主峰纯度持续降低,这极大限制了该样品的纯化和使用,同时对产品的纯化提出了较高的要求。本发明提供了一种针对美法仑杂质D的纯化的简单易行的方法。
发明内容
针对现有技术存在的问题,本发明提供一种美法仑杂质D的纯化方法。本发明提供一种美法仑杂质D的制备纯化路线,为美法仑原料药及其制剂的杂质分析及研究提供了便利,为美法仑的生产和用药安全提供检测方法及判定依据
为了实现上述目的,本发明提供一种美法仑杂质D的纯化方法,包括如下步骤:
步骤S1:配制流动相A、流动相B、稀释剂、样品液;
步骤S2:运用制备型高效液相色谱对样品液进行纯化,
条件如下:流速:10mL/min~40mL/min;柱温:10C~30℃;进样量:1~5ml;检测波长:200nm~220nm;色谱柱:十八烷基硅烷键合硅胶;
步骤S3:通过调节梯度洗脱中有机相的比例,进行梯度洗脱:
步骤S4:取样品溶液,注入制备高效液相色谱仪中,然后按照面积归一法,以样品色谱图中各色谱峰峰面积百分比进行计算。
优选的,所述步骤S3中洗脱梯度为:
在0到6min阶段,流动相A:流动相B为95:5~80:20;
在6到12min阶段,流动相A:流动相B为80:20~70:30;
在12min到18min阶段,流动相A:流动相B为70:30~60:40;
在18min到24min阶段,流动相A:流动相B为60:40~60:40;
在24min到26min阶段,流动相A:流动相B为60:40~5:95;
在26min到30min阶段,流动相A:流动相B为5:95~5:95。优选的,所述步骤S1中的流动相A选自经0.22um微孔过滤膜过滤的超纯水。
优选的,所述步骤S1中的流动相B选自色谱级乙睛。
优选的,所述步骤S1中的样品液的配制:称取美法仑杂质D,加稀释剂超声溶解至澄清溶液。
优选的,所述步骤S1中的稀释剂选自超纯水。
优选的,所述步骤S2中的流速:25ml/min;柱温:25℃;检测波长:210nm。
采用本发明的技术方案,具有以下有益效果:
在本技术发明中,梯度洗脱中有机相含量对美法仑杂质D的纯化有很大的影响。通过不同的比例组成进行梯度洗脱,可以达到美法仑杂质D与其他杂质的有效分离。最后确认梯度洗脱为在0到6min,流动相A:B为95:5~80:20;在6到12min,流动相A:B为80:20~70:30;在12min到18min,流动相A:B为70:30~60:40;在18min到24min,流动相A:B为60:40~60:40;在24min到26min,流动相A:B为60:40~5:95;在26min到30min,流动相A:B为5:95~5:95此梯度条件下进行洗脱,美法仑杂质D能分离出更多的杂质,仍能保持较好的分离度,分离度均大于1.5,符合要求。并且该梯度条件下,将单次纯化量由10mg/ml增加至100mg/ml,同样能得纯度≥95%的美法仑杂质D产品,样品峰与其他杂质分离度为1.61,符合要求,该方法操作简单,成本低。相较于目前国内外对美法仑杂质D纯化研究的空缺,本发明能为美法仑杂质D对照品的产品质量提供良好的方法。进而有效控制美法仑产品质量,为美法仑原料药及其制剂的杂质分析及研究提供便利,为美法仑的生产和要用安全提供检测方法和判断依据。
附图说明
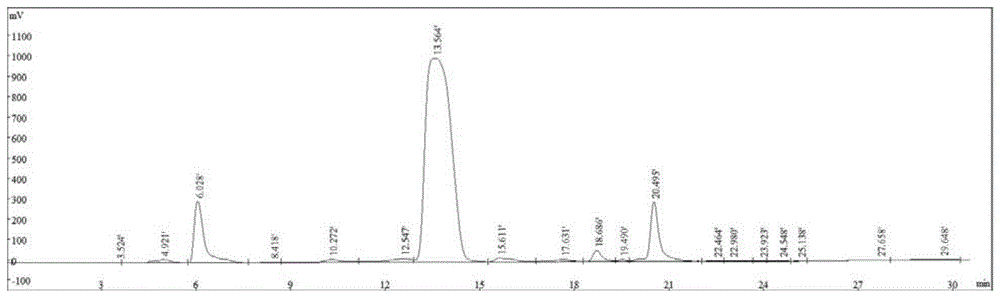
图1为本发明单次进样100mg/ml的制备纯化液相图。
图2为现制样品初始检测与放置0min、15min、3h、6h后的色谱纯度图。
具体实施方式
以下结合附图和具体实施例,对本发明进一步说明。
实施例1:
美法仑杂质D的纯化方法:
1)色谱条件:
仪器:制备高效液相色谱仪;
色谱柱:MorphlingTMAQ-C18,250*30mm,5um;
柱温:25℃;
流速:25ml/min;
检测波长:210nm;
流动相A:经0.22um微孔过滤膜过滤的超纯水;
流动相B:色谱级乙腈;
稀释剂:超纯水;
按照下表1进行梯度洗脱:
表1:梯度洗脱程序
2)样品配制:
样品溶液的配制:称取美法仑杂质D,加稀释剂超声溶解至澄清溶液,即得,美法仑杂质D待测溶液浓度为10mg/ml。
3)检测:
取待测溶液1ml,注入制备高效液相色谱仪中,色谱图如图1所示。
4)计算:
按照面积归一法,以样品色谱图中各色谱峰峰面积百分比计。
5)实验结果:
对实施例1中的主峰前后的杂峰进行分析,结果见表2。
表2:实施例1中纯化HPLC图分析结果
结论:从表2可以看出,在该色谱条件中美法仑杂质D与其他杂质的保留时间相差不大,分离度小于1.5,在上表1梯度洗脱下纯化时,该方法对美法仑杂质D不能较好的分离。
实施例2:
美法仑杂质D的纯化方法:
