用于治疗补体介导的病症的靶向给药
文献发布时间:2023-06-19 12:02:28
相关申请的交叉引用
本申请要求2018年12月17日提交的美国临时申请第62/780,573号和2019年7月22日提交的美国临时申请第62/877,193号的权益。这些申请通过引用以其整体并入。
技术领域
本发明属于施用小分子补体因子D (fD)抑制剂- (1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺的有利剂型和方法的领域-根据人临床试验,所述剂型和方法在人中提供了用于治疗补体介导的病症的特定的药代动力学和药效学特性。一方面,公开了提供所需药代动力学和药效学特征(包括C
背景技术
当免疫系统不能以正常方式运行时,就会发生免疫病症。炎症是保护性响应,其通常涉及免疫细胞、免疫系统、血管和分子介质。多种医学病症是由有害的免疫或炎性响应引起的,或者细胞无法对正常的免疫或炎性过程做出响应。
补体系统是先天免疫系统的一部分,其不适应宿主生命过程中的变化,但被适应性免疫系统募集和使用。例如,其有助于或补充抗体和吞噬细胞清除病原体的能力。这种复杂的调节途径允许对病原生物体快速响应,同时保护宿主细胞免受破坏。超过三十种蛋白质和蛋白质片段构成了补体系统。这些蛋白质通过调理作用(增强抗原的吞噬作用)、趋化作用(吸引巨噬细胞和中性粒细胞)、细胞裂解(破裂外源细胞膜)和凝集反应(使病原体聚集和结合在一起)起作用。
补体系统具有三种途径:经典途径、旁路途径和凝集素途径。补体因子D在补体级联的旁路途径的激活中起早期和核心作用。旁路补体途径的激活是由C3中硫酯键的自发水解产生C3(H
补体的功能障碍或过度激活与某些自身免疫性疾病、炎性疾病和神经变性疾病以及缺血再灌注损伤和癌症相关。例如,补体级联旁路途径的激活有助于C3a和C5a的产生,这两者都是强效过敏毒素,它们也在许多炎性病症中起作用。因此,在一些情况下,需要降低补体途径(包括旁路补体途径)的响应。
阵发性睡眠性血红蛋白尿症(Paroxysmal nocturnal hemoglobinuria) (PNH)是非恶性血液病症,其特征在于造血干细胞和后代成熟血细胞的放大,所述造血干细胞和后代成熟血细胞缺乏一些表面蛋白。PNH红细胞不能调节其表面补体激活,这产生了PNH的典型标志—补体介导的血管内贫血的慢性激活。目前,只有一种产品,即抗C5单克隆抗体依库珠单抗,在美国获批用于治疗PNH。然而,许多接受依库珠单抗治疗的患者仍然贫血,并且许多患者继续需要输血。另外,依库珠单抗治疗需要终生静脉内注射。因此,对开发补体途径的新型抑制剂存在未满足的需求。
与补体级联相关的其他病症包括但不限于非典型溶血性尿毒症综合征(aHUS)、溶血性尿毒症综合征(HUS)、C3肾小球病(C3G)或C3肾小球肾炎(C3GN)、腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析、神经脊髓炎(NMO)、重症肌无力(MG)、脂肪肝、非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化、肝衰竭、皮肌炎、肌萎缩侧索硬化、年龄相关性黄斑变性(AMD)、多发性硬化症、类风湿性关节炎以及对生物治疗剂(例如CAR T细胞疗法)做出响应的细胞因子或炎性响应。
由于因子D在补体旁路途径中的早期和重要作用以及其在经典和凝集素补体途径中的信号放大中的潜在作用,因此其是抑制或调节补体级联的有吸引力的靶标。因子D的抑制有效地中断了途径并削弱了膜攻击复合物的形成。虽然已经进行了开发因子D的抑制剂的初步尝试,但目前还没有临床批准的小分子因子D抑制剂。因子D抑制剂化合物的实例描述于以下公开内容中。
名称为“Indole compounds or analogues thereof useful for the treatmentof age-related macular degeneration”的Novartis PCT专利公布WO2012/093101描述了某些因子D抑制剂。其他因子D抑制剂描述于Novartis PCT专利公布:WO2012093101、WO2013/164802、WO2013/192345、WO2014/002051、WO2014/002052、WO2014/002053、WO2014/002054、WO2014/002057、WO2014/002058、WO2014/002059、WO2014/005150、WO2014/009833、WO2014/143638、WO2015/009616、WO2015/009977、WO2015/066241和WO2016088082中。
其他补体因子D抑制剂描述于Achillion Pharmaceuticals, Inc拥有的专利提交:美国专利第9,598,446号;第9,643,986号;第9,663,543号;第9,695,205号;第9,732,103号;第9,732,104号;第9,758,537号;第9,796,741号;第9,828,396号;第10,000,516号;第10,005,802号;第10,011,612号;第10,081,645号;第10,087,203号;第10,092,584号;第10,100,072号;第10,138,225号;第10,189,869号;第10,106,563号;第10,301,336号和第10,287,301号;国际公布第WO2019/028284号;第WO2018/160889号;第WO2018/160891号;第WO2018/160892号;第WO2017/035348号;第WO2017/035349号;第WO 2017/035351号;第WO2017/035352号;第WO 2017/035353号;第WO 2017/035355号;第WO2017/035357号;第WO2017/035360号;第WO2017/035361号;第WO2017//035362号;第WO2017/035415号;第WO2017/035401号;第WO2017/035405号;第WO2017/035413号;第WO2017/035409号;第WO2017/035411号;第WO2017/035417号;第WO2017/035408号;第WO2015/130784号;第WO2015/130795号;第WO2015/130806号;第WO2015/130830号;第WO2015/130838号;第WO2015/130842号;第WO2015/130845号和第WO2015/130854号以及美国专利公布第US2016-0361329号;第US 2016-0362432号;第US 2016-0362433号;第US 2016-0362399号;第US 2017-0056428号;第US 2017-0057950号;第US 2017-0057993号;第US 2017-0189410号;第US 2017-0226142号;第US 2017-0260219号;第US 2017-0298084号;第US 2017-0298085号;第US 2018-0022766号;第US 2018-0022767号;第US 2018-0072762号;第US2018-0030075号;第US 2018-0169109号;第US 2018-0177761号;第US 2018-0179185号;第US 2018-0179186号;第US 2018-0179236号;第US 2018-0186782号;第US 2018-0201580号;第US 2019-0031692号;第US 2019-0048033号;第US 2019-0144473号和第US 2019-0211033号中。
名称为“Therapeutic Inhibitory Compounds”的Lifesci Pharmaceuticals PCT专利公布WO2017/098328描述了在中心核心杂环中具有变化的各种因子D抑制剂。PCT专利公布WO2018/015818也称为“Therapeutic Inhibitory Compounds”,描述了不具有环状中央核心的因子D抑制剂。
名称为“Compounds useful in the complement, coagulation and kallikreinpathways and method for their preparation”的Biocryst Pharmaceuticals美国专利第6653340号描述了作为因子D的抑制剂的稠合双环化合物。由于因子D的抑制剂BCX1470缺乏特异性和半衰期短,因此停止了其开发。
名称为“Methods and compositions for the treatment ofglomerulonephritis and other inflammatory diseases”的Alexion PharmaceuticalsPCT专利公布WO1995/029697公开了针对补体途径的C5的抗体,其用于治疗肾小球肾炎和涉及补体系统的病理性激活的炎性疾患。Alexion Pharmaceutical的抗C5抗体依库珠单抗(Soliris®)和雷武珠单抗(ravulizumab)-cwvz (Ultomiris®)是目前市场上唯一的补体特异性抗体,也是唯一获批的阵发性睡眠性血红蛋白尿症(PNH)的治疗。
