一种降低肝脏中甘油三酯合成的方法
文献发布时间:2024-04-18 19:52:40
相关申请的交叉引用
本申请要求2021年5月28日提交的美国临时申请63/194,829的权益,该申请内容通过引用全部并入本申请。
技术领域
本申请涉及通过给药苯氧基烷基羧酸类化合物,例如MN-001和MN-002,来降低患者肝脏中甘油三酯合成和/或减少甘油三酯水平。
发明内容
一方面,本申请提供了一种减少受试者肝脏中甘油三酯合成的方法,该方法包括:向受试者给药有效剂量的式(I)所示化合物,其代谢物,或其药学上可接受的盐,其中m为2、3、4或5,n为3、4、5、6、7或8,X
另一方面,本申请提供了一种减少受试者肝脏中甘油三酯累积的方法,该方法包括:向受试者给药有效剂量的式(I)所示化合物,或其代谢物,或其药学上可接受的盐,其中m为2、3、4或5,n为3、4、5、6、7或8,X
在一些实施方案中,式(I)所示化合物是式(IA)所示的化合物:
在一些实施方案中,给药式(I)所示化合物的代谢产物,并且该代谢产物是式(IB)所示的化合物:
在一些实施方案中,受试者被诊断出患有高甘油三酯血症、非酒精性脂肪肝病(NAFLD)、非酒精性脂肪性肝炎(NASH)、胰岛素抵抗、糖尿病前期或糖尿病。在一些实施方案中,受试者被认为是健康的。在一些实施方案中,式(I)所示化合物经口服给药。在一些实施方案中,式(I)所示化合物每日服用一次、每日服用两次或每日服用三次。在一些实施方案中,式(I)化合物以液体或固体剂型服用。在一些实施方案中,式(I)化合物以固体剂型口服给药,并且式(I)化合物为正交晶系。在一些实施方案中,式(I)化合物按50mg/天至2,000mg/天的量,任选地分为一份、两份或三份进行服用。在一些实施方案中,式(I)所示的化合物以50mg、75mg、100mg、200mg、500mg、750mg或1,000mg的剂量每天一次、每天两次或每天三次进行服用。
附图说明
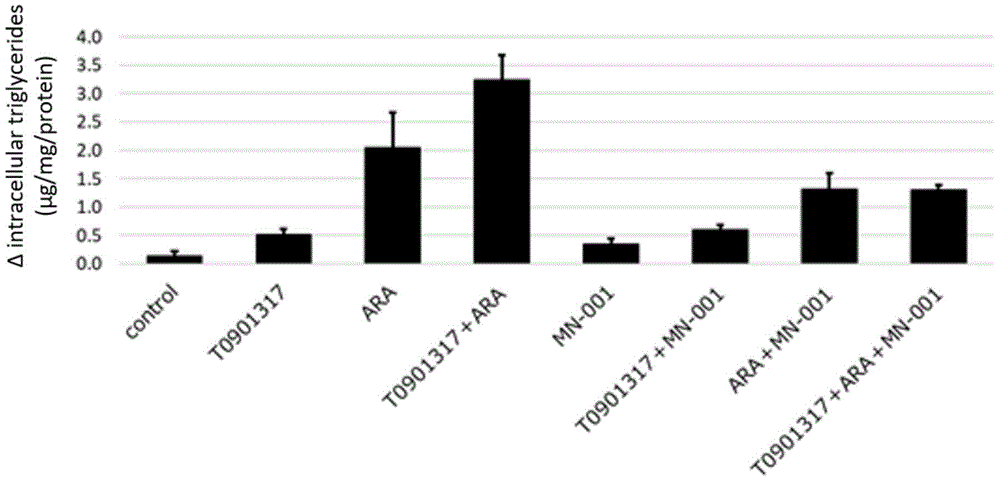
图1描述了HepG2细胞在与从花生四烯酸、T0901317和MN-001中选择的一种或多种物质共同孵化48小时后的细胞内甘油三酯水平的测量结果。Y轴:Δ细胞内甘油三酯(μg/mg/蛋白质)。
图2描述了HepG2细胞在与从花生四烯酸、T0901317和MN-001中选择的一种或多种物质共同孵化后的CD36 mRNA表达水平的测量结果。Y轴:相对于对照组的mRNA表达水平(任意单位)
图3描述了HepG2细胞在与从花生四烯酸、T0901317和MN-001中选择的一种或多种物质共同孵化后的ABCG1 mRNA表达水平的测量结果。Y轴:相对于对照组的mRNA表达水平(任意单位)
具体实施方式
定义
在本申请和所附权利要求中,除非上下文另有明确规定,否则单数形式“一”、“一个”和“该”包括复数指代。
在本申请中,"大约"一词为本领域普通技术人员所理解,并且根据使用的上下文而有所不同。如果在给定的上下文中,本领域普通技术人员对于该术语的用法不清楚,那么"大约"一词将表示特定术语的正负10%。
“给药”或“给予药物”(及其语法等效的表达方式)是指将药物给予患者,包括直接给药,如自我给药,和间接给药,如开具处方。例如,在本申请中使用时,指导患者自我给药和/或为患者提供处方的医生即为给予药物。
“C
“烷基”是指具有1-12个碳原子的单价无环烃基。烷基的非限制性实例包括甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基等。
“芳基”是指具有至多10个碳原子的单价芳族烃基。芳基的非限制性实例包括苯基和萘基。
“杂芳基”是指由1至10个碳原子和1至4个选自氧、氮、硫的杂原子组成的芳族基团,其中杂芳基中的氮和/或硫原子任选自氧化基团(例如,N-氧化物、-S(O)-或-S(O)
“环烷基”是指具有3-12个碳原子的一价非芳族环状烃基。环烷基的非限制性实例包括环丙基、环丁基、环戊基、环己基等。
