一种药物前体化合物及其制备方法和应用
文献发布时间:2023-06-19 19:27:02
技术领域
本发明涉及化学生物技术领域,具体涉及一种药物前体化合物及其制备方法和应用。
背景技术
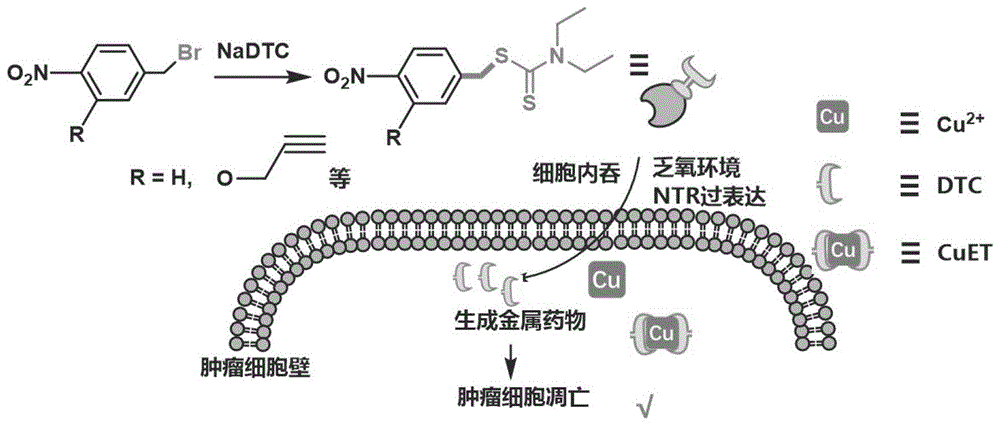
环境响应性药物释放是药物递送过程中提高递送精准度和释放可控性的有效策略。根据刺激源的来源区别,环境响应性可分为内源性响应和外源性响应。其中,内源性响应即刺激响应源来自肿瘤微环境相对于正常细胞和组织中环境物质的区别,如氧化还原水平、相关酶的表达水平、金属离子浓度富集差异等;而外源性响应即刺激响应源来自外加条件,如光、声、磁、热等。在药物前体化合物递送至特定部位后,经过不同的响应刺激源控制,使药效分子释放并达到有效药物浓度,即可产生响应毒性并用于对应的疾病治疗。硝基还原酶是肿瘤组织所处的乏氧条件下,细胞过量表达的一种可以通过硝基还原的方法代谢芳香族硝基化合物的物质。现有的研究主要集中于硝基还原酶探针的开发,用于肿瘤乏氧微环境的检测。同样地,其可以作为特殊的内源性响应刺激源,用于药物前体化合物的内源性响应释放和药物递送。
双硫仑是一种常用的戒酒药物,其分解产物二乙基二硫代氨基甲酸盐(酯)类化合物在较低的浓度下结合铜离子后得到的金属药物CuET,CuET对广泛的肿瘤细胞均具有高效的细胞毒性和抑制抗药性等功能。但其存在生理条件下易分解,循环周期较短等缺点,导致非特异性毒性高,肿瘤治疗效率低。因此,急需开发一种药物前体化合物解决上述问题。
发明内容
本发明提供了一种药物前体化合物及其制备方法和应用,解决了现有技术中CuET在生理条件下易分解,循环周期较短等缺点,无法精准可控释放导致的非特异性毒性高,肿瘤治疗效率低的问题。
一种药物前体化合物,其特征在于,所述前体化合物具有的化学结构式为式I:
R
优选的,所述R
将4-硝基苄溴溶于四氢呋喃,二乙基二硫代氨基甲酸钠三水合物溶于乙醇,两者混合后搅拌反应后,萃取和洗涤并除去溶剂后得到粗产物一,进行柱分离后得到前体化合物I;
优选的,所述4-硝基苄溴与二乙基二硫代氨基甲酸钠三水合物的摩尔比为1:(1.5~1.8);所述搅拌反应的温度为45~55℃;所述分离过程中,流动相为石油醚和乙酸乙酯的混合物,体积比为(10~9):1。
优选的,所述R
S1、在惰性气体保护条件下,将3-羟基-4-硝基苯甲醇和无水碳酸钾,溶于无水N,N’-二甲基甲酰胺,搅拌反应后,滴入溴丙炔,继续搅拌反应,萃取并除去溶剂得到粗产物二,进行柱分离后得到浅棕色粉末中间产物II;
S2、在惰性气体保护条件下,将中间产物II溶于四氢呋喃,冰浴冷却后,分次滴入三溴化磷,冰浴避光反应,萃取并除去溶剂得到粗产物三,进行柱分离,得到棕色粘液为中间产物III;
S3、将中间产物III溶于四氢呋喃,二乙基二硫代氨基甲酸钠三水合物溶于乙醇,将两者混合搅拌反应后,萃取并除去溶剂得到粗产物四,进行柱分离后得到淡黄色粉末前体化合物IV;
优选的,S1中,所述3-羟基-4-硝基苯甲醇、无水碳酸钾和溴丙炔的摩尔比为1:2:1.5;所述搅拌的温度为55~65℃,干燥时间为2~4h,所述溴丙炔滴入的时间为50~60min;所述继续搅拌反应的时间为24h;所述柱分离过程中,流动相为石油醚和乙酸乙酯的混合物,体积比为(4~2):1。
优选的,S2中,所述冰浴冷却的温度为2~5℃,时间为1~2h;所述中间产物II与三溴化磷的摩尔比为1:(1.5~1.8),所述三溴化磷滴入的时间为25~30min;所述冰浴反应的时间为2~4h;所述柱分离过程中,流动相为石油醚和乙酸乙酯的混合物,体积比为(8~4):1。