与补体介导的病症治疗相关的一个特殊困难是给药间隔期间抗补体活性的持续时间,以及在接受下一个疗程之前发生突破性溶血(breakthrough hemolysis)的可能性。例如,超过17天的依库珠单抗给药间隔可能与PNH患者发生突破性溶血的更大风险相关联。Nakayama 等人,Bio. Pharm. Bull. 2018; 39(2), 285-288。
鉴于由有害的免疫或炎性响应引起的多种医学病症,本发明的目的是提供治疗患有补体介导的病症的患者的剂量和方法,所述剂量和方法提供用于抑制旁路补体的所需药代动力学和药效学特征,所述药代动力学和药效学特征在治疗剂量的施用之间是持久的。
发明内容
本发明属于施用小分子补体因子D (fD)抑制剂- (1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1;参见下面的结构)的有利口服剂型和方法的领域,根据人临床试验,所述口服剂型和方法提供了随时间推移保持药物的有效性,同时最大限度地减小副作用的药代动力学C
化合物1
本发明之所以重要是因为补体级联的临界性和敏感性赋予其攻击和破坏其发觉是外来的、患病的或受感染的细胞的作用。如果剂量太高,可能会引起不希望的副作用,诸如抑制抗感染的能力或引起器官毒性。如果剂量太低,药物不能有效抵消旁路补体途径系统的过度活动或功能失调性活性。
令人惊讶地发现,化合物1在人中表现出昼夜代谢模式,这意味着其在白天的代谢明显高于夜间。这在给人施用药物之前是无法预测的。例如,表6提供了四种剂量(40 mgBID、80 mg BID、120 mg BID和200 mg BID,持续14天)下的C
化合物2
已发现,在每日两次约100 mg至200 mg,更具体地每日两次约120 mg至180 mg,甚至更具体地每日两次约120 mg至150 mg,或每日两次约150 mg至约200 mg,以及在一个实施方案中每日两次约120 mg的化合物1的口服剂量下,在剂量间隔结束时提供最佳最低平均血浆浓度(C
因此,作为下文详细描述的人临床试验的结果,令人惊讶地确定了口服给药方案,所述口服给药方案可以指导补体因子D (fD)抑制剂化合物1或其药学上可接受的盐的安全且有效的长期施用,所述化合物1或其药学上可接受的盐可用于治疗受试者,所述受试者患有补体功能障碍或过度激活,例如但不限于阵发性睡眠性血红蛋白尿症(PNH)、C3肾小球病(C3G)诸如致密沉积物病(DDD)和C3肾小球肾炎(C3GN),以及免疫复合物膜性增生性肾小球肾炎(IC-MPGN)。本文所述的剂量和方法提供了抑制旁路途径补体活性的所需药代动力学(PK)和药效学(PD)特征,例如,在120 mg BID至200 mg BID的剂量下,
在一些实施方案中,提供的剂量提供了约50 ng/mL至450 ng/mL的最小平均血浆浓度(C
在一些实施方案中,提供的剂量在BID给药方案的昼夜周期中,在较低的C
此外,当以120 mg BID和更高剂量(例如,200 mg BID)给药时,化合物1实现了接近完全和持续的旁路途径(AP)抑制,其中如通过AP溶血和AP Wieslab测定所测量的,在120mg BID时在最低平均血浆浓度(C
一方面,对于补体介导的病症的治疗,以在治疗期间提供约50 ng/ml至200 ng/ml的的最低平均血浆浓度(C
一方面,对于补体介导的病症的治疗,以在治疗期间提供约225 ng/ml至450 ng/ml的的最低平均血浆浓度(C
一方面,以本文所述的特定剂量施用化合物1。在一些实施方案中,施用化合物1使得单剂量提供如本文所述的特定PK血液分布。在一些实施方案中,施用于受试者的剂量为约25 mg至约275 mg。在一些实施方案中,所施用的剂量为约40 mg至约160 mg。在一些实施方案中,所施用的剂量至少约为40 mg、至少约为50 mg、至少约为60 mg、至少约为75 mg、至少约为90 mg、至少约为100 mg、至少约为125 mg、至少约为150 mg、至少约为160 mg、至少约为170 mg、至少约为175 mg、至少约为180 mg、至少约为190 mg、至少约为200 mg、至少约为210 mg、至少约为220 mg、至少约为230 mg、至少约为240 mg、至少约为250 mg、至少约为260 mg或至少约为275 mg。在一些实施方案中,在治疗期间以每天两次120mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次150 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次175 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次200 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次220mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天两次240 mg的剂量施用化合物1。在一些实施方案中,在治疗期间以每天一次240 mg的剂量施用化合物1。
因此,本文提供的某些实施方案包括但不限于:
A) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供为约65 ng/mL至95 ng/mL的两种不同的昼夜C
B) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供约为90 ng/mL+/- 10%的两种不同的昼夜C
C) 口服剂型,其包含对降低旁路补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人血浆中提供为约65 ng/mL至95 ng/mL的C
D) 口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人血浆中提供约为90 ng/mL+/- 10%的C
E) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供至少为65 ng/mL的两种不同的昼夜C
F) 口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服剂型在人中提供至少为65 ng/mL的人血浆C
G) BID口服剂型,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述BID口服剂型在人血浆中提供约为100 ng/mL+/- 10%的两种不同的昼夜C
H) 口服给药方案,其包含对降低补体D途径的活性有效的量的(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)或其药学上可接受的盐,所述口服给药方案在人血浆中提供至少为100 ng/mL+/- 10%的C
I) 上述A)至H)中任一项的口服给药方案,其中所述剂型包含约100 mg至200 mg。
J) 上述I)的口服剂量,其包含约120 mg。
K) 上述I)的口服剂量,其包含约200 mg。
L) 上述A)至K)中任一项的口服剂型,其中在患有阵发性睡眠性血红蛋白尿症(PNH)的患者中测量人血浆中的C
M) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C
N) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C
O) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C
P) 上述A)至K)中任一项的口服剂型,其中在患者中测量人血浆中的C
Q) 一种治疗患有补体D相关病症的患者的方法,所述方法包括施用有效量的上述A)至K)中任一项的口服剂型。
R) 上述Q)的方法,其中所述患者患有阵发性睡眠性血红蛋白尿症(PNH)。
S) 上述Q)的方法,其中患者患有选自脂肪肝诸如非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化或肝衰竭的病症。
T) 上述Q)的方法,其中患者患有选自肌萎缩侧索硬化、类风湿性关节炎、补体旁路途径(AP)相关肾病、组分3肾小球病(C3G)病症、C3肾小球肾炎(C3GN)、致密沉积物病(DDD)、膜性增生性肾小球肾炎(MPGN)和免疫复合物膜性增生性肾小球肾炎(IC-MPGN)的病症。
U) 上述Q)的方法,其中患者患有选自肌萎缩侧索硬化、类风湿性关节炎、补体旁路途径(AP)相关肾病和肾小球病的病症。
V) 上述Q)的方法,其中患者患有选自年龄相关性黄斑变性(AMD)、视网膜变性、眼部疾病、地图样萎缩、早期或新生血管性年龄相关性黄斑变性、自身免疫性干眼病和环境性干眼病的病症。