“杂环基”是指环内具有1至10个碳原子和1至4个选自氧、氮、硫的杂原子的单价非芳香族环状基团,其中杂环基中的氮和/或硫原子任选自氧化基团(例如,N-氧化物、-S(O)-或-S(O)
“氨基”是指-NH
“烷基氨基”是指-NHR
“二烷基氨基”是指-N(R
“包含”表示所述的方法和组合物包括所列举的要素,但不排除其他要素。“基本上由......组成”用于定义方法和组合物时,表示排除对于所述目的的组合物具有本质性意义的任何其他要素。“由......组成”表示排除多于痕量元素的其他成分和用于给药本发明的组合物的实质性方法步骤或制备组合物或实现预期结果的工艺步骤。由这些过渡术语和短语定义的实施方案,都属于本发明的范围。
本申请所使用的“有效剂量”是指这样的一种剂量,当给予患者时,能够产生预期的治疗效果,例如,缓解、改善、减轻或消除患者的医疗症状的一个或多个表现。完全的治疗效果不一定会在给予一次剂量(或剂量方案)后出现,可能只有在给予一系列剂量后才会出现。因此,有效剂量可以分一次或多次给予。
“药学上可接受的”是指对患者(包括人类患者)无毒并适合给药的。
“药学上可接受的盐”指的是对患者无毒并适合给药的盐类。非限制性的例子包括碱金属盐、碱土金属盐和各种伯、仲、叔铵盐。当式(I)化合物的酯包含阳离子时,例如,当该酯包含氨基酸酯时,其盐类可以包括各种羧酸盐、磺酸盐和无机酸盐。盐的某些非限制性实例包括钠盐、钾盐和钙盐。
“保护基团”是指一些已知的官能团,当它们与另一个官能团结合时,可以使该受保护的官能团对于在化合物的其他部分上进行的反应和相应的反应条件呈惰性,并且可以在去除保护基团的条件下恢复原来的官能团。保护基团选自与分子的其余部分相容的基团。“羧酸保护基团”在苯氧基烷基羧酸的合成过程中保护其羧基官能团。“羧酸保护基”是指在苯氧基烷基羧酸的合成过程中保护羧基官能团的保护基。羧酸保护基的非限制性实例包括苄基、对甲氧基苄基、对硝基苄基、烯丙基、二苄基和三苯甲基。更多的羧酸保护基的例子可以在标准的参考书中找到,例如Greene和Wuts编著的《有机合成中的保护基》(第二版,1991年,John Wiley&Sons出版社)和McOmie编著的《有机化学中的保护基》(1975年,PlenumPress出版社)。本申请所述的羧酸的保护和去除保护的方法可以在文献中找到,特别是可在上述提及的Greene和Wuts所著书籍及其引用的参考文献中找到。
“治疗”医疗状况或患者是指采取措施以获得有益或期望的结果,包括临床结果。对于本申请所述的某些方面和某些实施方案而言,有益或期望的临床结果包括但不限于减少、缓解或改善肝脏中甘油三酯水平升高所导致或相关的一个或多个表现或负面影响,改善一个或多个临床结果,减少疾病程度,延缓或减缓疾病进展,改善、减轻或稳定疾病状态,以及其他本申请所述的有益结果。
肝脏中甘油三酯的累积和甘油三酯的合成
肝脏是脂肪酸代谢的中心器官。脂肪酸是通过肝细胞从血浆中摄取以及通过从头生物合成而在肝脏中累积。在营养过剩和肥胖的情况下,肝脏的脂肪酸代谢发生改变,常导致甘油三酯(TG)在肝细胞内积累。TG在肝脏中的长期积累,可以导致并预示肝脏疾病,例如脂肪肝、非酒精性脂肪性肝病(NAFLD)和非酒精性脂肪性肝炎(NASH)。然而,TG在肝脏中的积累不一定与血清TG水平的升高相关,反之亦然。例如,高甘油三酯血症早期患者可能表现出血清TG水平升高,但直到疾病进一步发展之前,没有TG在肝脏中积累。患有代谢紊乱的患者,如胰岛素抵抗、前期糖尿病或糖尿病,如果代谢紊乱没有得到适当的治疗,也可能表现出TG在肝脏中积累。这种TG在肝脏中的积累,可以在没有血清TG水平升高的情况下通过TG的从头合成增加而产生。由于TG在肝脏中的积累是肝脏疾病的危险因素,因此需要治疗选择来预防和/或治疗TG在肝脏中的积累和合成。
方法
一方面,本申请提供了一种减少受试者肝脏中甘油三酯合成的方法,该方法包括以下步骤、或基本上由以下步骤组成,或由以下步骤组成:向受试者给药有效剂量的式(I)所示化合物,其代谢物,或其药学上可接受的盐,其中m为2-5的整数,n为3-8的整数,X
