一种雷贝拉唑钠肠溶制剂有关物质的测定方法
文献发布时间:2023-06-19 11:32:36
技术领域
本发明涉及药物分析技术领域,具体涉及一种雷贝拉唑钠肠溶制剂有关物质的测定方法。
背景技术
雷贝拉唑钠是第二代质子泵抑制剂,其通过特异性地一直H+/K+-ATP酶系统的作用而抑制胃酸分泌,临床上适用于治疗消化性溃疡、反流性食管炎和胃泌素瘤。雷贝拉唑钠的常见不良反应为头痛,其他少见的不良反应有无力、便秘、头晕、恶心、腹痛即尿异常,可能由其中的杂质引起。雷贝拉唑钠合成过程中的起始原料、中间体、终产物类似物及降解产物等均可能成为其杂质,从而影响产品的质量。因此,对原料药及制剂进行质量控制是药物研发工作中的重点和难点。
目前,对雷贝拉唑钠的质量控制方法一般是采用美国药典和欧洲药典收载的雷贝拉唑钠原料药的有关物质分析方法,该分析方法存在以下问题:1、以雷贝拉唑钠原料药为检测对象,而如雷贝拉唑钠肠溶片等制剂由于受到辅料和制备工艺的影响,其杂质谱与原料药不一致,使得该分析方法无法直接用于制剂的检测;2、现有的分析方法耐受性不好,杂质A与主峰的分离度受色谱柱品牌影响较大,部分品牌的C18色谱柱分离度不符合规定;3、现有技术流动相有3种,在梯度洗脱3相混合时产生气泡,使保留时间约11min处出现倒峰,且大小不一,由于杂质F在该位置出峰,因此干扰杂质F的检测。
因此,寻找一种能够适用于雷贝拉唑钠肠溶制剂有关物质的测定方法对于控制雷贝拉唑钠的质量、提高药品疗效、降低毒副作用迫在眉睫。
发明内容
为了克服现有技术的不足,本发明的目的在于提供一种雷贝拉唑钠肠溶制剂有关物质的测定方法,该测定方法能够确保雷贝拉唑钠肠溶片的所有杂质峰、辅料峰、主峰得到有效分离,从而实现有关物质的定量分析,从而保证雷贝拉唑钠肠溶片的质量可控性。
为解决上述问题,本发明所采用的技术方案如下:
一种雷贝拉唑钠肠溶制剂有关物质的测定方法,该方法采用高效液相色谱法测定雷贝拉唑钠肠溶片中的有关物质,色谱条件为:色谱柱以十八烷基硅烷键合硅胶为填充剂,采用磷酸氢二钾溶液为流动相A、甲醇为流动相B、乙腈为流动相C进行梯度洗脱。
作为本发明优选的实施方式,所述流动相A的浓度为3.5g/L~5.2g/L,用磷酸调节pH值至7.0±0.2。
作为本发明优选的实施方式,所述流动相A、流动相B及流动相C的梯度洗脱程序如下:
作为本发明优选的实施方式,所述色谱柱的规格为C18,4.6mm×250mm,5μm柱。
作为本发明优选的实施方式,所述色谱柱的柱温为40~50℃。
作为本发明优选的实施方式,紫外检测器的检测波长为280±2nm;流动相的流速为0.8~1.2ml/min。
作为本发明优选的实施方式,本发明的测定方法具体包括如下步骤:
(1)供试品溶液的配制:取本品细粉适量,精密称定,加入氢氧化钠溶液和甲醇,摇匀,制成每1ml含有雷贝拉唑钠及有关物质0.5mg的供试品溶液;
(2)对照品溶液的配制:精密称取10mg雷贝拉唑钠对照品,加入溶剂溶解并稀释至刻度,摇匀,制成每1ml含有雷贝拉唑钠及有关物质1μg的对照品溶液;
(3)灵敏度溶液的配制:精密移取对照品溶液5ml,置20ml量瓶中,加溶剂稀释至刻度,摇匀,即得;
(4)系统适用性溶液的配制:
取雷贝拉唑钠、杂质A、杂质B、杂质C、杂质F对照品适量,加溶剂溶解并稀释成每1ml含雷贝拉唑钠0.5mg、杂质A 10μg、杂质B 2.5μg、杂质C 2.5μg、杂质F 2.5μg的系统适用性溶液;