W) 上述Q)至V)中任一项的方法,其中将所述剂型施用一个月或以上。
X) 上述Q)至V)中任一项的方法,其中将所述剂型施用至少6个月。
Y) 上述A)至K)中任一项的口服剂型,其提供了小于约2000 ng/mL的C
Z) 上述A)至K)中任一项的口服剂型,其提供了小于约1000 ng/mL的C
AA) 上述A)至K)中任一项的口服剂型,其用于治疗患有补体D相关病症的患者。
BB) 一种制备用于治疗患有补体D相关病症的患者的药物的方法,所述方法包括制备上述A)至K)中任一项的口服剂型。
CC) 上述BB)的口服剂型用于治疗上述R)至V)中任一项所列的任何病症。
DD) 上述CC)的方法,其用于治疗上述R)至V)中所列的任何病症。
附图简述
图1是表示在施用40 mg、80 mg和120 mg的单剂量的化合物1或安慰剂后通过AP溶血测量的AP活性百分比的图。第1至第3组在第1天第0小时被施予单剂量的化合物1。y轴代表相对于对照的AP活性的抑制百分比。x轴代表从第一次施用化合物1后的时间。
图2是表示化合物1的血浆浓度与通过AP溶血测定评价的血清AP活性的抑制之间的关系的PK-PD分析的图。y轴代表AP活性的抑制百分比。x轴代表化合物1的血浆浓度。
图3是表示两个端点之间AP活性的百分比的线性回归的图。
图4A和图4B是表示对于40 mg的化合物1剂量的从0小时至144小时(第7天)的平均血浆化合物I的浓度-时间曲线(线性[图4A]和半对数[图4B]标度)的图。
图5A和图5B是表示对于80 mg的化合物1剂量的从0至144小时(第7天)的平均血浆化合物1的浓度-时间曲线(线性[图5A]和半对数[图5B]标度)的图。
图6A和图6B是表示对于120 mg的化合物1剂量的从0至144小时(第7天)的平均血浆化合物1的浓度-时间曲线(线性[图6A]和半对数[图6B]标度)的图。
图7A和图7B是表示剂量比例分析的从0至24小时的平均血浆化合物I的浓度-时间曲线图(线性标度[图7A]和半对数标度[图7B])的图。
图8A和图8B是表示通过AP Wieslab测定评估的按随机治疗组计的在给药后24小时内(图8A)和随时间推移(图8B)的平均旁路途径功能活性(相对于阳性对照的百分比)的图。
图9A和图9B是表示按随机治疗组计的在给药后24小时内(图9A)和随时间推移(图9B)的的Bb血浆浓度相对于基线的平均改变的图。
图10表示稳态建模的图形显示,其表明90%的AP活性的抑制(IC90)在C
图11表示每天两次以120 mg施用的化合物1的预计稳态的图形显示。
图12是将化合物1和Danicopan的多递增剂量抑制与S型模型拟合的图形显示。y轴是对旁路途径的抑制百分比。x轴是以ng/mL为单位的药物血浆浓度。化合物1的模拟90%抑制水平为88 ng/mL。Danicopan的模拟90%抑制水平为235 ng/mL。
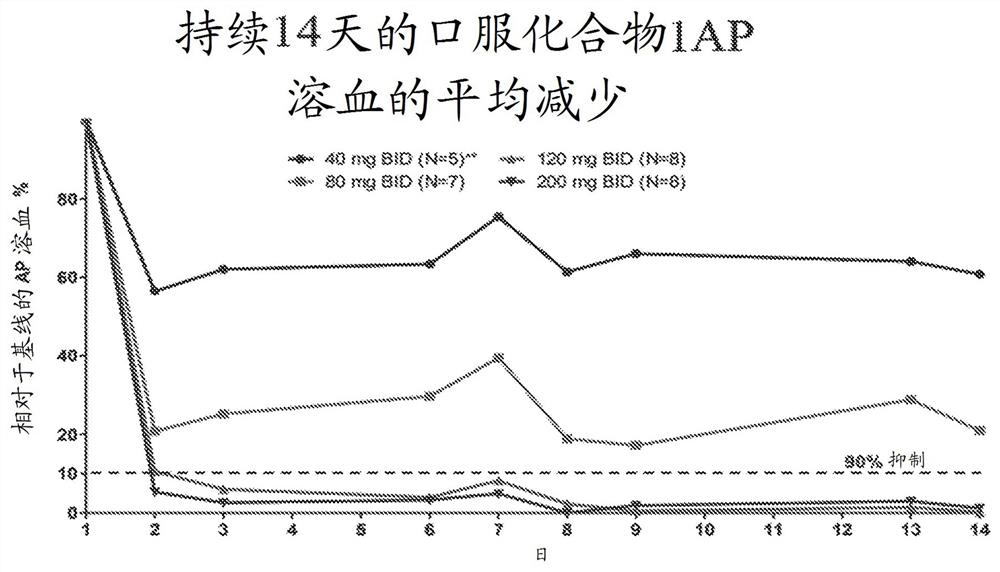
图13是服用40 mg、80 mg、120 mg或200 mg的化合物1达14天的患者的旁路途径溶血的平均减少的图形显示。y轴是相对于基线的AP溶血百分比。x轴是治疗的天数,其中第1天是给药的第一天。每天在0小时时收集数据点。40 mg BID研究中的两名受试者因非安全相关原因而停药,第3名受试者因在第7天漏服剂量而从分析中移除。
图14是在空腹的受试者(n=6)中以240 mg的单剂量、在进食的受试者(n=6)中以120 mg的单剂量施用的化合物1以及安慰剂(n=4)的平均血清旁路途径溶血的图形显示。在从第1天(给药前)至第7天的预定时间点收集的样品中评估血清AP溶血。x轴是以小时为单位的时间,y轴是AP活性百分比。
图15是在空腹的受试者(n=6)中以240 mg的单剂量、在进食的受试者(n=6)中以120 mg的单剂量施用化合物1后以及施用安慰剂(n=4)后的前24小时内,如通过ELISA测定法对所有受试者测定的平均血浆Bb浓度的图形显示。水平虚线网格线表示:正常值上限(1.42 µg/ml);正常值下限(0.48 µg/ml);和定量下限(LLOQ) (0.33 µg/ml)。x轴是以小时为单位的时间,y轴是以µg/mL为单位测量的平均血浆Bb浓度。
图16是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血浆浓度的图形显示。在从第1天(给药前)至第16天的预定时间点收集的样品中评估每个队列的血浆浓度。x轴是以小时为单位的时间,y轴是以ng/mL为单位的平均血浆浓度。
图17是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径的溶血活性的图形显示。显示了每个队列的完整的21天时间过程的活性。在第1天(0小时至12小时)、第7天(0小时至12小时)和第14天(0小时至16小时)进行了密集采样;在指定的日期于0小时时(早晨的PK谷)收集所有其他血清样品。x轴是以天为单位的时间,y轴是AP活性%。
图18是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径的溶血活性的图形显示。在第1天(0小时至12小时)进行了密集采样。x轴是以小时为单位的时间,y轴是AP活性百分比。
图19是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径溶血活性的图形显示。在第7天(0小时至12小时)进行了密集采样。x轴是以小时为单位的时间,y轴是AP活性百分比。
图20是以40 mg BID (n=8)、80 mg BID (n=7)、120 mg BID (n=8)、200 mg BID(n=8)施用的化合物1以及安慰剂BID (n=14)的平均血清旁路途径的溶血活性的图形显示。在第14天(0小时至12小时)进行了密集采样。x轴是以小时为单位的时间,y轴是AP活性百分比。
图21A是根据血浆化合物1的浓度和相应血清样品中AP Wieslab活性的抑制百分比进行的PK-PD评估的图形显示。使用简单的E
图21B是根据血浆化合物1的浓度和相应血清样品中AP Wieslab活性的抑制百分比进行的PK-PD评估的图形显示。使用四参数S型模型进行非线性回归分析。最佳拟合值和95%置信区间以ng/mL为单位。x轴是以ng/mL为单位测量的化合物1血浆浓度,y轴是以百分比测量的AP活性的抑制百分比。
具体实施方式
补体的功能障碍或过度激活与某些自身免疫性疾病、炎性疾病和神经变性疾病以及缺血再灌注损伤和癌症相关。例如,补体级联的旁路途径的激活有助于C3a和C5a的产生,这两者都是强效过敏毒素,它们也在许多炎性病症中起作用。因此,在一些情况下,需要降低补体途径(包括旁路补体途径)的响应。已发现,在每日两次约100 mg至200 mg,更具体地每日两次约120 mg至180 mg,甚至更具体地每日两次约120 mg至150 mg,或每日两次约150mg至约200 mg,以及在一个实施方案中每日两次约120 mg的化合物1的口服剂量下,在剂量间隔结束时提供最佳最低平均血浆浓度(C
或其药学上可接受的组合物、盐、同位素类似物或前药,所述口服剂型被施用于受试者(优选为人)以用于治疗由补体功能障碍或过度激活介导的病症(“补体介导的病症”),使得获得本文所述的特定PK和/或PD血液分布。已发现,给人受试者服用化合物1至为约65ng/mL至95 ng/mL的平均血浆浓度水平提供了对AP的强效抑制。制备化合物1的方法描述于PCT 公布第开2017/035353号(通过引用并入本文)。
“如本文中所用,AUC”(量*时间/体积)意指血浆浓度-时间曲线下的面积。
如本文中所用,“AUC(0-
如本文中所用,“AUC
如本文中所用,“AUEC”是指药效(PD)效应曲线下的面积。
如本文中所用,“AUEC
如本文中所用,“%AUEC
如本文中所用,“A
如本文中所用,“BID”是指“每日两次(bis en die)”或每天两次。
如本文中所用,“C
如本文中所用,“C
如本文中所用,“CSR”意指临床研究报告。
如本文中所用,“持续时间
如本文中所用,“EC
如本文中所用,“最低平均血浆浓度”意指C
如本文所用,“t
如本文中所用,“%CV”意指变异系数。
如本文中所用,“AP”意指旁路补体途径。
如本文中所用,“Bb”意指补体因子B的Bb片段。
如本文中所用,“ELISA”意指酶联免疫吸附测定。
如本文中所用,“FD”意指补充因子D。
如本文中所用,“MAD”意指多递增剂量。
如本文中所用,“N”意指患者总数。
如本文中所用,“PD”意指药效学。
如本文中所用,“PK”意指药代动力学。
如本文中所用,“SAD”意指单一递增剂量。
如本文中所用,“SD”意指标准偏差。
适用于本发明的药物组合物/组合的术语“载体”是指与化合物I一起提供的稀释剂、赋形剂或媒介物。
“剂型”意指活性剂的施用单位。剂型的实例包括片剂、胶囊剂、混悬剂、液体剂、乳剂、颗粒剂、球剂(spheres)、颊用形式(buccal)、舌下剂(sublingual)、凝胶剂、粘膜剂(mucosal)等。
