一种2-乙酰基四氢吡啶的制备方法
文献发布时间:2024-04-18 20:01:23
技术领域
本发明属于香精、香料技术领域,具体涉及一种2-乙酰基四氢吡啶的制备方法。
背景技术
公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
2-乙酰基四氢吡啶(简称ATHP)是一种易挥发的小分子杂环化合物,被认为是谷物类特征香气化合物之一,具有面包烧烤中的香气,面包皮和爆玉米花的特征香气。其包含2-乙酰基-3,4,5,6-四氢吡啶、2-乙酰基-1,4,5,6-四氢吡啶两组分,两组分为互变异构体,两组分沸点分别为205℃、269℃,两组分的结构式如下:
目前,关于ATHP的制备方法有多种,其主要存在过程繁琐、条件苛刻、收率低、使用危险试剂或有毒试剂(LDA、叠氮化钠、氢氰酸等),安全性差等问题。专利CN109503469A公开了一种2-乙酰基吡啶的制备方法,以2-吡啶甲酸为原料,经酰氯化反应得到化合物Ⅱ,化合物Ⅱ与丙二酸二烃基酯缩合得到化合物Ⅲ,化合物Ⅲ经过水解反应得到2-乙酰基吡啶。但该制备方法所得化合物并非是ATHP,其只是构造了乙酰基基团,并没有通过加氢还原得到ATHP。
综上,迫切需要开发新的、安全性高的、产品收率高的、可操作性强和易于工业化的2-乙酰基四氢吡啶制备方法。
发明内容
为了解决现有技术的不足,本发明的目的是提供一种2-乙酰基四氢吡啶的制备方法,该制备方法的原料易得、产品收率高,可操作性性强,易于工业化。
为了实现上述目的,本发明的技术方案为:
本发明的第一个方面,提供一种2-乙酰基四氢吡啶的制备方法,包括如下步骤:
2-吡啶甲酸与二甲羟胺盐酸盐进行缩合反应合成中间体I;
中间体I经加氢还原,合成中间体II;
中间体II与甲基格氏试剂进行格氏反应,得到2-乙酰基四氢吡啶;
所述中间体I、中间体II的结构式如下所示:
;/>
上述反应的方程式如下:
本发明通过缩合反应构建了Weinreb酰胺(中间体Ⅰ);再通过控制反应时间、温度和压力,确保加氢还原吡啶环时停留在亚胺,避免过度还原;最后通过格氏反应,且控制格氏反应停留在乙酰基,得到2-乙酰基四氢吡啶。整个工艺简单,反应条件温和,选择性好,产品收率和纯度高,易于工业化生产。
在本发明的一些实施例中,所述缩合反应具体包括如下步骤:
向2-吡啶甲酸有机溶液中依次加入N,N'-羰基二咪唑(CDI)、二甲羟胺盐酸盐(化学名称:N,O-二甲基羟胺盐酸盐)和三乙胺,常温下进行缩合反应,反应后,得到中间体I。
在本发明的一些实施例中,所述2-吡啶甲酸有机溶液的溶剂选自四氢呋喃、二氯甲烷、二氯乙烷、甲基叔丁基醚或2-甲基四氢呋喃中的一种,优选为二氯甲烷。
在本发明的一些实施例中,所述2-吡啶甲酸有机溶液中,2-吡啶甲酸与有机溶剂的质量比为0.2-0.3:1。
在本发明的一些实施例中,所述2-吡啶甲酸、N,N'-羰基二咪唑、二甲羟胺盐酸盐和三乙胺的质量比为1-3:5:1-3:1-3。
在本发明的一些实施例中,向2-吡啶甲酸有机溶液中加入N,N'-羰基二咪唑后,室温下搅拌反应1-2 h,再依次加入二甲羟胺盐酸盐和三乙胺,继续保温反应至反应停止。
在本发明的一些实施例中,反应停止后,过滤并收集滤液,滤液采用饱和氯化铵溶液、饱和碳酸氢钠溶液、去离子水各洗一次,收集有机相加入无水硫酸镁干燥,过滤,滤液负压蒸干有机溶剂,得到中间体I油状物。所得中间体I不需要提纯,可直接用于下步加氢还原反应。