另一方面,本申请提供了一种减少受试者肝脏中甘油三酯累积的方法,该方法包括以下步骤、或基本上由以下步骤组成,或由以下步骤组成:向受试者给药有效剂量的式(I)所示化合物,或其代谢物,或其药学上可接受的盐,其中式(I)所示化合物的定义同上。
在一些实施方案中,受试者被诊断出患有高甘油三酯血症、非酒精性脂肪肝病(NAFLD)、非酒精性脂肪性肝炎(NASH)、胰岛素抵抗、糖尿病前期或糖尿病。在一些实施方案中,受试者被认为是健康的。
本申请所述的“其代谢物”是指与式(I)化合物表现出基本相似的治疗活性的代谢物。这些代谢产物的非限制性例子包括这样的化合物,其中式(I)所示的化合物的–COCH
含有这种1-羟乙基基团的代谢产物在1-羟乙基基团的1-位上含有一个不对称中心。相应的对映异构体和其混合物,包括外消旋混合物,都包含在本申请所述的式(I)化合物的代谢产物中。
在一些实施方案中,式(I)化合物是式(IA)化合物(或MN-001):
在一些实施方案中,式(I)化合物和式(IA)化合物的代谢物是式(IB)化合物(或MN-002):
在一些实施方案中,化合物经口服给药。在一些实施方案中,化合物每日服用一次、每日服用两次或每日服用三次。在一些实施方案中,化合物以液体或固体剂型服用。在一些实施方案中,化合物以固体剂型口服给药,并且以基本上不含其它多晶型的正交晶系形式存在。
在一些实施方案中,化合物按50mg/天至5,000mg/天的量,任选地分为一份、两份或三份进行服用。在一些实施方案中,化合物以25mg、50mg、75mg、100mg、150mg、200mg、250mg、500mg、750mg、1000mg、1500mg或2000mg的剂量,按照每天一次、每天两次或每天三次的方式进行服用。在一些实施方案中,该化合物的给药剂量为50mg每日一次(qd),50mg每日两次(bid),50mg每日三次(tid),100mg qd,100mg bid,100mg tid,500mg qd,500mg bid,500mg tid,750mg qd,750mg bid,750mg tid,或者前5天500mg tid,后5天750mg bid。在一些实施方案中,该化合物的给药时间至少为1周、2周、3周、4周、8周、12周或无限期。
在一些实施方案中,式(I)化合物、其代谢物或其药学上可接受的盐是本申请所述方法中使用的唯一活性剂。
合成
式(I)化合物的合成方法和某些生物活性已记载于美国专利No.4,985,585中,该专利的全部内容通过引用的方式并入本文。例如,式(IA)化合物通过式(II)的苯酚反应来制备得到:
其中,R为具有式(III)结构的羧酸保护基,
提供式(IC)化合物:
酸保护基团或R基团的非限制性实例包括C
式(IC)所示的受保护的羧酸经过脱保护反应得到了式(IA)所示的化合物。根据本申请的内容可知,在一些实施方案中,根据本申请式(IC)化合物是有效的。脱保护方法的非限制性实例包括,在H
反应在惰性有机溶剂中进行,惰性有机溶剂包括但不限于丙酮、甲乙酮、二乙酮或二甲基甲酰胺。亲核取代反应可以在室温以下至溶剂的回流温度之间的温度下进行,反应中存在无机碱,如碳酸钾或碳酸钠,并可以选择性地存在碘化钾。反应需进行足够的时间,以便用薄层色谱和1H-NMR等常用方法可确定产物的含量。本申请中使用的其他化合物是按照本申请描述的方法或适当地替换起始原料制备的,和/或按照本领域技术人员所熟知的方法制备的。另请参见专利号为5,290,812的美国专利(通过引用将其全部内容并入本申请)
式(IA)化合物在受控条件下重新结晶,得到一种基本纯的正交晶系多晶形,称为A型晶体(例如90%或更高,优选至少95%的A型晶体)。多晶形物A及其制备方法已记载于专利号为No.7,060,854和No.7,064,146的美国专利中,其全部内容通过引用并入本申请。式(I)化合物的所有多晶形物都是活性的,但优选A型多晶形物。在某些条件下,A型多晶形物的溶解度和生物利用度优于其他多晶形物,因此A型多晶形物可能可以提供改进的固体制剂。