“患者”或“宿主”或“受试者”是需要治疗或预防(包括但不限于通过调节补体因子D途径)本文具体描述的任何病症的人或非人动物。通常宿主是人。“患者”或“宿主”或“受试者”还指例如哺乳动物、灵长类动物(例如,人)、牛、绵羊、山羊、马、狗、猫、兔、大鼠、小鼠、鱼、鸟等。
如本文中所用,术语“药学上可接受的盐”是指在合理的医学判断范围内,适用于与受试者(例如,人受试者)接触而没有过度毒性、刺激、过敏响应等,与合理的益处/风险比相称,并且对其预期用途有效的那些盐,以及在可能的情况下,目前公开的主题的化合物的两性离子形式。
因此,术语“盐”是指本公开主题的化合物的相对无毒的无机和有机酸加成盐。这些盐可以在化合物的最终分离和纯化期间原位制备,或通过将纯化的化合物以其游离碱的形式与适合的有机或无机酸单独反应以及分离由此形成的盐来制备。药学上可接受的碱加成盐可用金属或胺诸如碱金属和碱土金属氢氧化物或有机胺形成。用作阳离子的金属的实例包括但不限于钠、钾、镁、钙等。合适的胺的实例包括但不限于N,N'-二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、N-甲基葡糖胺和普鲁卡因。
盐可由无机酸硫酸、焦硫酸、重硫酸、亚硫酸、重亚硫酸、硝酸、磷酸、磷酸一氢酸、磷酸二氢酸、偏磷酸、焦磷酸、氯酸、溴酸、碘酸诸如盐酸、硝酸、磷酸、硫酸、氢溴酸、氢碘酸、磷酸等制备。代表性盐包括氢溴酸盐、盐酸盐、硫酸盐、重硫酸盐、硝酸盐、乙酸盐、草酸盐、戊酸盐、油酸盐、棕榈酸盐、硬脂酸盐、月桂酸盐、硼酸盐、苯甲酸盐、乳酸盐、磷酸盐、甲苯磺酸盐、柠檬酸盐、马来酸盐、富马酸盐、琥珀酸盐、酒石酸盐、萘酸盐、甲磺酸盐、葡庚糖酸盐、乳糖酸盐、月桂基磺酸盐和羟乙基磺酸酯等。盐也可以由有机酸,诸如脂族单羧酸和二羧酸、苯基取代的链烷酸、羟基链烷酸、链烷二酸、芳族酸、脂族和芳族磺酸等制备。代表性盐包括乙酸盐、丙酸盐、辛酸盐、异丁酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、延胡索酸盐、马来酸盐、扁桃酸盐、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、邻苯二甲酸盐、苯磺酸盐、甲苯磺酸盐、苯乙酸盐、柠檬酸盐、乳酸盐、马来酸盐、酒石酸盐、甲磺酸盐等。药学上可接受的盐可包括基于碱金属和碱土金属(诸如钠、锂、钾、钙、镁等)的阳离子,以及无毒的铵、季铵和胺阳离子,包括但不限于铵、四甲铵、四乙铵、甲胺、二甲胺、三甲胺、三乙胺、乙胺等。还设想了氨基酸的盐,诸如精氨酸盐、葡糖酸盐、半乳糖醛酸盐等。参见,例如,Berge等人,J. Pharm. Sci., 1977, 66, 1-19,其通过引用并入本文。
“药物组合物”是包含至少一种活性剂和至少一种其他物质诸如载体的组合物。“药物组合”是至少两种活性剂的组合,所述至少两种活性剂可以组合成单一剂型,或者以单独的剂型一起提供,并说明所述活性剂将一起用于治疗本文所述的任何病症。
“药学上可接受的赋形剂”意指可用于制备药物组合物/组合,通常是安全的、无毒的,并且在生物学上和其他方面都适合于向宿主(通常是人)施用的赋形剂。在一些实施方案中,使用对于兽医用途是可接受的赋形剂。
除非另有说明,否则在整个说明书和权利要求书中,给定的化学式或名称应涵盖所有光学和立体异构体,以及其中存在此类异构体和混合物的外消旋混合物。
在下文和本文中的一般描述中,无论何时使用涉及化合物1的任何术语,都应该理解为包括药学上可接受的盐或组合物,除非另有说明或与本文不一致。
如本文所预期的并且出于本文公开范围的目的,本文所述的所有范围包括出现在确定范围内的任何和所有数值。例如,如本文所设想的,1至10或1与10之间的范围将包括数值1、2、3、4、5、6、7、8、9、10及其分数。
本发明提供了特定剂型和使用所述剂型治疗患有补体介导的病症的受试者的方法,所述剂型提供补体因子D (fD)抑制剂(1R,3S,5R)-2-(2-(3-乙酰基-5-(2-甲基嘧啶-5-基)-1H-吲唑-1-基)乙酰基)-N-(6-溴-3-甲基吡啶-2-基)-5-甲基-2-氮杂双环[3.1.0]己烷-3-甲酰胺(化合物1)的血液分布范围。
一方面,向患有补体介导的病症的受试者施用化合物1,使得单剂量的化合物1提供如本文所述的特定的PK和/或PD血液分布。在一些实施方案中,施用化合物1使得在治疗期间,在BID给药方案的昼夜周期中,为约50 ng/mL至约200 ng/mL的最低平均血浆浓度(C
一方面,化合物1以剂型提供,例如以诸如固体剂型或基于液体凝胶的剂型等口服剂型提供,所述剂型提供最大平均血浆浓度(C
一方面,化合物1以剂型提供,例如以诸如固体剂型或基于液体凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的AUC
对每个采样的12小时给药间隔计算另外两个药效学活性测量值:持续时间
一方面,化合物1以剂型提供,例如以诸如固体剂型或充液胶囊或基于液体凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的持续时间
一方面,化合物1以剂型提供,例如以诸如固体剂型或充液胶囊或基于凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的最大血浆浓度%AUEC
一方面,化合物1以剂型提供,例如以诸如固体剂型或充液胶囊或基于凝胶的剂型等口服剂型提供,所述剂型提供如本文所述的最大血浆浓度的时间(t
在一些实施方案中,施用化合物1,使得单剂量提供如本文所述的特定PK和/或PD血液特性。在一些实施方案中,施用于受试者的剂量为约25 mg至约275 mg。在一些实施方案中,施用的剂量至少约为25 mg、至少约为30 mg、至少约为35 mg、至少约为40 mg、至少约为45 mg、至少约为50 mg、至少约为55 mg、至少约为60 mg、至少约为65 mg、至少约为70mg、至少约为75 mg、至少约为80 mg、至少约为85 mg、至少约为90 mg、至少约为95 mg、至少约为100 mg、至少约为110 mg、至少约为120 mg、至少约为130 mg、至少约为140 mg、至少约为150 mg、至少约为160 mg、至少约为170 mg、至少约为180 mg、至少约为190 mg、至少约为200 mg、至少约为210 mg、至少约为220 mg、至少约为230 mg、至少约为240 mg、至少约为250 mg、至少约为260 mg、至少约为270mg或至少约为275 mg。在一些实施方案中,以至少约为120mg的单剂量施用化合物1。在一些实施方案中,以至少约为240 mg的单剂量施用化合物1。在一些实施方案中,以120 mg +/-10%的单剂量施用化合物1。在一些实施方案中,在治疗期间每天施用化合物1一次。在一些实施方案中,在治疗期间每天施用化合物1两次(BID)。在一些实施方案中,在治疗期间每天施用化合物1至少两次。在一些实施方案中,在治疗期间每天施用化合物1三次或更多次。在一些实施方案中,每天以至少约为120 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为150 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为175 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为200 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为210 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为225 mg的单剂量施用化合物1两次。在一些实施方案中,每天以至少约为250 mg的单剂量施用化合物1两次。在一些实施方案中,每天施用化合物1两次,每次剂量间隔大约12小时。在一些实施方案中,每天施用化合物1两次,每次剂量间隔约12小时,持续28天。在一些实施方案中,每天施用化合物1两次,持续至少约4周、至少约5周、至少约6周或至少约12周。在一些实施方案中,每天施用化合物1两次,持续至少约3周、至少约6周或至少约9周。
化合物1可作为纯化学品施用。或者,化合物1可作为药物组合物施用,如本文所述,所述药物组合物包含对于需要这种治疗的宿主(通常是人)有效的量的化合物1。因此,本公开提供了纯净的药物组合物,其包含以一定量的化合物或药学上可接受的盐存在的化合物1的剂型以及至少一种药学上可接受的载体,以实现本文所述的血液分布范围。药物组合物可包含化合物或盐作为唯一的活性剂,或者在替代实施方案中,包含所述化合物和至少一种另外的活性剂。药物组合物还可包含一定摩尔比的活性化合物和另外的活性剂。
载体包括赋形剂和稀释剂,并且必须具有足够高的纯度和足够低的毒性,以使其适合施用于向接受治疗的患者施用。载体可以是惰性的,或者其可具有其自身的药物益处。与化合物1结合使用的载体的量足以为每单位剂量的化合物的施用提供物质的实际量。