在本发明的一些实施例中,所述加氢还原包括如下步骤:
向含有中间体I的醇溶液中加入催化剂,氢气气氛下进行加氢还原反应,反应完毕后得到中间体II。
在本发明的一些实施例中,所述醇选自甲醇、乙醇或异丙醇中的一种。
在本发明的一些实施例中,所述含有中间体I的醇溶液中,中间体I与醇的质量比为0.3-0.4:1。
在本发明的一些实施例中,所述催化剂为钯碳。经验证,钌碳加氢还原得到的是哌啶基团,雷尼镍没法进行加氢还原,只有钯碳作为催化剂才能控制加氢还原吡啶环停留在亚胺,避免了过度还原。
在本发明的一些实施例中,所述中间体I和催化剂的质量比为9-11:1。
在本发明的一些实施例中,所述加氢还原反应,反应压力为0.08-0.1 MPa,反应温度为40-45℃。反应时间为30-35 h,优选为30-32 h。本发明采用钯碳为催化剂,通过控制反应压力、时间和温度,确保加氢还原吡啶环时停留在亚胺,避免过度还原。
在本发明的一些实施例中,反应完毕后,过滤出催化剂,滤液脱干有机溶剂后,得到中间体Ⅱ油状物。所得中间体Ⅱ不需要提纯,可直接用于下步格氏反应。
在本发明的一些实施例中,所述格氏反应包括如下步骤:
向中间体II有机溶液中加入甲基格氏试剂,在保护性气氛下,进行格氏反应,反应完毕后得到2-乙酰基四氢吡啶。
具体的,将中间体II加入有机溶剂中,得到中间体II有机溶液,加毕氮气破空于冷浴中降温,降温至反应温度时加入甲基格氏试剂,保温反应。
在本发明的一些实施例中,所述中间体II有机溶液的溶剂选自四氢呋喃、二氯甲烷、二氯乙烷、甲基叔丁基醚或2-甲基四氢呋喃中的一种,优选为四氢呋喃。
在本发明的一些实施例中,所述中间体II有机溶液中,中间体II和有机溶剂的质量比为0.1-0.2:1。
在本发明的一些实施例中,所述甲基格氏试剂选自甲基溴化镁或甲基碘化镁中的一种。优选的,所述甲基格氏试剂的浓度为2-4 mol/L。
在本发明的一些实施例中,所述中间体II与所述甲基格氏试剂的用量比为120-160:380-450,g/mL。
在本发明的一些实施例中,所述格氏反应,反应温度为-10-0℃。反应时间为4-5h。
在本发明的一些实施例中,所述格氏反应结束后,向反应体系内加入饱和氯化铵溶液淬灭,静置分液,收集有机相,二氯甲烷萃取水相,合并有机相加入无水硫酸镁干燥,过滤,负压蒸干有机溶剂,得到2-乙酰基四氢吡啶粗品油状物。所述粗品油状物可进一步精馏纯化,得到2-乙酰基四氢吡啶纯品。
本发明的有益效果为:
本发明提供了一种2-乙酰基四氢吡啶的制备方法,包括如下步骤:2-吡啶甲酸与二甲羟胺盐酸盐进行缩合反应合成中间体I;中间体I经加氢还原,合成中间体II;中间体II与甲基格氏试剂进行格氏反应,得到2-乙酰基四氢吡啶。本发明通过控制2-吡啶甲酸与二甲羟胺盐酸盐的缩合反应构建了Weinreb酰胺;再通过控制反应时间、温度和压力,加氢还原吡啶环停留在亚胺,避免过度还原;最后通过格氏反应,且格氏反应停留在乙酰基,得到2-乙酰基四氢吡啶。整个工艺简单,反应条件温和,选择性好,产品收率和纯度高,易于工业化生产。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