(5)检测:分别精密量取供试品溶液和对照品溶液各10μL,分别注入液相色谱仪,记录色谱图,供试品溶液色谱图中如有杂质峰,按外标法以峰面积计算杂质含量。
其中,所述步骤(1)的具体操作如下:取本品细粉适量,精密称定,置100ml量瓶中,加0.05mol/L的氢氧化钠溶液40ml,超声振摇10min,加入40ml甲醇并超声20min,冷却至室温后加甲醇定容至刻度,摇匀,制成每1ml含有雷贝拉唑钠及有关物质0.5mg的供试品溶液。
作为本发明优选的实施方式,所述步骤(2)和(4)中的溶剂为甲醇与0.05mol/L氢氧化钠溶液按60:40的体积比混合的混合溶液。
相比现有技术,本发明的有益效果在于:
本发明所提供的测定方法能够确保雷贝拉唑钠肠溶片的所有杂质峰、辅料峰、主峰得到有效分离,确保所有杂质能被检出并准确测定,从而实现有关物质的定量分析,从而保证雷贝拉唑钠肠溶片的质量可控性。与现有技术相比,就相同色谱柱而言,本发明杂质A与主峰的分离度增加了3倍,有效克服了部分C18色谱柱杂质A与主峰分离度不符合规定的问题,同时解决了现有技术基线不稳定、在保留时间约11min处出现倒峰而干扰杂质F检测的问题。本发明对雷贝拉唑钠肠溶片中主要的工艺杂质及降解杂质分别制定了科学合理的限度,且限度符合ICH指导原则,确保产品的安全性;本发明与现行版中国药典及其他法定标准中的有关物质方法相比,限度更加科学合理。
附图说明
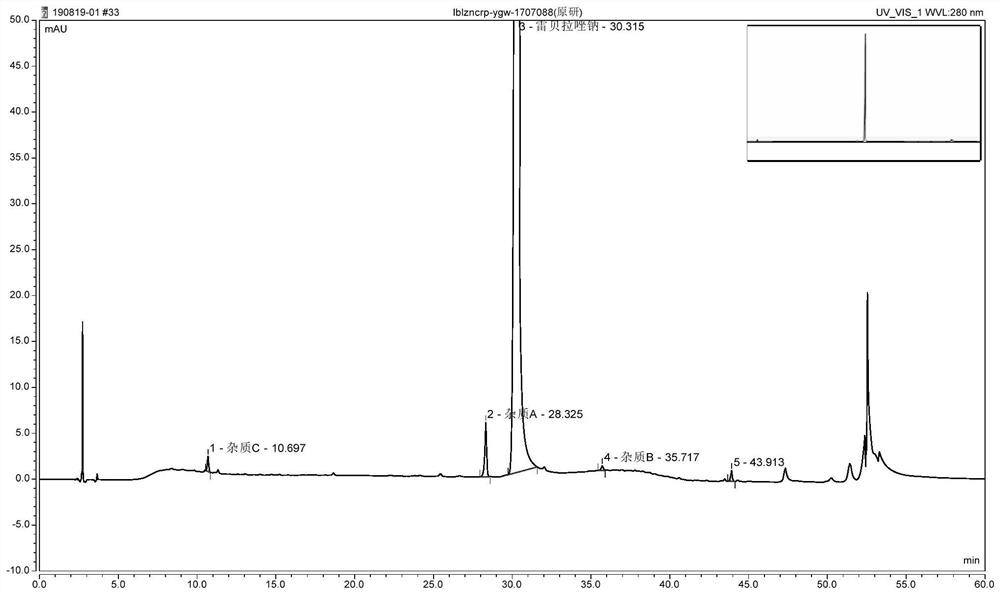
图1为实施例1中雷贝拉唑钠与各已知杂质的分离度色谱图;
图2为实施例2中雷贝拉唑钠与各已知杂质的分离度色谱图;
图3为实施例3中雷贝拉唑钠与各已知杂质的分离度色谱图;
图4为色谱条件优化专属性研究中空白辅料与各已知杂质分离度色谱图;
图5为色谱条件优化专属性研究中空白辅料与供试品未经强降解的图谱;
图6为色谱条件优化专属性研究中供试品经强光降解的图谱;
图7为色谱条件优化专属性研究中供试品经高温降解的图谱;
图8为色谱条件优化专属性研究中供试品经强氧化降解的图谱;
图9为色谱条件优化专属性研究中供试品经强碱降解的图谱;
图10为色谱条件优化专属性研究中供试品经强酸降解的图谱;
图11为色谱条件优化中各杂质定量限图谱;
图12为色谱条件优化中各杂质检测限图谱;
图13为方法学考察中雷贝拉唑钠及各杂质的线性考察色谱图;
图14为方法学考察中雷贝拉唑钠及各杂质的准确度考察色谱图;
图15为比较例中中国药典方法杂质峰分离情况色谱图;
图16为比较例中日本药典方法杂质峰分离情况色谱图;
图17为比较例中USP/EP方法杂质峰分离情况色谱图;
图18为比较例中本发明方法杂质峰分离情况色谱图。
具体实施方式
下面结合附图和具体实施方式对本发明作进一步详细说明。
实施例1~3:
一种雷贝拉唑钠肠溶制剂有关物质的测定方法,具体包括以下步骤:
(1)溶剂的配制:取氢氧化钠2.0g,加水溶解并稀释至1000ml,作为0.05M氢氧化溶液;取甲醇1500ml,与上述0.05M氢氧化钠溶液1000ml混合均匀,作为溶剂。