载体类别包括但不限于粘合剂、缓冲剂、着色剂、稀释剂、崩解剂、乳化剂、调味剂、助流剂、润滑剂、防腐剂、稳定剂、表面活性剂、压片剂和润湿剂。一些载体可被列于不止一个类别中,例如植物油可在一些制剂中用作润滑剂,而在另一些制剂中用作稀释剂。示例性药学上可接受的载体包括糖、淀粉、纤维素、黄蓍胶粉、麦芽、明胶;滑石粉和植物油。药物组合物中可包含任选的活性剂,其基本上不干扰本发明化合物的活性。
可配制用于口服施用的药物组合物/组合。这些组合物可包含实现所需结果的任何量的活性化合物,例如0.1重量% (wt.%)至99重量% (wt.%)的化合物,通常至少约为5重量%的化合物。一些实施方案包含至少约25重量%至至少约50重量%或至少约5重量%至至少约75重量%的化合物。
如本文中所设想的,提供如本文所述的PK和/或PD血液分布的剂型可用于治疗补体介导的病症。通过达到和/或维持本文所述的某些血液分布,可抑制旁路补体途径,提供可用于治疗由有缺陷或过度活跃的补体反应导致的病症的给药方案。
在一些实施方案中,施用化合物1,使得在治疗期间,在BID给药方案的昼夜周期中,为约50 ng/mL至约200 ng/mL的最低平均血浆浓度(C
在一些实施方案中,施用化合物1,使得在治疗期间达到约10小时至约12小时的持续时间
在一些实施方案中,施用化合物1,使得在治疗期间达到约80%至约100%的%AEUC
在一些实施方案中,施用化合物1,使得在治疗期间达到小于约3000 ng/mL的C
在一些实施方案中,施用化合物1,使得在治疗期间达到为约6000 ng*hr/mL至20000 ng*hr/mL的AUC
在一些实施方案中,提供了用于治疗与宿主的补体级联功能障碍相关联的病症的方法,所述方法包括施用化合物1以实现如本文所述的特定PK和/或PD血液分布。在一些实施方案中,提供了用于抑制受试者的旁路补体途径激活的方法,所述方法包括施用化合物1以实现如本文所述的特定PK和/或PD血液分布。在一些实施方案中,提供了调节受试者的因子D活性的方法,所述方法包括施用化合物1以实现如本文所述的特定 PK和/或PD血液分布。
在一些实施方案中,所述病症是阵发性睡眠性血红蛋白尿症(PNH)。一方面,以足够的量向患有PNH的受试者施用化合物1,其中在治疗过程中,乳酸脱氢酶(LDH)水平(血管内溶血的生物标志物)从施用前测量的基线LDH水平相对于基线降低至少约30%、至少约40%、至少约50%、至少约60%、至少约70%、至少约80%或超过80%。一方面,以足够的量向患有PNH的受试者施用化合物1,其中在治疗过程中,血红蛋白水平从施用前测量的基线水平相对于基线升高至少约10%、至少约15%、至少约20%、至少约25%或超过25%。
在一些实施方案中,所述病症是膜性增生性肾小球肾炎(MPGN)。MPGN是影响肾脏肾小球或过滤器的疾病。直到最近,当潜在病因可鉴定时,膜性增生性肾小球肾炎(MPGN)在临床上被归类为原发性MPGN、特发性MPGN或继发性MPGN。主要根据电子致密沉积物的超微结构外观和位置,将原发性MPGN进一步分为三种类型—I型、II型和III型。然而,临床和组织病理学方案都存在问题,因为它们都不是基于疾病的发病机制。对补体在MPGN发病机制中的作用的更好理解导致提议重新分类为免疫球蛋白介导的疾病(由经典补体途径驱动的)和非免疫球蛋白介导的疾病(由旁路补体途径驱动的)。这种重新分类导致了改进的诊断临床算法,并出现了一组新的疾病,称为C3肾小球病,最好的代表是致密沉积物病(DDD)和C3肾小球肾炎(C3GN)。
在一些实施方案中,所述病症是C3肾小球病。C3肾小球病是一组导致肾脏功能障碍的相关疾患。C3肾小球病的主要特征包括尿中的蛋白质水平高(蛋白尿)、尿中带血(血尿)、尿量减少、血液中蛋白质水平低以及身体许多部位肿胀。受影响的个体的血液中的补体组分3(或C3)的水平可能特别低。与C3肾小球病相关的肾脏问题往往会随着时间的推移而恶化。大约一半的受影响的个体在他们的诊断后10年内发展为终末期肾病(ESRD)。ESRD是危及生命的疾患,其会阻止肾脏有效地过滤身体中的液体和废物。
在一些实施方案中,所述疾病是致密沉积物病(DDD)。在一些实施方案中,所述疾病是C3肾小球肾炎(C3GN)。
在一些实施方案中,所述疾病是免疫复合物膜性增生性肾小球肾炎(IC-MPGN))。IC-MPGN是肾脏疾病,其与C3G共有许多临床、病理、遗传和实验室特征。高达40%的IC-MPGN患者没有可鉴定的潜在病因,被认为患有特发性IC-MPGN。患有特发性IC-MPGN的受试者可具有低的C3水平和正常的C4水平,类似于在C3G中观察到的那些,以及许多与异常旁路途径的活性相关的相同遗传或获得性因素。那些具有低的C3和正常的C4的受试者很可能具有显著的旁路途径的过度活性。C3Nef在MPGN 1型患者中的存在被确认为与其在C3GN患者中的存在一样频繁。在IC-MPGN患者中发现了编码旁路途径蛋白(包括 fH和fI)的基因的突变。尽管在肾活检中存在IgG、IgM和C1q (在一小部分病例中)的免疫阳性荧光染色,但大约46%的IC-MPGN病例表现出降低的C3水平和正常的C4水平。这些数据表明旁路途径在IC-MPGN中失调。
在一些实施方案中,施用化合物1,使得LDH水平在治疗期间从高于或等于正常值上限(ULN)的1.5倍的水平降低。在一些实施方案中,施用化合物1,使得Hgb水平在治疗期间升高。
可由本文所述的化合物1或其盐或组合物治疗或预防的其他病症还包括但不限于:
(i) 腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析、神经脊髓炎(NMO)、脂肪肝、非酒精性脂肪性肝炎(NASH)、肝脏炎症、肝硬化、肝衰竭、皮肌炎、肌萎缩侧索硬化、年龄相关性黄斑变性(AMD)、类风湿性关节炎以及响应于生物治疗剂(例如CAR T细胞疗法)的细胞因子或炎性反应、阵发性睡眠性血红蛋白尿症(PNH)、遗传性血管性水肿、毛细血管渗漏综合征、非典型溶血性尿毒症综合征(aHUS)、溶血性尿毒症综合征(HUS)、腹主动脉瘤、血液透析并发症、溶血性贫血或血液透析;
(ii) 重症肌无力(MG)、多发性硬化、神经病学病症、格林巴利综合征(GuillainBarre Syndrome)、中枢神经系统疾病和其他神经变性疾患、肾小球肾炎(包括膜性增生性肾小球肾炎)、SLE肾炎、增生性肾炎、肝纤维化、组织再生和神经再生或巴勒奎尔-西蒙斯综合征(Barraquer-Simons Syndrome);
(iii) 脓毒症、全身炎性响应综合征(SIRS)、不适当或不良补体激活的病症、IL-2治疗期间的白介素2诱导的毒性、炎性病症、自身免疫性疾病的炎症、系统性红斑狼疮(SLE)、克罗恩病(Crohn’s disease)、类风湿性关节炎、炎性肠病、狼疮性肾炎、关节炎、免疫复合物病症和自身免疫性疾病、系统性狼疮或红斑狼疮的炎性作用;
(iv) 缺血/再灌注损伤(I/R损伤)、心肌梗塞、心肌炎、缺血再灌注后疾患、球囊血管成形术、动脉粥样硬化、心肺旁路术或肾旁路术中的泵后综合征、肾局部缺血、主动脉重建后肠系膜动脉再灌注、抗磷脂综合征、自身免疫性心脏病、缺血再灌注损伤、肥胖或糖尿病;
(v) 阿尔茨海默痴呆症、中风、精神分裂症、创伤性脑损伤、创伤、帕金森病(Parkinson's disease)、癫痫、移植排斥、胎儿丢失的预防、生物材料反应(例如,在血液透析、植入物中)、超急性同种异体移植排斥、异种移植排斥、移植、银屑病、烧伤、热损伤,包括烧伤或冻伤;
(vi) 哮喘、过敏、急性呼吸窘迫综合征(ARDS)、囊性纤维化、成人呼吸窘迫综合征、呼吸困难、咳血、慢性阻塞性肺病(COPD)、肺气肿、肺栓塞和梗塞、肺炎、纤维性粉尘病(纤维性粉尘病)、惰性粉尘和矿物质(例如,硅、煤尘、铍和石棉)、肺纤维化、有机尘病、化学损伤(由于刺激性气体和化学品,例如氯、光气、二氧化硫、硫化氢、二氧化氮、氨和盐酸)、烟雾损伤、热损伤(例如,烧伤、冻伤)、支气管收缩、过敏性肺炎、寄生虫病、古德帕斯彻氏综合征(Goodpasture's Syndrome)(抗肾小球基底膜肾炎)、肺血管炎、寡免疫性血管炎(Pauci-immune vasculitis)或免疫复合物相关炎症。
在一些实施方案中,化合物1可以与至少一种另外的治疗剂组合或交替提供,例如用于治疗本文所列的病症。在一些实施方案中,化合物1可以与补体系统的至少一种另外的抑制剂或具有不同生物学作用机制的第二活性化合物(例如,C5抑制剂、C3抑制剂、补体因子B抑制剂或泛补体抑制剂)组合或交替施用以实现如本文所述的特定PK和/或PD血液分布。
C5抑制剂是本领域已知的。在一些实施方案中,C5抑制剂是靶向C5的单克隆抗体。在一些实施方案中,C5抑制剂是依库珠单抗(Soliris