例如,可通过将式(IA)化合物在25-40℃下,溶解在按重量计算的5至10份乙醇中,得到一种黄色至橙色的溶液,从而获得A型晶体。向乙醇溶液加入1-10份水,并在20-25℃下搅拌约15-60分钟,然后在5-10℃下额外搅拌1-4小时,优选2.0-3.0小时,得到一种灰白色的悬浮液。向这种悬浮液中加入5-15份水,并在5-10℃下额外搅拌1-4小时,优选为1.5-2.0小时。通过真空过滤分离出一种白色至灰白色的固体产物,并将滤饼用水洗涤并在25-40℃下真空干燥12-24小时。
本申请所使用的以对映异构形式存在的化合物,如式(I)化合物的某些代谢产物(例如,式IB化合物),可通过光学分辨的方法得到两种对映体。这种光学分辨方法包括但不限于,通过形成例如(S)-(-)-1-(1-萘基)乙胺与相应的羧酸化合物的碱的非对映体盐,或者通过手性柱色谱分离对映体。这些化合物的中间体也存在对映异构形式,可以用类似的方法分离。
给药和制剂
本申请使用的化合物可以通过口服、静脉注射、肌肉注射、皮下注射或经皮给药的方法给药。有效剂量水平可以有很大的变化,例如,每天约100至4000mg。在一些实施方案中,每日剂量的范围是250至2,000mg,分为一次、两次或三次给药。在一些实施方案中,剂量是1000mg,每天两次。在一些实施方案中,合适的剂量包括1000mg qd、1000mg bid和750mgtid。
实际用量将取决于受治疗患者的情况。正如本领域技术人员所知,治疗医师将会考虑许多影响活性物质作用的因素,例如患者的年龄、体重、性别、饮食和病情,以及给药的时间、速率和途径。本领域技术人员可使用常规的剂量测定试验来确定给定的条件下的最佳剂量。
本申请使用的化合物可以制成任何药用可接受的形式,包括液体、粉末、霜剂、乳剂、片剂、锭剂、栓剂、混悬液、溶液等。含有本申请所使用的化合物的治疗性组合物通常会按照已知和确定的做法,与一种或多种药学上可接受的成分一起配制。一般来说,片剂是利用载体如改性淀粉,单独或与10%重量的羧甲基纤维素(Avicel)混合制成的。该制剂在片剂成型过程中以1000至3000磅的压力进行压制。片剂的平均硬度优选为约1.5至8.0kp/cm
口服用的制剂可以制成硬胶囊,其中本申请所使用的具有治疗活性的化合物与惰性固体稀释剂如碳酸钙、磷酸钙或高岭土混合,或者制成软胶囊,其中本申请所使用的具有治疗活性的化合物与油性介质如液体石蜡或橄榄油混合。合适的载体包括碳酸镁、硬脂酸镁、滑石粉、糖、乳糖、果胶、糊精、淀粉、明胶、阿拉伯胶、甲基纤维素、羧甲基纤维素钠、低熔点蜡、可可脂等。
本申请所使用的化合物可以与药学上可接受的辅料制成水性悬浮液,所述辅料可为悬浮剂,如羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、海藻酸钠、聚乙烯吡咯烷酮、黄蓍胶和阿拉伯胶;分散剂或润湿剂,如天然磷脂,如卵磷脂,或碱性氧化物与脂肪酸的缩合产物,如聚氧乙烯硬脂酸酯,或环氧乙烷与长链脂族醇的缩合产物,如十七氧乙烯氧基乙醇,或乙氧化乙烯与由脂肪酸和六元醇衍生的部分酯的缩合产物,如聚氧乙烯山梨醇单油酸酯或乙氧化乙烯与由脂肪酸和六元醇缩水合物衍生的部分酯的缩合产物,如聚氧乙烯山梨糖单油酸酯。这种水性悬浮液还可以含有一种或多种防腐剂,如对羟基苯甲酸乙酯或对羟基苯甲酸正丙酯,一种或多种着色剂,一种或多种调味剂和一种或多种甜味剂,如甘油、山梨醇、蔗糖、糖精、环己烷磺酸钠或环己烷磺酸钙。
合适的制剂还包括缓释剂型,如美国专利号为4,788,055;4,816,264;4,828,836;4,834,965;4,834,985;4,996,047;5,071,646;及5,133,974的美国专利中所描述的那些,其内容通过引用整体并入本文。
适合口服给药的其他形式包括液体制剂,包括乳液、糖浆、酊剂、水溶液或在使用前不久转化为液体制剂的固体制剂。乳液可以在溶液中制备,例如在水性丙二醇溶液中,或者可以含有乳化剂,例如卵磷脂、山梨酸单油酸酯或阿拉伯胶等。水溶液可以通过将活性成分溶解在水中,并添加适当的着色剂、香料、稳定剂和增稠剂来制备。固体制剂除了活性成分外,还可以含有着色剂、调味剂、稳定剂、缓冲剂、人造和天然甜味剂、分散剂、增稠剂、增溶剂等。