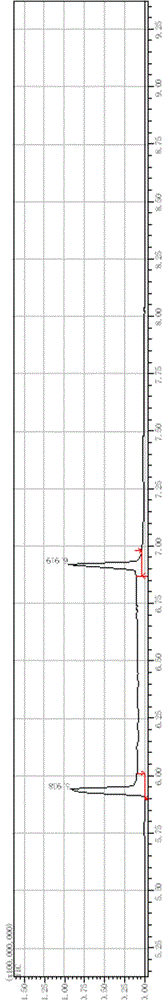
图1是本发明实施例3所得2-乙酰基四氢吡啶的气质联用气相图;
图2是本发明实施例3所得2-乙酰基四氢吡啶中2-乙酰基-3,4,5,6-四氢吡啶(5.938 min)的气质联用质谱图,其中A为测试得到的2-乙酰基-3,4,5,6-四氢吡啶的质谱图,B为数据库中与之匹配的2-乙酰基-3,4,5,6-四氢吡啶的质谱图,且两幅图中分子量大于200(即横坐标在200之后)时,无峰;
图3是本发明实施例3所得2-乙酰基四氢吡啶中2-乙酰基-1,4,5,6-四氢吡啶(6.919 min)的气质联用质谱图,其中A为测试得到的2-乙酰基-1,4,5,6-四氢吡啶的质谱图,B为数据库中与之匹配的2-乙酰基-1,4,5,6-四氢吡啶的质谱图,且两幅图中分子量大于200(即横坐标在200之后)时,无峰;
图4是本发明实施例3所得的2-乙酰基四氢吡啶的H谱图;
图5是本发明实施例3所得的2-乙酰基四氢吡啶的C谱图;
图6为本发明实施例1所得的中间体Ⅰ油状物的气质联用气相图;
图7为本发明实施例1所得的中间体Ⅰ油状物的气质联用质谱图,且质谱图中分子量大于200(即横坐标在200之后)时,无峰;
图8为本发明实施例1所得的2-乙酰基四氢吡啶纯品的气相色谱图,该图中横坐标(即保留时间)大于12min时,无峰。
具体实施方式
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
如无特殊说明,以下实施例所用试剂均为常规产品,可通过购买得到。
实施例1
2-乙酰基四氢吡啶的合成
反应路线如下:
具体合成方法如下:
(1)合成中间体I
向1 L反应瓶中依次投入510 g二氯甲烷、123 g 2-吡啶甲酸、250 g CDI,开启搅拌,25-30℃反应1-2 h,依次加入117 g二甲羟胺盐酸盐、116 g三乙胺,继续保温反应3-4h,薄层检测2-吡啶甲酸无残留,反应停止。
过滤反应液,收集滤液,滤液采用饱和氯化铵溶液、饱和碳酸氢钠溶液、去离子水各水洗一次,收集有机相加入无水硫酸镁干燥,过滤,滤液负压蒸干有机溶剂,得到149 g中间体I油状物,收率为89.7%,经气相色谱检测,所得中间体I的纯度大于99%。
所述中间体Ⅰ油状物的气质检测结果如图6和图7所示,通过碎片与产品最终分子量确定为中间体I,且通过后续验证合成到产品也说明了中间体I的准确性。
(2)合成中间体II
向1 L高压釜中依次投入448 g甲醇、14.9 g钯炭(干基)、149 g中间体I,氢气置换,控制压力0.08~0.1 MPa,升温控制温度40~45℃,反应30~32 h,反应完毕,过滤钯炭,脱干有机溶剂得到145 g中间体II油状物,收率为95%,经气相色谱检测,所得中间体II的纯度大于99%。
(3)合成2-乙酰基四氢吡啶
向2 L反应瓶中依次投入1160 g四氢呋喃、145 g中间体II,加毕氮气破空于冷浴中降温,温度降至-5℃时加入426 mL甲基溴化镁格氏试剂(3 mol.L
上述粗品油状物进一步精馏纯化得到86.4 g 2-乙酰基四氢吡啶纯品,收率为81%,经气相色谱检测,如图8和表1所示,所得2-乙酰基四氢吡啶纯品的纯度大于99%。
表1 2-乙酰基四氢吡啶纯品纯度检测的气相色谱数据
实施例2