在一些实施方案中,C5抑制剂可以是但不限于:重组人微型抗体,例如Mubodina®(单克隆抗体,Adienne Pharma and Biotech, Bergamo, Italy;参见美国专利第7,999,081号)、coversin (小动物蛋白,Volution Immuno-pharmaceuticals, Geneva,Switzerland;参见例如Penabad等人Lupus, 2012, 23(12):1324-6)、LFG316 (单克隆抗体,Novartis, Basel, Switzerland和Morphosys, Planegg, Germany;参见美国专利第8,241,628号和第8,883,158号); ARC-1905 (聚乙二醇化的RNA适体,Ophthotech,Princeton, NJ and New York, NY;参见Keefe等人,Nature Reviews Drug Discovery,9, 537-550)、RA101348和RA101495 (大环肽,Ra Pharmaceuticals, Cambridge, MA)、SOBI002 (亲和体(affibody),Swedish Orphan Biovitrum, Stockholm, Sweden)、ALN-CC5 (Si-RNA, Alnylam Pharmaceuticals, Cambridge, MA)、ARC1005 (适体,NovoNordisk, Bagsvaerd, Denmark)、SOMAmers (适体,SomaLogic, Boulder, Co)、SSL7 (细菌蛋白毒素,参见,例如Laursen等人Proc. Natl. Acad. Sci. U.S.A., 107(8):3681-6)、MEDI7814 (单克隆抗体,MedImmune, Gaithersburg, MD)、金精三羧酸、金精三羧酸衍生物(Aurin Biotech, Vancouver, BC,参见美国专利申请公布2013/003592)、RG6107 (抗C5循环抗体,Roche Pharmaceuticals, Basel, Switzerland)、ALXN1210和ALXN5500 (单克隆抗体,Alexion Pharmaceuticals, New Haven, CT)、TT30 (融合蛋白,AlexionPharmaceuticals, New Haven, CT)、REGN3918 (单克隆抗体,Regeneron, Tarrytown,NY)、ABP959 (依库珠单抗生物仿制药,Amgen, Thousand Oaks, CA)或其组合。
在一些实施方案中,C5抑制剂是重组人微型抗体,例如Mubodina®。Mubodina®是由Adienne Pharma and Biotech开发的完全人源重组抗体C5。Mubodina®描述于美国专利第7,999,081号中。
在一些实施方案中,C5抑制剂是coversin。Coversin是重组蛋白,其源自在
在一些实施方案中,C5抑制剂是特度鲁单抗(Tesidolumab)/LFG316. 特度鲁单抗是Novartis和Morphosys开发的单克隆抗体。特度鲁单抗描述于美国专利第8,241,628号和第8,883,158号中。
在一些实施方案中,C5抑制剂是ARC-1905。ARC-1905 是Ophthotech开发的聚乙二醇化的RNA适体。ARC-1905描述于Keefe等人Nature Reviews Drug Discovery, 9:537-550中。
在一些实施方案中,C5抑制剂是RA101348。RA101348是Ra Pharmaceuticals开发的大环肽。
在一些实施方案中,C5抑制剂是RA101495。RA101495是Ra Pharmaceuticals开发的大环肽。
在一些实施方案中,C5抑制剂是SOBI002。SOBI002是Swedish Orphan Biovitrum开发的亲和体。
在一些实施方案中,C5抑制剂是ARC1005。ARC1005是Novo Nordisk开发的适体。
在一些实施方案中,C5抑制剂是C5的SOMAmers。SOMAmers是SomaLogic开发的适体。
在一些实施方案中,C5抑制剂是SSL7。SSL7是Laursen等人Proc. Natl. Acad.Sci. U.S.A., 107(8):3681-6中描述的细菌蛋白质毒素。
在一些实施方案中,C5抑制剂是MEDI7814。MEDI7814是MedImmune开发的单克隆抗体。
在一些实施方案中,C5抑制剂是金精三羧酸。在另一个实施方案中,C5抑制剂是金精三羧酸衍生物。这些金精衍生物由Aurin Biotech开发,并在美国专利申请公布第2013/003592号中进一步描述)。
在一些实施方案中,C5抑制剂是RG6107/SKY59。RG6107/SKY59是由RochePharmaceuticals开发的抗C5循环抗体。
在一些实施方案中,C5抑制剂是ALXN1210。在另一个实施方案中,C5抑制剂是ALXN5500。ALXN1210和ALXN5500是Alexion Pharmaceuticals开发的单克隆抗体。
在一些实施方案中,C5抑制剂是TT30。TT30是Alexion Pharmaceuticals开发的融合蛋白。
在一些实施方案中,C5抑制剂是ABP959。ABP959是Amgen开发的依库珠单抗生物仿制药单克隆抗体。
在一些实施方案中,C5抑制剂是抗C5 siRNA。抗C5 siRNA是由AlnylamPharmaceuticals开发的。
在一些实施方案中,C5抑制剂是Erdigna
在一些实施方案中,C5抑制剂是培戈-阿伐普他(avacincaptad pegol)/Zimura
在一些实施方案中,C5抑制剂是SOBI005。SOBI005是Swedish Orphan Biovitrum开发的蛋白质。
在一些实施方案中,C5抑制剂是ISU305。ISU305是ISU ABXIS开发的单克隆抗体。
在一些实施方案中,C5抑制剂是REGN3918。REGN3918是Regeneron开发的单克隆抗体。
本文提供了用于治疗受试者的PNH的方法,所述方法包括向受试者施用有效量的C3抑制剂与有效量的选自式I或式II的CFD抑制剂的组合或交替施用有效量的所述C3抑制剂和有效量的所述CFD抑制剂。
C3抑制剂是本领域已知的。在一些实施方案中,化合物1与康普他汀(compstatin)和/或康普他汀类似物组合或交替施用。康普他汀和康普他汀类似物是已知的并且被发现是有用的C3抑制剂,参见美国专利第9,056,076号、第8,168,584号、第9,421,240号、第9,291,622号、第8,580,735号、第9371365号、第9,169,307号、第8,946,145号、第7,989,589号、第7,888,323号、第6,319,897号以及美国专利申请公布第2016/0060297号、第2016/0015810号、第2016/0215022号、第2016/0215020号、第2016/0194359号、第2014/0371133号、第2014/0323407号、第2014/0050739号、第2013/0324482号和第2015/0158915号。在又一实施方案中,康普他汀类似物是4(1MeW)POT-4. 4(1MeW)POT-4由Potentia开发。在又一实施方案中,康普他汀类似物是AMY-201。AMY-201是由Amyndas Pharmaceuticals开发的。
在一些实施方案中,化合物1可以与C3抑制剂组合,所述抑制剂包含但不限于:H17(单克隆抗体,EluSys Therapeutics, Pine Brook, NJ)、米考西普(基于CR1的蛋白)、sCR1(基于CR1的蛋白,Celldex, Hampton, NJ)、TT32 (基于CR-1的蛋白,AlexionPharmaceuticals, New Haven, CT)、HC-1496 (重组肽)、CB 2782 (酶,CatalystBiosciences, South San Francisco, CA)、APL-2 (peg化的合成周期肽,ApellisPharmaceuticals, Crestwood, KY)或其组合。
在一些实施方案中,C3抑制剂是H17。H17是EluSys Therapeutics正在开发的人源化单克隆抗体。H17描述于Paixao-Cavalcante等人J. Immunol. 2014, 192(10):4844-4851中。
在一些实施方案中,C3抑制剂是米考西普。米考西普是由InflazymePharmaceuticals开发的基于CR1的蛋白。
在一些实施方案中,C3抑制剂是sCR1。 sCR1是由Celldex开发的CR1蛋白的可溶性形式。
在一些实施方案中,C3抑制剂是TT32。TT32是Alexion Pharmaceuticals开发的基于CR-1的蛋白。
在一些实施方案中,C3抑制剂是HC-1496。HC-1496是InCode开发的重组肽。
在一些实施方案中,C3抑制剂是CB 2782。CB 2782是源自Catalyst Biosciences开发的人膜型丝氨酸蛋白酶1 (MTSP-1)的新型蛋白酶。
在一些实施方案中,C3抑制剂是APL-2。APL-2是Apellis Pharmaceuticals开发的APL-1的聚乙二醇化形式。