本申请所使用的化合物可以制成适用于注射的剂型(例如,通过注射,如推注或连续输注),并且可以在安瓿、预填充注射器、小体积输注或添加了防腐剂的多剂量容器中以单位剂量形式呈现。这些组合物可以采用悬浮液、溶液或乳状液等形式,在油性或水性载体中制备,例如在水性聚乙二醇溶液中制备。油性或非水性载体、稀释剂、溶剂或赋形剂的实例包括丙二醇、聚乙二醇、植物油(例如橄榄油)和可注射的有机酯(例如油酸乙酯),并且可以含有配方剂,例如防腐剂、润湿剂、乳化剂或悬浮剂、稳定剂和/或分散剂。或者,活性成分也可以以粉末形式存在,其通过无菌分离无菌固体得到或通过在使用前与适当的载体(如无菌水、无热原水)配制,从溶液中冻干得到。
本申请所使用的化合物可制成软膏剂、乳霜剂、乳液或透皮贴剂,局部给药于表皮。例如,软膏剂和乳霜剂可通过在水性或油性基质中加入适当的增稠剂和/或胶凝剂制备得到。乳液可通过水性或油性基质配制且一般还含有一个或多个乳化剂、稳定剂、分散剂、悬浮剂、增稠剂或着色剂。适用于口腔局部给药的制剂包括在调味基质中含有活性成分的锭剂,基质通常为蔗糖、阿拉伯胶或黄蓍胶;在惰性基质如明胶和甘油或蔗糖和阿拉伯胶中含有活性成分的锭剂;以及在合适的液体载体中含有活性成分的漱口剂。
本申请所使用的化合物可制成栓剂,用于给药。在这种制剂中,先将低熔点的蜡熔化,如脂肪酸甘油酯或可可脂的混合物,并且通过如搅拌的方式均匀地分散活性成分。然后将熔融的均匀混合物倒入尺寸合适的模具中,使其冷却并凝固。
本申请所使用的化合物可制成阴道给药的制剂。除了活性成分外,该制剂还含有本领域已知的合适的载体,如子宫托、卫生棉条、霜剂、凝胶、糊剂、泡沫或喷雾剂。
本申请所使用的化合物可制成鼻腔给药的制剂。溶液或悬浮液可通过常规方法例如滴管、吸管或喷雾器直接给药于鼻腔。该制剂可以以单剂量或多剂量形式提供。患者可通过滴管或移液器给药适当的、预定体积的溶液或悬浮液。喷雾可通过如计量雾化喷雾泵等方式给药。
本申请所使用的化合物可制成气雾剂,该气雾剂可给药于包括鼻腔的呼吸道。该化合物一般具有较小的粒径,例如5微米或更小。这样的粒径可通过本领域已知的技术手段获得,例如微粉化。活性成分与合适的推进剂一起以压力包装的形式提供,所述推进剂如氯氟烃(CFC)(例如二氯二氟甲烷、三氯氟甲烷或二氯四氟乙烷),二氧化碳或其他适当的气体。气雾剂还可含有表面活性剂,如卵磷脂。药物的剂量可由计量阀控制。活性成分也可以干粉的形式提供,例如将化合物与适当的粉末基质混合,所述粉末基质如乳糖、淀粉、淀粉衍生物(如羟丙基甲基纤维素和聚乙烯吡咯烷酮)。粉末载体可在鼻腔内形成凝胶。粉末组合物可以以单位剂量的形式存在,例如将其装入如明胶或泡罩包装的胶囊或药筒中,因此粉末可通过吸入器给药。
根据需要,可以制备具有肠溶涂层的制剂,以实现活性成分的持续或控制释放给药。一种可用于本发明目的的常用控释制剂包括惰性核心(例如糖球)、第一层、涂有内部药物的第二层、以及外膜或控制药物从内层释放的第三层。
核心可以是水溶性或可溶胀的材料,也可以是通常用作核心的任何此类材料或制成珠子或颗粒的其他药学上可接受的水溶性或可溶胀材料。核心可以是由蔗糖/淀粉(蔗糖球NF)、蔗糖晶体制成的球体,或由赋形剂(如微晶纤维素和乳糖)组成并挤出和干燥得到的球体。
第一层中的基本不溶于水的材料通常是“胃肠道不溶性”或“胃肠道部分不溶性”的成膜聚合物(分散或溶解在溶剂中)。例如,乙基纤维素、醋酸纤维素、醋酸丁酯纤维素、聚甲基丙烯酸酯(如乙基丙烯酸/甲基丙烯酸甲酯共聚物(
含有活性成分的第二层可以由活性成分(药物)与作为粘合剂的聚合物组成,或者不含作为粘合剂的聚合物。粘合剂通常是亲水性的,但可以是水溶性的或不溶于水的。含有活性药物的第二层中所使用的示例性聚合物是亲水性聚合物,例如聚乙烯吡咯烷酮、聚烯烃醇(如聚乙二醇)、明胶、聚乙烯醇、淀粉及其衍生物、纤维素衍生物,例如羟丙基甲基纤维素(HPMC)、羟丙基纤维素、羧甲基纤维素、甲基纤维素、乙基纤维素、羟乙基纤维素、羧乙基纤维素、羧甲基羟乙基纤维素、丙烯酸聚合物、聚甲基丙烯酸酯或任何其他药学上可接受的聚合物。第二层中药物与亲水性聚合物的比例通常在1:100到100:1(w/w)之间。