具体实施参照实施例1,与实施例1不同的是,梯度含有机相比例不同:参照下表3。
表3:实施例2梯度洗脱程序
实验结果:
对实施例2色谱图中的主峰前后的杂峰进行分析,结果见表4。
表4:实施例2中纯化HPLC图分析结果
结论:从表4可以看出,在该色谱条件中美法仑杂质D与其他杂质的保留时间相差不大,但主峰分离度大于1.5,在上表3梯度洗脱下纯化时,该方法对美法仑杂质D能有效的分离。
实施例3:
美法仑杂质D的纯化方法:
具体实施参照实施例1,与实施例1不同的是,梯度含有机相比例不同:参照下表5。
表5:实施例3梯度洗脱程序
实验结果:
对实施例3色谱图中的主峰前后的杂峰进行分析,结果见表6。
表6:实施例3中纯化HPLC图分析结果
结论:从表6可以看出,在该色谱条件中美法仑杂质D与其他杂质的保留时间相差明显,相比于实施例2,主峰后的杂峰保留时间由8.35后移至9.50,主峰分离度由2.18增加至3.15,在上表5梯度洗脱下纯化时,该方法对美法仑杂质D有较好的分离。
实施例4:
美法仑杂质D的纯化方法:
具体实施参照实施例1,与实施例1不同的是,梯度含有机相比例不同:
表7:实施例4梯度洗脱程序
实验结果:
对实施例4色谱图中的主峰前后的杂峰进行分析,结果见表8。
表8:实施例3中纯化HPLC图分析结果
结论:从表8可以看出,在该色谱条件中美法仑杂质D与其他杂质的保留时间相差明显,相比于实施例3,主峰后的杂峰保留时间由9.50后移至13.55,增加10.56和12.77两个新的杂质峰,主峰分离度由3.15增加至3.44,在上表7梯度洗脱下纯化时,该方法对美法仑杂质D有较好的分离。
实施例5:
美法仑杂质D的纯化方法:
具体实施参照实施例1,与实施例1不同的是,梯度含有机相比例不同:
表9:实施例4梯度洗脱程序
实验结果:
对实施例5色谱图中的主峰前后的杂峰进行分析,结果见表10。
表10:实施例3中纯化HPLC图分析结果
结论:从表10可以看出,在该色谱条件中美法仑杂质D与其他杂质的保留时间相差明显,相比于实施例4,主峰保留时间由7.48后移至12.82,主峰前面杂质峰增加,主峰后的杂峰保留时间移至14.39,主峰分离度降低至3.09,但分离度还是大于1.5,在上表9梯度洗脱下纯化时,该方法对美法仑杂质D有较好的分离。
实施例6:
美法仑杂质D的纯化方法:
具体实施参照实施例1,与实施例1不同的是,梯度含有机相比例不同:参照下表11。
表11:实施例4梯度洗脱程序
实验结果:
对实施例6色谱图中的主峰前后的杂峰进行分析,结果见表12。
表12:实施例6中纯化HPLC图分析结果
从表12可以看出,在该色谱条件中美法仑杂质D与其他杂质的保留时间相差明显,相比于实施例5,主峰保留时间有移至13.54,主峰前后的杂质峰均增加,新增杂质峰只占比1%。但是主要的杂峰保留时间移至20.55,主峰分离度降低至2.90,但分离度还是大于1.5,在上表11梯度洗脱下纯化时,该方法对美法仑杂质D有较好的分离,符合美法仑杂质D的纯化要求。
通过实施例1~6的检测发现,梯度洗脱中有机相含量对美法仑杂质D的纯化有很大的影响。通过不同的比例组成进行梯度洗脱,可以达到美法仑杂质D与其他杂质的有效分离。最后确认梯度洗脱为在0到6min,流动相A:B为95:5~80:20;在6到12min,流动相A:B为80:20~70:30;在12min到18min,流动相A:B为70:30~60:40;在18min到24min,流动相A:B为60:40~60:40;在24min到26min,流动相A:B为60:40~5:95;在26min到30min,流动相A:B为5:95~5:95。此梯度条件下进行洗脱,美法仑杂质D能分离出更多的杂质,仍能保持较好的分离度,分离度均大于1.5,符合要求。并且该梯度条件下,将单次纯化量由10mg/ml增加至100mg/ml,同样能得纯度≥95%的美法仑杂质D产品,样品峰与其他杂质分离度为1.61,符合要求,而且纯化后得到的白色粉末状的美法仑杂质D。该粉末状固体在常温环境(湿度<50%)环境中可以稳定称量而未见明显潮解,同时该粉末样品在密封、避光、-20℃冷冻保存条件下5天后检测未见明显分解。该方法操作简单,成本低。为美法仑、盐酸美法仑原料和制剂的质量控制,提供了很大的便利。
以上所述仅为本发明的优选实施例,并非因此限制本发明的专利范围,凡是在本发明的发明构思下,利用本发明说明书及附图内容所作的等效结构变换,或直接/间接运用在其他相关的技术领域均包括在本发明的专利保护范围内。
- 一种地铁车站基坑的实时监测预警系统及其监测预警方法
- 一种地铁深基坑明挖防坍塌施工方法
- 一种地铁基坑施工坍塌危险自动化监测预警装置
- 一种基坑施工坍塌危险自动化监测预警装置