CFB抑制剂是本领域已知的。在一些实施方案中,化合物1可以与CFB抑制剂组合,所述CFB抑制剂包括但不限于:抗-FB SiRNA (Alnylam Pharmaceuticals, Cambridge,MA)、TA106 (单克隆抗体,Alexion Pharmaceuticals, New Haven, CT)、LNP023 (小分子,Novartis, Basel, Switzerland)、SOMAmers (适体,SomaLogic, Boulder, CO)、拜卡西奥单抗(bikaciomab)(Novelmed Therapeutics, Cleveland, OH)、complin (例如,Kadam等人, J. Immunol. 2010, DOI:10.409/jimmunol.10000200)、Ionis-FB-L
在一些实施方案中,CFB抑制剂是抗FB siRNA。抗FB siRNA是AlnylamPharmaceuticals开发的。
在一些实施方案中,CFB抑制剂是TA106。TA106是Alexion Pharmaceuticals开发的单克隆抗体。
在一些实施方案中,CFB抑制剂是LNP023。LNP023是Novartis开发的CFB的小分子抑制剂。LNP023和相关抑制剂描述于Maibaum等人Nat. Chem. Biol. 2016, 12:1105-1110中。
在一些实施方案中,CFB抑制剂是complin。Complin是肽在Kadam等人J. Immunol.2010 184(12):7116-24中描述的肽抑制剂。
在一些实施方案中,CFB抑制剂是Ionis-FB-L
补体成分的泛抑制剂是本领域已知的。在一些实施方案中,所述抑制剂是FUT-175。
实施例
单一递增剂量(SAD)研究是化合物1的首次人体研究。主要目的是证明化合物1(一种经口施用的补体因子D抑制剂)的单一递增口服剂量在健康的志愿者中的安全性和耐受性。次要目的包括评价药代动力学(PK)特性以及化合物1 PK与药效学(PD)特征之间的关系,即补体旁路途径(AP)活性的抑制(PK/PD)。
在4个剂量组中对18名健康志愿者进行给药和评价,其中10名受试者接受安慰剂(表1)。第一剂量组具有6名活性剂受试者和6名安慰剂受试者;随后的组各自具有6名活性剂受试者和2名安慰剂受试者。给药后对每组随访28天。直到第28天的最后一次预定随访,对所有受试者的安全性进行监测。血液和尿液样品在从第1天至第4天的预定时间点收集以确定化合物1的血浆和尿液浓度,并从第1天至第7天的预定时间点收集以确定补体相关活性。测定了通过AP Wieslab测定法测量的血清AP活性以及血浆Bb、因子D水平和几种其他PD生物标志物的结果。
表1. ACH228-001中的剂量组(SAD研究)
PBO=安慰剂
兔红细胞(RBC)、不含Ca++和Mg++ (GVB0)的明胶佛罗那缓冲液(gelatin veronalbuffer)和100 mM MgCl2 - 100 mM EGTA (MgEGTA)购自Complement Technology, Inc,(Tyler, TX)。 GVB0-MgEGTA缓冲液是通过将GVB0和100 mM MgEGTA以9:1的比率混合制备的。RBC在购买的两周内使用;每天在测定前通过在800 x g和4℃下离心3分钟收集细胞,并将其重悬于等体积的新鲜冷GVB0 - MgEGTA中至密度为5×108个细胞/mL。本研究中使用的商业ELISA试剂盒的信息包含在下面的每个特定测定中。
对于人血清的制备,通过标准临床程序将静脉血收集到Gold Top SST真空采血管(vacutainer) (BD Vacutainer)中。在室温下凝固30分钟或更长时间后,将管以约1,300 xg在4℃下离心15分钟以将血清与凝块分离。然后将血清等分到预冷的冷冻小瓶(50-200 µl/小瓶)中,于-80℃下储存。每个小瓶仅解冻一次并用于每次补体相关测定,丢弃剩余样品而不进一步使用。为了制备血浆,将静脉血收集到Lavender Top, K2EDTA真空采血管中,并轻轻倒转几次,使血液样品与抗凝剂完全混合。将管置于冰浴中30分钟,然后在4℃下以约1300 x g离心15分钟以将血浆与血细胞分离。将血浆在预冷的低温储存管中分成等分试样,并在采集血样后一小时内储存在-80℃的冰箱中。对于递送,将含有血清或血浆的管在干冰存在的情况下进行运送,而无需额外的冻融循环。
溶血(%) = ((Hm – Bk) – (Sp – Bf)) / ((NH - BkNH) – (Sp – Bf)) × 100%
其中Hm = 溶血孔中的A
使用以下两个模型,利用时间匹配的血浆化合物1的浓度值和血清AP抑制值,通过利用GraphPad Prism (La Jolla, CA)进行的非线性回归来评价PK-PD关系:
1. 简单的E
Y = E
其中X =血浆化合物1的浓度,Y =相应血清样品中的抑制百分比,E
2. 四参数S型模型,用于确定EC
Y = E
其中X、Y、E
对于所有受试者,在从第1天至第7天的特定方案确定的时间点收集血液样品,以使用AP溶血测定和AP Wieslab测定来测定
对于施用了40 mg、80 mg或120 mg的单剂量的化合物1的所有受试者观察到AP活性的强效抑制(图1)。具体而言,所有组中的受试者在化合物1给药后均实现了对AP活性的完全抑制。在第1组中,根据AP溶血测定,大约90%或更高的抑制在给药后维持了至少4小时。在第2组和第3组中,大约90%或更高的抑制在给药后维持了至少8小时。相反地,在安慰剂受试者中未观察到AP活性的显著变化(图1)。使用简单的E
另外,对于第3组中化合物1-治疗的受试者和安慰剂受试者,通过本文所述的AP溶血测定法测量的所有血清样品中的AP活性与AP Wieslab数据密切相关(相关系数,r =0.83)(图3)。使用以下简单的E
对于包含组1至组3 SAD PK群体的18名受试者,总共306个血浆样品(在给药前和给药后0.5小时、1小时、1.5小时、2小时、2.5小时、3小时、4小时、6小时、8小时、10小时、12小时、16小时、24小时[第2天]、48小时[第3天]、72小时[第4天]和144小时[第7天]从每名受试者收集的17个样品)可用于血浆中的PK测定。
在包含PK群体的所有18名受试者中,给药前血浆化合物1的浓度低于LLOQ (即,<0.100 ng/mL)。给药后化合物1被迅速吸收,其中所有18名受试者在评价的第一个给药后采样时间点(即,给药后0.5小时)具有可测量的血浆化合物1的浓度。随机化治疗组的平均血浆化合物1的浓度相对时间曲线的图分别显示于图4A (40 mg)、图5A (80mg)、图6A (120mg)(线性标度)和图4B (40 mg)、图5B (80 mg)和图6B (120 mg)(半对数标度)中,汇总统计数据提供于表2中。最大平均血浆浓度(78.03 ± 19.65 ng/mL在40 mg组中,224.05 ±108.27 ng/mL在80 mg组中,以及282.83 ± 89.15 ng/mL在120 mg组中)发生在给药后1至3小时之间。基于平均浓度曲线,在单剂量施用至多120 mg后,血浆化合物1的浓度似乎以剂量比例的方式增加。在所有剂量组中,在给药后72小时的时间点最后一次检测到血浆化合物1的浓度。
评价化合物1的单一递增剂量以确定所研究的40 mg至120 mg的剂量范围内的血浆和尿液PK参数。每个标称采样时间点时的血浆和尿液化合物1的浓度的汇总统计数据按随机治疗组呈现在表2中。
血浆化合物1的C
PK群体中包含的所有受试者(n = 18;每6名受试者随机接受40 mg、80 mg和120mg的化合物1)被纳入剂量比例分析。血浆化合物1的C
化合物1的肾排泄低,其中几何平均剂量恢复为未改变的化合物1 (范围为所施用的剂量的0.17%至0.26%)。各剂量组间的肾清除率值也相似,其中几何平均值的范围为0.16L/h至0.19 L/h。
表2. 血浆和尿液中化合物1的药代动力学参数估计值的总结
缩写:Ae
一直至第7天(以天为单位报告的)和给药后24小时内(通过随机治疗组和整体)的平均补体AP功能活性(相对于阳性对照的百分比)的图形显示分别见于图8B和图8A。如通过Wieslab测定所评估的,对于施用了40 mg、80 mg或120 mg的单剂量的化合物1的所有受试者均观察到对AP功能活性的强效抑制。
早在给药后0.5小时,对于所有化合物1治疗的受试者均注意到>80%的对AP功能活性的抑制(或相对于阳性对照≤20%的AP功能活性结果),所有受试者在给药后2小时实现了> 90%的对AP功能活性的抑制。在给药后1.5小时(在40 mg剂量组中)至3小时(在80 mg和120 mg剂量组中),达到了AP功能活性的最低点;对于40 mg、80 mg和120 mg剂量组,平均(± SD) AP功能活性分别降至最低水平1.25 (± 1.153)%、0.13 (± 0.097)%和0.09 (±0.102)%。对于40 mg剂量组,90%或更高的平均抑制在给药后维持了至少6小时。对于所研究的较高剂量组(即,120 mg和240 mg),90%或更高的平均抑制维持了更长的时间段(即,给药后持续至少10小时)。