第三层或外膜中所使用的用来控制药物释放的聚合物可以选自水不溶性聚合物或具有pH依赖性溶解度的聚合物,例如,乙基纤维素、羟丙基甲基纤维素酞酸酯、醋酸纤维素酞酸酯、醋酸纤维素三甲酸酯、聚甲基丙烯酸酯或其混合物,且可选择性地与上述提到的那些增塑剂结合。
如果需要,控制释放层除了上述聚合物外,还可以包含具有不同溶解性特征的另一种或多种物质,以调节控制释放层的渗透性,从而调节释放速率。可以与例如乙基纤维素等一起使用、用作改性剂的示例性聚合物包括:HPMC、羟乙基纤维素、羟丙基纤维素、甲基纤维素、羧甲基纤维素、聚乙二醇、聚乙烯吡咯烷酮(PVP)、聚乙烯醇、具有pH依赖性溶解度的聚合物,例如醋酸纤维素酞酸酯或氨基甲基丙烯酸共聚物和甲基丙烯酸共聚物,或其混合物。如果需要的话,还可以在控制释放层中加入诸如蔗糖、乳糖和药用级表面活性剂等添加剂。
本申请还提供了制剂的单位剂型。在这种形式中,制剂被分为含有适量活性成分(例如但不限于式(I)化合物、其代谢物或药学上可接受的盐)的单位剂量。单位剂型可以是包装好的制剂,包装中含有分开数量的制剂,例如包装的片剂、胶囊和瓶装或安瓿装的粉末。单位剂型也可以是胶囊、片剂、扁囊剂或锭剂,或者是包装好的适当数量的这些剂型。
其他合适的药用载体及其制剂在《Remington:The Science and Practice ofPharmacy 1995》中有描述,该书由E.W.Martin编辑,Mack Publishing Company出版,第19版,Easton,PA。
实施例
实施例1
HepG2细胞(取自人肝癌细胞样本)与选自10μM花生四烯酸(ARA)、1μM LXR激动剂T0901317(“T1317”)和10μM MN-001中的一种或多种物质共同培养48小时。T1317刺激LXR-SREBP-1c ChREPB信号,增加肝脏中的甘油三酯(TG)合成,加重脂肪肝。处理后测量细胞内甘油三酯水平(图1)。
与对照组相比,T1317使TG合成增加了3.8倍;ARA使TG合成增加了15.3倍;T1317和ARA的组合使TG合成增加了24.3倍。
添加MN-001抑制了HepG2细胞中ARA和/或T1317引起的TG的积累(TG合成)。更具体地说,MN-001的添加抑制了T1317对TG合成的1.7倍的增加,并且,MN-001的添加抑制了ARA或T1317和ARA的组合对TG合成的3.7倍的增加。这些结果可能与抑制VLDL分泌从而抑制TG还原和脂肪肝形成有关。
MN-001对TG代谢相关分子mRNA表达的影响:(1)脂肪酸转位酶/CD36,其参与肝脏中肝细胞摄取ARA,和(2)ABCG1,其通过介导细胞胆固醇向HDL转移而调节组织脂质水平,可通过测量从HepG2细胞中提取的RNA和使用实时PCR对其进行研究。为此,从HepG2细胞(用选自10μM花生四烯酸(ARA)、1μM T1317和10μM MN-001中的一种或多种处理48小时后)中提取了RNA,并将其转化为cDNA。
转化为cDNA后,通过qPCR测量了CD36 mRNA的表达水平(图2)。游离脂肪酸通过含有CD36的受体复合物被吸收到细胞中,CD36在胰岛素抵抗状态下上调。饱和脂肪酸(例如棕榈酸、硬脂酸)和果糖诱导脂肪生成通路,并通过增加CD36表达和调节ChREBP(调控脂肪合成)来促进脂肪积累。在本实验中,ARA使CD36表达相对于对照组增加了1.8倍,MN-001使CD36表达相对于对照组下调了0.6倍,将MN-001添加到ARA中与仅添加ARA相比,抑制CD36表达下调了0.6倍。更具体地说,MN-001处理使CD36表达在无ARA和有ARA的情况下分别降低了39%和43%,从而表明MN-001抑制了ARA的细胞更新。