(2)供试品溶液的配制:避光操作,临用新制。取三个批次的本品研细,取细粉适量(约相当于雷贝拉唑钠50mg)作为实施例1~3,分别进行如下操作:精密称定,置100ml量瓶中,加0.05mol/L的氢氧化钠溶液40ml,超声振摇10min,加入约40ml甲醇并超声20min,冷却至室温后加甲醇定容至刻度,摇匀,作为供试品溶液。
(3)对照品溶液的配制:避光操作。精密称取约10mg雷贝拉唑钠对照品,置100ml量瓶中,加入溶剂溶解并稀释至刻度,摇匀,作为雷贝拉唑钠对照品贮备液。精密移取1ml对照品贮备液,置100ml量瓶中,加溶剂稀释至刻度,摇匀,即得。
(4)灵敏度溶液的配制:避光操作。精密移取对照品溶液5ml,置20ml量瓶中,加溶剂稀释至刻度,摇匀,即得。
(5)系统适用性溶液的配制:
取雷贝拉唑钠、杂质A、杂质B、杂质C、杂质F对照品适量,加溶剂溶解并稀释成每1ml含雷贝拉唑钠0.5mg、杂质A 10μg、杂质B 2.5μg、杂质C 2.5μg、杂质F 2.5μg的系统适用性溶液;
其中,杂质A为2-[[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]-甲基磺酰基]-1H-苯并咪唑,分子式为C
杂质B为2-[[4-(3-甲氧基丙氧基)-3-甲基-2-吡啶基]-甲硫基]-1H-苯并咪唑,分子式为C
杂质C为1-(1H-苯并咪唑-2-基)-3-甲基-4-氧代-1,4-二氢吡啶-2-羧酸,分子式为C
杂质F为1H-苯并咪唑-2-硫醇,分子式为C
(6)色谱条件如下:
用十八烷基硅烷键合硅胶为填充剂(BDS Hypersil
流动相A、流动相B及流动相C的梯度洗脱程序如下:
(7)系统适用性试验:分别精密量取系统适用性溶液、灵敏度溶液、对照品溶液各10μl,注入液相色谱仪。系统适用性溶液中,杂质A峰与杂质F峰、杂质A与雷贝拉唑峰的分离度均应不小于1.5。灵敏度溶液中,主成分信噪比应不小于10,对照品溶液重复进样6次,主成分峰面积RSD不大于5.0%。
(8)测定法:精密量取对照品溶液和供试品溶液各10μl,分别注入液相色谱仪,记录色谱图。供试品溶液的色谱图中如有杂质峰,按外标法以峰面积计算各杂质的含量,小于灵敏度溶液主峰面积(0.05%)的杂质峰以及辅料峰忽略不计。
式中:A
(9)结果如表1和图1~3所示。
表1实施例1~3的雷贝拉唑钠肠溶制剂有关物质的测定结果
由表1和图1~3可知,实施例1~3中的主要杂质为杂质A,其含量在0.43%~0.48%之间,总杂在0.43%~0.67%之间。其中实施例1(1707088批)的生产日期比实施例2~3早两年,可见本发明的测定方法能准确检出储存过程中降解产生的微量的杂质B和杂质C,实施例2与实施例3的生产日期较为接近,各杂质的检测结果基本一致,表明本方法重复性良好。
实验例
通过实验例对本发明所公开的方法进行色谱条件筛选、方法学考察,能有效的说明本发明的可靠性、实用性和稳定性。
实验例1雷贝拉唑钠肠溶制剂有关物质测定色谱条件筛选
(1)基本色谱条件
用十八烷基硅烷键合硅胶为填充剂(BDS Hypersil
流动相A、流动相B及流动相C的梯度洗脱程序如下:
(2)检测波长的确定
雷贝拉唑钠肠溶片质量标准仅被中国药典收载,但雷贝拉唑钠原料的质量标准在各药典中均有收载,各药典中有关物质方法的检测波长略有不同,其中,中国药典与日本药典有关物质方法流动相均为等度洗脱,检测波长为290nm,美国药典与欧洲药典有关物质方法流动相均为梯度洗脱程序,检测波长为280nm。经检测对比雷贝拉唑钠及杂质A、B、C、D、E、F、G、H、I、K的紫外吸收光谱,大部分的最大吸收波长在280nm~290nm附近。本发明方法为确保方法的专属性、便捷性,使用梯度洗脱程序,检测波长选择280nm。