优选的,S3中,所述中间产物III与二乙基二硫代氨基甲酸钠三水合物的摩尔比为1:(1.5~1.8);所述搅拌反应的温度为45~55℃;所述柱分离过程中,流动相为石油醚和乙酸乙酯的混合物,体积比为(4~2):1。
优选的,当R
S1:在惰性气体保护条件下,将4-(溴甲基)苯基氨基甲酸叔丁酯溶于四氢呋喃,二乙基二硫代氨基甲酸钠三水合物溶于乙醇,两者混合搅拌反应后,萃取并除去溶剂后得到粗产物五,进行柱分离后得到中间产物V;
S2:将中间产物V溶于二氯甲烷,向溶液中滴入三氟乙酸,两者混合搅拌反应后,萃取并洗涤除去溶剂后得到粗产物六,进行柱分离后得到化合物VI;
优选的,S1中,所述4-(溴甲基)苯基氨基甲酸叔丁酯与二乙基二硫代氨基甲酸钠三水合物的摩尔比为1:(1.5~1.8);所述搅拌反应的温度为45~55℃;所述柱分离过程中,流动相为石油醚和乙酸乙酯的混合物,体积比为(5~2):1;
S2中,所述溶剂体积与三氟乙酸的体积比为1:1;所述搅拌反应的温度为20~35℃;所述反应时间为1~2h;所述柱分离过程中,流动相为石油醚和乙酸乙酯的混合物,体积比为(4~1):1。
本发明的第二个目的在于将所述药物前体化合物在制备抗肿瘤药物中的应用。
与现有技术相比,本发明的有益效果:
(1)本发明的合成方法产率可观,制备过程简单易操作,实用性强,硝基邻位修饰炔丙基醚,不影响硝基的还原性及硝基对位的二硫代苄酯的酶响应断裂性能,有助于进一步的化学和结构修饰。
(2)本发明所合成的药物前体化合物,当R
附图说明
图1为前体化合物I的核磁共振氢谱图;
图2为中间产物II的核磁共振氢谱图;
图3为中间产物III的核磁共振氢谱图;
图4为前体化合物IV的核磁共振氢谱图;
图5为前体化合物IV的质谱图;
图6为中间产物V的核磁共振氢谱图;
图7为前体化合物VI的核磁共振氢谱图;
图8为乏氧条件下硝基还原酶响应细胞毒性的工艺步骤图;
图9为前体化合物I在试管内硝基还原酶响应后,在不同铜离子条件下,与细胞孵育的细胞毒性;
图10为正常培养条件下,二乙基二硫代氨基甲酸钠三水合物(DTC)、前体化合物I、前体化合物IV和前体化合物VI的细胞毒性;
图11为正常培养并在铜离子存在条件下,DTC、前体化合物I、前体化合物IV和前体化合物VI的细胞毒性;
图12为乏氧培养条件下,前体化合物IV在不同铜离子条件下,与细胞孵育的细胞毒性。
具体实施方式
下面将结合本发明实施例中的数据,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
需要说明的是,本发明中所使用的专业术语只是为了描述具体实施例的目的,并不是旨在限制本发明的保护范围,除非另有特别说明,本发明以下各实施例中用到的各种原料、试剂、仪器和设备均可通过市场购买得到或者通过现有方法制备得到。
实施例1
一种药物前体化合物,制备方法包括以下步骤:
称取1.08g4-硝基苄溴溶解于10mL四氢呋喃(THF),称取1.69g二乙基二硫代氨基甲酸钠(DTC)三水合物溶解于10mL乙醇,将两者混合后,50℃搅拌反应12h;反应结束后,旋蒸除去溶剂,加入70mL去离子水和等体积(70mL)的乙酸乙酯萃取,有机相用饱和食盐水洗涤3次后,加入3g无水Na
将产物I溶解后,通过核磁共振氢谱对其结构进行表征,谱图如图1所示,
实施例2
一种药物前体化合物,制备方法包括以下步骤:
S1、称取1.69g3-羟基-4-硝基苯甲醇和2.76g无水碳酸钾(K