此外,还通过qPCR测量了ABCG1 mRNA的表达水平(图3)。ABCG1是一种在脂质负载的巨噬细胞中高度表达并介导胆固醇从细胞内流出到HDL的蛋白。ABCG1缺乏会导致巨噬细胞和肝细胞内胆固醇大量积累。在本实验中,MN-001使ABCG1表达相对于对照组增加了17倍,而ARA+MN-001使ABCG1表达相对于对照组增加了9.7倍。
实施例2
进行了多中心、双盲、随机、安慰剂对照的临床二期试验研究以评估MN-001的疗效、安全性和耐受性。患有非酒精性脂肪肝(NAFLD)、2型糖尿病(DM)和高甘油三酯血症的患者被告知并被邀请参与该研究。该研究包括筛选期(长达-8周),随后的治疗期(24周),以及随访访问(在最后一次给药后大约一周)。
这项试验的样本人群为21岁以上、75岁以下的男性和女性参与者,他们都经MRI扫描(MRI-PDFF>8%)确诊为NAFLD,同时患有2型糖尿病和高甘油三酯血症。
该研究的主要目标是:
·第24周时,通过基于磁共振质子密度脂肪分数(MRI-PDFF)测量的肝脏脂肪含量的变化
·第24周时,空腹血清甘油三酯相对于基线的变化
次要目标是:
·评估MN-001的安全性和耐受性
·评估MN-001对血脂的影响
оHDL-C,LDL-C,总胆固醇水平
筛选期(长达8周)
为完成筛选评估,共分配了长达8周的时间。在筛选期间,将对参与者的研究资格进行评估。进行以下评估:MRI-PDFF(磁共振成像质子密度脂肪分数)、病史(包括当前和既往用药回顾)、简要体检(包括身高和体重)、生命体征和12-ECG。采集血液样本用于全血细胞计数(CBC)、全套代谢功能检测组合(CMP)、空腹血脂组化验和凝血组化验(PT/INR)。进行尿液分析,并对绝经前的女性参与者进行血清人绒毛膜促性腺激素(β-hCG)妊娠试验。
治疗期(24周)
参与者接受MN-001 250mg口服片剂,每日两次或相应的安慰剂每日两次的治疗,持续24周。在双盲治疗(DBT)期间,评估药物的安全参数,并记录合用药物。
结束研究访问
所有完成研究的患者在治疗结束后一周左右(±3天)返回进行随访。记录生命体征、体重、为临床安全实验室采集的血液样本,以及同时用药(CMs)和不良事件(AEs)。
一些实施方案
实施方案1.一种减少受试者肝脏中甘油三酯合成的方法,该方法包括:向受试者给药有效剂量的式(I)所示化合物:
其代谢物,或其药学上可接受的盐,其中m为2、3、4或5,n为3、4、5、6、7或8,X
实施方案2.实施方案1中的方法,其中式(I)所示化合物为式(IA)所示化合物。
实施方案3.实施方案1中的方法,其中给药式(I)所示化合物的代谢产物,该代谢产物为式(IB)所示化合物。
实施方案4.实施方案1-3中的任一方法,其中受试者被诊断为高甘油三酯血症、非酒精性脂肪肝(NAFLD)、非酒精性脂肪性肝炎(NASH)、胰岛素抵抗、前期糖尿病或糖尿病。
实施方案5.实施方案1-3中的任一方法,其中受试者被认为是健康的。
实施方案6.实施方案1-5中的任一方法,其中式(I)所示化合物口服给药。
实施方案7.实施方案1-6中的任一方法,其中式(I)所示化合物每日给药一次、两次或三次。
实施方案8.实施方案1-7中的任一方法,其中式(I)所示化合物以液体或固体剂型给药。
实施方案9.实施方案1-8中的任一方法,其中式(I)所示化合物以固体剂型口服给药,并且式(I)所示化合物以正交晶型存在。
实施方案10.实施方案1-9中的任一方法,其中式(I)化合物按50mg/天至2,000mg/天的量,任选地分为一份、两份或三份进行服用。
实施方案11.实施方案1-10中的任一方法,其中式(I)化合物以50mg、75mg、100mg、200mg、500mg、750mg或1,000mg的剂量每天一次、每天两次或每天三次进行服用。
实施方案12.一种减少受试者肝脏中甘油三酯累积的方法,该方法包括:向受试者给药有效剂量的式(I)所示化合物:
或其代谢物,或其药学上可接受的盐,其中m为2、3、4或5,n为3、4、5、6、7或8,X
实施方案13.实施方案12中的方法,其中式(I)所示化合物为式(IA)所示化合物。