(3)专属性试验
以高温、强酸、强碱、氧化、光照破坏实验考察上述色谱条件能否分离雷贝拉唑钠及其可能存在的杂质称取本品适量(约相当于雷贝拉唑钠50mg的量)分别置于7个100ml的容量瓶中,分别按下表方法制备各强降解条件下的供试品溶液。另取空白辅料贮备液10ml,置100ml的容量瓶中,同供试品溶液平行操作,按表2所示的方法制备各强降解条件的空白辅料溶液。分别精密量取各破坏后的供试品溶液、空白辅料溶液及对照品溶液10μl,注入液相色谱仪,记录色谱图,结果见表3及图4~图10。
表2强降解试验方法
表3专属性试验结果
表3及图4~10表明,各破坏条件下杂质均有所增加,各杂质分离度符合要求,主峰纯度均大于950,物料平衡在97.9%~100.6之间,说明本色谱条件有良好的专属性。
实验例2方法学考察
(1)定量限
A、雷贝拉唑钠对照品贮备液:精密称取雷贝拉唑钠对照品25mg置于50ml量瓶中,加入稀释剂溶解并稀释至刻度,摇匀,精密移取上述溶液1ml置于50ml量瓶中,加入稀释剂溶解并稀释至刻度,摇匀,即得。
B、杂质A对照贮备液:精密称取杂质A 5mg置于100ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,即得。
C、混合杂质贮备液:精密称取杂质B、杂质C、杂质F各5mg置于同一200ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,即得。
D、混合对照品溶液:精密量取雷贝拉唑钠对照品贮备液、杂质A对照品贮备液1ml、混合杂质贮备液各2ml置于20ml量瓶中,加入稀释剂溶解并稀释至刻度,摇匀,即得。
E、精密量取混合对照品溶液10μl,注入液相色谱仪,记录色谱图,观察雷贝拉唑钠及各杂质峰的信噪比,并使用空白辅料进一步稀释,过滤,取续滤液,进样检测。当信噪比≥10时,作为相应成分的定量限。连续进样6次,计算主成分及各杂质峰面积的RSD,结果见表4和图11;
表4定量限试验结果
表4及图11所示的结果表明,各杂质含量在0.03%以上时能被定量检出。
(2)检测限
取定量限溶液用空白辅料溶液逐级稀释,滤过,取续滤液进样测定。当信噪比≥3时,作为相应成分的检测限。结果见表5及图12。
表5检测限试验结果
表5及图12所示的结果表明,各杂质含量在0.007%以上时能被检出。
(3)线性考察
在不同日期,分别由2个分析员在不同仪器上进行线性试验。
S1、精密量取定量限项下的雷贝拉唑钠对照品贮备液、杂质A对照贮备液、混合杂质贮备液,按表6所示方法,置相应体积的量瓶中,加空白溶剂溶液稀释定容至刻度,摇匀,过滤,取续滤液,制得系列浓度的线性测试溶液。
表6线性试验溶液的配制
S2、精密量取各线性测试溶液10μl进样检测,记录色谱图,结果见图13。分别以雷贝拉唑钠及各杂质对照浓度(μg/ml)为横坐标,以相应峰面积为纵坐标,按最小二乘法进行线性回归,线性回归方程及相关系数(r)见表7。
表7和图13表明,杂质A、B、C、F以及雷贝拉唑钠在定量限浓度至限度200%浓度范围内,线性回归相关系数均大于0.999,线性关系良好。