2-乙酰基四氢吡啶的合成
反应路线如下:
具体合成方法如下:
(1)合成中间体I
向1 L反应瓶中依次投入510 g四氢呋喃、123 g 2-吡啶甲酸、250 g CDI,开启搅拌,25-30℃反应1-2 h,依次加入117 g二甲羟胺盐酸盐、116 g三乙胺,继续保温反应3-4h,薄层检测2-吡啶甲酸无残留,反应停止。
过滤反应液,收集滤液,滤液采用饱和氯化铵溶液、饱和碳酸氢钠溶液、去离子水各水洗一次,收集有机相加入无水硫酸镁干燥,过滤,滤液负压蒸干有机溶剂,得到138 g中间体I油状物,收率为83.1%,经气相色谱检测,所得中间体I的纯度大于99%。
(2)合成中间体II
向1 L高压釜中依次投入141 g乙醇、13.8 g钯炭(干基)、138 g中间体I,氢气置换,控制压力0.08~0.1 MPa,升温控制温度40~45℃,反应30~32 h,反应完毕,过滤钯炭,脱干有机溶剂得到134 g中间体II油状物,收率为94.8%,经气相色谱检测,所得中间体II的纯度大于99%。
(3)合成2-乙酰基四氢吡啶
向2 L反应瓶中依次投入1080 g四氢呋喃、134 g中间体II,加毕氮气破空于冷浴中降温,温度降至-5℃时加入394 mL甲基碘化镁格氏试剂(3 mol.L
上述粗品油状物进一步精馏纯化得到69.5g2-乙酰基四氢吡啶纯品,收率为70.5%,经气相色谱检测,所得2-乙酰基四氢吡啶纯品的纯度大于99%。
实施例3
2-乙酰基四氢吡啶的合成
反应路线如下:
具体合成方法如下:
(1)合成中间体I
向1 L反应瓶中依次投入510 g二氯甲烷、123 g 2-吡啶甲酸、202 g CDI,开启搅拌,25-30℃反应1-2 h,依次加入109 g二甲羟胺盐酸盐、106 g三乙胺,继续保温反应3-4h,薄层检测2-吡啶甲酸无残留,反应停止。
过滤反应液,收集滤液,滤液采用饱和氯化铵溶液、饱和碳酸氢钠溶液、去离子水各水洗一次,收集有机相加入无水硫酸镁干燥,过滤,滤液负压蒸干有机溶剂,得到154.3 g中间体I油状物,收率为92.9%,经气相色谱检测,所得中间体I的纯度大于99%。
(2)合成中间体II
向1 L高压釜中依次投入463 g甲醇、15.4 g钯炭(干基)、154.3 g中间体I,氢气置换,控制压力0.08~0.1 MPa,升温控制温度40~45℃,反应30~32 h,反应完毕,过滤钯炭,脱干有机溶剂得到150.2 g中间体II油状物,收率为95.1%,经气相色谱检测,所得中间体II的纯度大于99%。
(3)合成2-乙酰基四氢吡啶
向2 L反应瓶中依次投入1200 g四氢呋喃、150.2 g中间体II,加毕氮气破空于冷浴中降温,温度降至-5℃时加入442 mL甲基溴化镁格氏试剂(3 mol.L
上述粗品油状物进一步精馏纯化得到89.5 g 2-乙酰基四氢吡啶纯品,收率为81.1%,经气相色谱检测,所得2-乙酰基四氢吡啶纯品的纯度大于99%。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种空调器的导风组件、空调器室内机和空调器
- 加热片组件、空调器室外机、空调器
- 空调器的壳体组件及具有其的空调器
- 一种空调器出风口组件的驱动装置和空调器
- 一种下支架、蒸发器组件及空调器
- 一种下支架、蒸发器组件及空调器