实施方案14.实施方案12中的方法,其中给药式(I)所示化合物的代谢产物,该代谢产物为式(IB)所示化合物。
实施方案15.实施方案12-14中的任一方法,其中受试者被诊断为高甘油三酯血症、非酒精性脂肪肝(NAFLD)、非酒精性脂肪性肝炎(NASH)、胰岛素抵抗、前期糖尿病或糖尿病。
实施方案16.实施方案12-14中的任一方法,其中受试者被认为是健康的。
实施方案17.实施方案12-16中的任一方法,其中式(I)所示化合物口服给药。
实施方案18.实施方案12-17中的任一方法,其中式(I)所示化合物每日给药一次、两次或三次。
实施方案19.实施方案12-18中的任一方法,其中式(I)所示化合物以液体或固体剂型给药。
实施方案20.实施方案12-19中的任一方法,其中式(I)所示化合物以固体剂型口服给药,并且式(I)所示化合物以正交晶型存在。
实施方案21.实施方案12-20中的任一方法,其中式(I)化合物按50mg/天至2,000mg/天的量,任选地分为一份、两份或三份进行服用。
实施方案22.实施方案12-21中的任一方法,其中式(I)化合物以50mg、75mg、100mg、200mg、500mg、750mg或1,000mg的剂量每天一次、每天两次或每天三次进行服用。
虽然已经说明和描述了一些实施方案,但是应当理解,在不脱离本申请权利要求所限定的更为宽泛的技术时,可根据本领域普通技术对实施方案进行变更和修改。
本申请示例性描述的实施方式可以在缺少本申请中未具体说明的一个或多个要素、一个或多个限制的情况下适当地实施。因此,例如,“包括”、“包含”、“含有”等术语应应予以进行宽泛地理解且不构成限定。此外,本申请中使用的术语和表达方式是作为描述的术语而不是限制性的术语,在使用这些术语和表达方式时,并无意排除所示和所述的特征或其部分的任何等效物,而是应认识到在所要求保护的技术范围内可能有各种修改。此外,“基本上由......组成”一词将被理解为包括那些具体记载的要素和那些不实质性影响所要求保护的技术的基本特征和新颖特征的附加元素。短语“由......组成”排除任何未指定的元素。
本申请不限于其中所描述的具体实施方式。在不偏离本发明的实质和范围的情况下,实施例可以进行许多修改和变化,这对于本领域的技术人员来说是显而易见的。除了本申请列举的之外,本申请范围内的具有等效功能的方法和组合,根据前文的描述对于本领域技术人员来说将是显而易见的。这些修改和变化落入所附权利要求的范围内。本申请仅受权利要求以及这些权利要求所享有的等效物的完整范围所限制。应当理解,本发明不限于特定的方法、试剂、化合物或组合物,它们当然可以变化。还应当理解,本文中使用的术语仅是为了描述具体实施方式,而不是为了进行限制。
此外,如果披露的特征或方面是用马库什权利要求来描述的,那么,本领域技术人员将会认识到,该披露也相应地描述了马库什权利要求中的任何单个成员或子群体。
如本领域技术人员所理解的那样,为了任何和所有的目的,特别是在提供书面描述方面,本文申请的范围也包括其可能的所有子范围和子范围的组合。任何列出的范围都可以很容易地被认为已进行了充分的描述,并允许将同一范围分解为相等的一半、三分之一、四分之一、五分之一、十分之一等和使得相同的范围可以被分解成至少相等的二分之一、三分之一、四分之一、五分之一、十分之一等。作为非限制性示例,本文讨论的每个范围都可以很容易地被分解成下三分之一、中三分之一和上三分之一等。如本领域技术人员所理解的那样,例如“最多”,“至少”,“大于”,“小于”等类似的语言,都包括所述的数字,并且指的是随后可以被分解为如上所述的子范围的范围。最后,如本领域技术人员所理解的,一个范围包括每个单独的成员。
本说明书中提到的所有出版物、专利申请、已授权专利和其他文件均通过引用的方式并入本文,如同每个单独的出版物、专利申请、已授权专利或其他文件均被具体且单独地通过引用的方式完整地并入本文。通过引用的方式并入本文的定义,若与本申请中的定义相矛盾,则排除该引入的定义。
其他实施方案在所附权利要求中阐述。