表7线性试验结果
(4)准确度
S1、精密量取杂质A贮备液,混合杂质对照品贮备液,雷贝拉唑钠对照品贮备液按下表的方法移取该溶液置相应的量瓶中,加空白辅料溶液稀释至刻度,混匀,过滤,取续滤液。各浓度梯度平行制备3份。
表8准确度试验溶液的配制
S2、精密量取上述溶液各10μl,分别注入液相色谱仪,记录色谱图,按下式计算主成分及各杂质的含量和回收率。结果如表9和图14所示。
回收率=实际测得量/理论加入量×100%
表9准确度试验结果
表9和图14结果表明,各杂质及雷贝拉唑钠在50%、100%、150%浓度水平的回收率在90.0%至110.0%之间,说明该方法测定结果准确性良好。
(5)精密度--重复性
S1、取本品适量(约相当于雷贝拉唑钠25mg的量),精密称定,置于50ml量瓶中,加入16ml0.05mol/L氢氧化钠溶液振摇超声10min溶解后,再加入16ml甲醇振摇超声20min,精密量取杂质A对照贮备液、混合杂质贮备液各5ml于该量瓶中,用甲醇定容至刻度,摇匀,过滤,取续滤液,作为供试品溶液,平行制备6份。避光操作,临用新制。
S2、精密量取上述供试品溶液及对照品溶液10μl,分别注入液相色谱仪,记录色谱图,计算各杂质的结果,结果如表10所示。
表10重复性试验结果
表10结果表明,供试品重复测定6次各杂质结果的RSD在8.5%以下,本测定方法的重复性良好。
(6)精密度--中间精密度
由不同分析人员在不同日期使用不同仪器对同一批号的雷贝拉唑钠肠溶片进行上述重复性试验,结果见表11。
表11中间精密度试验结果
表11结果表明,不同人员不同日期分别重复测定6次供试品,各杂质结果的RSD在8.1%以下,本测定方法的中间精密度良好。
(7)溶液稳定性
取对照品溶液和供试品溶液,6℃储存条件下,分别于0、8、16、24、36、48小时进行测定,计算不同时间点对照品溶液中雷贝拉唑钠峰面积的RSD,计算不同时间点供试品溶液中各杂质及总杂的含量及RSD,结果见表12。
表12溶液稳定性试验结果
表12结果表明,对照品溶液在6℃条件下48小时内是稳定的,供试品溶液在6℃条件下48小时内也是稳定的。
比较例
在各国药典中,仅有中国药典收载雷贝拉唑钠肠溶片,但中国药典、日本药典、美国药典及欧洲药典均收载了其原料药雷贝拉唑钠的质量标准。取雷贝拉唑钠10种杂质对照品(包括杂质A、杂质B、杂质C、杂质D、杂质E、杂质F、杂质G、杂质H、杂质I、杂质K对照品),分别按照中国药典、日本药典、美国药典、欧洲药典及本发明的有关物质测定方法进行检测分析,结果见图15~图18。
图15~图18结果表明:(1)中国药典方法为等度洗脱方法,杂质D峰与杂质F峰、杂质B峰与杂质I峰重叠,无法分离,方法专属性较差;(2)日本药典方法为等度洗脱方法,杂质A峰与杂质E峰、杂质H与主成分峰、杂质F与杂质K均无法分离,方法专属性较差;(3)美国药典与欧洲药典方法相同,采用梯度洗脱方法,杂质B峰与杂质I峰重叠,无法分离,方法专属性较差;(4)本发明的有关物质方法上述10个杂质的色谱峰以及雷贝拉唑钠主峰均能有效分离,方法专属性符合规定。
上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
- 一种雷贝拉唑钠肠溶制剂有关物质的测定方法
- 一种替米考星肠溶制剂有关物质的检测方法