S2、称取1.66g中间产物II在氩气保护条件下溶解于10mL无水THF,在4℃冰浴下冷却1h,向溶液中滴加1.35mL三溴化磷(PBr
S3、称取1.89g中间产物III溶解于THF,称取2.36g二乙基二硫代氨基甲酸钠三水合物溶解于乙醇,将两者混合后,50℃搅拌反应12h;反应结束后,旋蒸除去溶剂,加入70mL去离子水和等体积的乙酸乙酯萃取,有机相用饱和食盐水洗涤3次后,加入无水Na
实施例3
一种药物前体化合物,制备方法包括以下步骤:
S1、称取1.43g 4-(溴甲基)苯基氨基甲酸叔丁酯溶解于THF,称取1.69g二乙基二硫代氨基甲酸钠三水合物溶解于乙醇,将两者混合后,50℃搅拌反应12h;反应结束后,旋蒸除去溶剂,加入70mL去离子水和等体积的乙酸乙酯萃取,有机相用饱和食盐水洗涤3次后,加入无水Na
S2、称取1.42g中间产物VI,溶解于6mL二氯甲烷中,然后向其中加入等体积三氟乙酸,室温下搅拌反应1h。反应结束后,旋蒸除去溶剂。加入50mL二氯甲烷,有机相分别用等体积水、饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤三次后,有机相中加入无水硫酸钠干燥2h,过滤,旋蒸除去溶剂得到固体粗产物VI,以石油醚:乙酸乙酯(v/v)=(4~1):1为流动相进行柱分离,得到0.84g黄绿色油状物,为化合物VI,产率为83%;将化合物VI溶解后,通过核磁共振氢谱对其结构进行表征,核磁氢谱如图7所示,
对比例1
一种药物前体化合物,制备方法包括以下步骤:
称取1.13g药物前体化合物I溶解于15mL甲醇,随后依次加入2.6g锌粉和3.96g冰醋酸混合均匀,60℃搅拌反应12h;反应结束后,过滤除去不溶杂质,旋蒸除去溶剂得到固体粗产物V;向固体粗产物V中加入10mL去离子水,用1mol/L氢氧化钠水溶液调节溶液pH至9左右;过滤除去新产生的不溶物,收集滤液并加入50mL去离子水,再加入50mL乙酸乙酯萃取3次后,有机相加入无水Na
通过将前体化合物I还原制备前体化合物VI时,可能是由于反应条件的影响,无法提纯出前体化合物VI,但是得到的中间产物可以异构化,有效重构释放出DTC分子并结合铜离子产生明显的细胞毒性。
为了说明本发明提供的药物前体化合物的相关性能,将实施例1~3提供的药物前体化合物及硝基还原产物进行细胞毒性测试;
下述中将实施例1提供的药物前体化合物配置的溶液标记为I;将实施例1提供的药物前体化合物配置的溶液中加入浓度为50μM的氯化铜溶液,标记为Cu-I;将实施例2提供的药物前体化合物配置的溶液标记为IV;将实施例2提供的药物前体化合物配置的溶液中加入铜离子标记为Cu-IV;将实施例3提供的药物前体化合物VI配置的溶液标记为VI;将实施例3提供的药物前体化合物VI配置的溶液中加入铜离子标记为Cu-VI。
为了探讨本发明提供的药物前体化合物的酶响应性能及产生的细胞毒性,通过噻唑蓝(MTT)法对不同条件下孵育后的细胞活性进行检测。分别称取28.4mg药物前体化合物I、33.8mg药物前体化合物IV和25.4mg硝基还原产物VI,溶解于1mL二甲亚砜(DMSO)中备用。
将HeLa细胞接种到96孔板中,每孔2×10
细胞与样品孵育24h后,弃去培养基,并用pH为7.4,浓度为0.01M的PBS缓冲液洗涤2次后,向每个孔中加入含有MTT(100μL,0.5mg/mL)的新鲜培养基,并在正常培养条件下孵育4h。弃去上清液,用PBS缓冲液洗涤,然后加入DMSO(100μL)以溶解甲瓒,用酶标仪检测在490nm、560nm和720nm紫外吸光度,通过比值计算可间接反映活细胞力水平。
前体化合物I在试管内硝基还原酶响应后,在不同铜离子条件下,与细胞孵育的细胞毒性如图9所示。数据显示,在NTR和NADH的共同作用下,前体化合物I在铜离子存在的条件下能够显示出明显的细胞毒性,当前体化合物I的浓度为10μM时,同等条件加入NTR组细胞存活率仅为约12%,而不加NTR的对照组,细胞存活率为约57%。该结果表明,基于前体化合物I的结构具有NTR的响应性,且响应后结合铜离子后能产生有效的细胞毒性。
表1前体化合物I试管内还原后的细胞存活率数据
作为对照,正常培养条件下,二乙基二硫代氨基甲酸钠(DTC)、前体化合物I、前体化合物IV和前体化合物VI的细胞毒性如图10所示。结果显示,在无铜离子存在的条件下,DTC和前体化合物I、IV、VI对HeLa细胞的杀伤率均在23%以下,其中前体化合物IV最大为23%。即表明正常培养且无铜离子条件下,这四种化合物的细胞相容性较好。
表2正常培养条件下细胞存活率数据
/>
在以上条件中加入金属铜离子,即在正常培养并有铜离子存在条件下,DTC、前体化合物I、前体化合物IV和前体化合物VI的细胞毒性如图11所示。数据显示,加入铜离子后,各组的细胞毒性都有所增加,其中DTC和前体化合物VI组细胞存活率分别降至65%和10%,说明前体化合物VI在细胞孵育条件下能有效重构释放出DTC分子并结合铜离子产生明显的细胞毒性,但由于DTC本身稳定性不强,在被细胞摄取前可能分解而降低其与铜离子结合能产生的药效。而前体化合物I和IV未有响应源刺激,加入铜离子后在最高浓度为5μM时,细胞存活率仍高于65%。
表3 正常培养并在铜离子存在条件下细胞存活率数据
HeLa细胞在乏氧条件下应激性产生硝基还原酶,在乏氧培养及不同铜离子条件下,前体化合物IV的细胞毒性如图12所示。结果显示,随着前体化合物IV的浓度增加,细胞毒性随之增加,增加趋势较弱。而在加入铜离子的条件下,细胞存活率由85%以上降低至18%以下,即说明前体化合物在细胞的乏氧应激产生的硝基还原酶的作用下能够有效进行还原反应,但反应产物只有结合铜离子生成金属药物CuET才能抑制肿瘤细胞活性。
表4 乏氧培养条件下前体化合物IV的细胞存活率数据
综上可知,前体化合物I和IV在外加硝基还原酶或细胞乏氧应激产生的硝基还原酶条件下能有效进行结构中的硝基还原,所释放的二乙基二硫代氨基甲酸酯产物在有效结合铜离子的条件下,能对肿瘤细胞产生强烈的活性抑制作用。
需要说明的是,本发明中涉及数值范围时,应理解为每个数值范围的两个端点以及两个端点之间任何一个数值均可选用,由于采用的步骤方法与实施例相同,为了防止赘述,本发明描述了优选的实施例。尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例做出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 一种红树内生真菌来源的1,4-萘醌类化合物及其制备方法和在制备抗炎药物中的应用
- 一种三萜类化合物及其制备方法和在制备抗癌药物中的应用
- 一种抗结核化合物及其在制备抗结核药物中的应用以及一种抗结核药物组合物
- 一种乙酰化二苯并-1,3-二氧杂环辛类化合物的制备方法与作为抗菌药物的应用
- 一种索拉菲尼与青蒿素类化合物的组合药物及其在制备抗癌药物中的应用
- 用以制备18F放射性药物的固体载体上所连接的前体化合物及其制备方法与应用
- 用以制备18F放射性药物的固体载体上所连接的前体化合物及其制备方法与应用