一直至第7天(以天为单位报告的)和给药后24小时内(通过随机治疗组和整体)的平均血浆Bb浓度的图形显示可分别见于图9B和图9A。如图9A和图9B所示,相对于基线和安慰剂治疗组,所有评价的化合物1剂量组在单剂量施用后Bb血浆浓度均显著降低。尽管在单剂量组中评价的受试者数量相对较少(n=6),但观察到的变化幅度似乎是剂量依赖性的。在基线时,40 mg、80 mg和120 mg剂量组的Bb血浆浓度在测定的正常参考范围内(即,0.49 µg/mL至1.42 µg/mL);40 mg、80 mg和120 mg剂量组的平均(± SD)基线值分别为0.80 (±0.195) µg/mL、0.78 (±0.128) µg/mL和0.70 (±0.095) µg/mL。Bb血浆浓度在给药后6小时(在40 mg剂量组中)至8小时(在80 mg和120 mg剂量组中)达到最低点。给药后6小时,40mg剂量组中的平均(± SD) Bb血浆浓度已降至0.51 (±0.091) µg/mL。给药后8小时,平均Bb血浆浓度对于80 mg剂量组已降至0.48 (±0.056) µg/mL,并且对于120 mg剂量组降至0.40 (±0.029) µg/mL。对于80 mg和120 mg剂量组,平均Bb血浆浓度分别到给药后10小时(对于80 mg剂量组)和给药后24小时(第2天)(对于120 mg剂量组)增加到高于LLN(以用于测定),到给药后48小时(第3天)恢复至基线值。在研究期间,受试者在Bb血浆浓度方面都未出现治疗突发异常。
基于
本文报道的采用AP溶血测定的
具有两个额外的单剂量队列进行的化合物1的多递增剂量(MAD)研究也已完成。主要目的是证明多递增口服剂量的化合物1 (一种口服施用的补体因子D抑制剂)在健康志愿者中的安全性和耐受性。次要目的包括评价药代动力学(PK)分布以及化合物1 PK与药效学(PD)特征之间的关系,即补体旁路途径(AP)的活性的抑制(PK/PD)。在MAD研究中,在于空腹状态下施用化合物1 (40 mg、80 mg、120 mg和200 mg BID)的四个剂量组中持续14天评价了化合物1的多剂量药代动力学和药效学(表3)。每组中8名受试者接受活性药物;第一剂量组(40 mg)中8名受试者中接受安慰剂,而在其他三个剂量组中的每一组中,2名受试者接受安慰剂。以240 mg (空腹的受试者)和120 mg (进食的受试者)的单剂量向两个额外队列施用化合物1 (表3)。两个单剂量队列各自包括6名接受活性药物的受试者和两名接受安慰剂的受试者。在给药前的选定日(在局部低谷)以及在第1天、第7天和第14天以0.5小时至2小时的间隔收集用于PK和PD评估的血浆和血清样品以用于更详细的评估。
表3. 具有额外的单剂量队列的化合物1 MAD研究中的剂量组
a - 两名第1组受试者在第6天之前因非治疗突发不良事件的原因停药;从第1天至第3天可获得PK和PD结果。
240 mg单剂量的药代动力学结果与来自SAD研究的数据一致,并证明在40 mg至240 mg的单剂量范围内,暴露(C
表4. 240 mg (空腹)和120 mg (进食)的单剂量施用后化合物1的药代动力学参数的总结
来自化合物1的MAD临床研究中额外单剂量队列的药效学结果表明对补体AP活性的持续剂量依赖性抑制。在从第1天(给药前)至第7天的预定时间点收集的样品中评估血清AP溶血活性。240mg剂量组中的
在40 mg、80 mg、120 mg和200 mg多剂量队列中进行初始剂量后,平均暴露(C
图16是四个剂量组的平均浓度图,并且表5总结了所有组的平均多剂量药代动力学参数。
稳定状态下给药前血浆浓度有一致的趋势,早晨比夜晚高,一些早晨的谷约为夜晚的谷的两倍。40 mg、80 mg、120 mg和200 mg BID组的平均给药前浓度在为15.6 ng/mL-23.5 ng/mL、45.9 ng/mL-83.7 ng/mL、80.6 ng/mL-153 ng/mL和247 ng/mL-392 ng/mL的相应范围内(表6)。
表5. 重复剂量施用14天后化合物1的药代动力学参数的总结
表6. 重复剂量施用后化合物1的平均给药前稳态(谷值)血浆浓度(ng/mL)
旁路途径溶血的平均减少如图13所示。在研究持续期间,120 mg和200 mg BID患者组的AP溶血均减少了90%以上。平均而言,这种减少超过95% (参见图13)。
在从第1天(给药前)至第21天的预定时间点,在从所有队列收集的样品中评估血清AP溶血活性。在第1天(0小时至12小时)、第7天(0小时至12小时)和第14天(0小时至16小时)进行密集采样;在指定日期的0小时(早晨的PK谷)时收集所有其他血清样品。显示了每个队列在第1-21天(图17)、第1天(图18)、第7天(图19)和第14天(图20)的完整的时间过程中的活性。还在预定的时间点测量了通过AP Wieslab测定法
多剂量ACH-5228施用后,血清AP活性显示出快速且剂量依赖性的抑制。每个队列中活性剂受试者间的平均给药前(第1天,0小时)AP活性的范围为69.43% (40 mg)至73.29%(80 mg)。在第7天和第14天(稳态PK条件),给药后的活性降低至4.00% (40 mg)和0.00%(第2组至第4组)的最小值。然而,在40 mg BID和80 mg BID时,这种AP抑制伴随着在PK谷值(0小时和12小时时间点)时的显著恢复。相反,在120 mg BID时,平均AP活性在第7天在从0小时至10小时以及在第14天在从0小时至12小时的所有时间点时均保持低于10%。在200 mgBID时,平均AP活性在第7天和第14天在从0小时至12小时的所有时间点时均保持低于10%。到第17天,即最后一次给药后三天,所有剂量的AP活性都恢复到大约给药前水平。相比之下,安慰剂受试者中的AP活性与为84.97%的给药前值相比几乎没有变化,其中所有时间点间的最小值为76.84%。
将如通过利用患者样品进行的
表7显示了在第7天和第14天的0小时和12小时时间点时的AP溶血活性的经计算的抑制百分比,以及从第6天至第14天的所有0小时和12小时时间点(代表在稳态PK的条件下的PK谷值)共同的AP溶血活性的经计算的抑制百分比。在40 mg BID和80 mg BID时,在0小时和12小时时间点观察到部分抑制。在120 mg BID时,所有0小时时间点时的抑制超过90%(97.2%的抑制),并且所有12小时时间点也几乎如此(87.8%的抑制)。在200 mg BID时,所有0小时和12小时时间点时的抑制超过90% (分别为97.6%和95.1%)。这些结果与AP Wieslab测定得出的结果一致,其中在120 mg BID和200 mg BID时,所有0小时和12小时时间点时的经计算的抑制百分比超过90%。
表7. 0小时和12小时时间点(PK谷值):第7天、第14天和第6天至第14天时的多剂量队列中对血清AP溶血活性的抑制
将每个时间点时的多剂量个体的血清AP活性以同一个体的给药前活性(第1天,0小时,定义为100%)作归一化。将每个时间点时的对这种给药前归化一化的AP活性的抑制计算为相对于给药前活性的降低(即,100% -给药前归一化的活性)。这些AP抑制值用于计算以下两个PD参数,以定量第7天和第14天的AP抑制的积分幅度和持续时间。
首先,将从0小时至12小时的AP抑制的积分幅度计算为从0小时至12小时的效应曲线下面积(AUEC) (AUEC0-12)。通过线性梯形法计算 AUEC0-12,其中将从0小时至12小时的相邻时间点之间的线性函数下的面积相加。另外,将%AUEC0-12计算为以其间隔的最大可能值作归一化的AUEC0-12;100%的最大%AUEC0-12表明从0小时至12小时连续完全(100%)抑制。在40 mg BID和80 mg BID时,低于90%的经计算的%AUEC0-12值反映了在剂量之间观察到的AP溶血活性的显著恢复。在120 mg BID和200 mg BID时,平均%AUEC0-12值(第7天和第14天,组合分析)分别为98.2%和99.1%,表明在整个12小的时期间几乎完全抑制AP (表8)。这些值与从AP Wieslab测定得出的结果一致,其中在120 mg BID和200 mg BID时,%AUEC0-12值(第7天和第14天,组合分析)分别为98.6%和99.3%。
表8. 多剂量队列中从0小时至12小时的AP溶血抑制的积分幅度(AUEC0-12):第7天和第14天
其次,AP抑制的持续时间计算为0小时与12小时之间流逝的时间(持续时间
表9. 多剂量队列中从0小时至12小时的AP溶血抑制的持续时间(持续时间
使用两种模型(图21A和图21B)在所有时间点和多剂量队列中的个体之间评价时间匹配的血浆化合物1的浓度(PK)与血清AP溶血活性(PD)的抑制之间的PK-PD关系。使用简单的E
利用两种模型的分析证明了给药后AP活性的浓度依赖性抑制。使用E
在1期SAD研究中单剂量化合物1施用后,使用AP溶血测定确认了对血清AP活性的快速且几乎完全的抑制。在240 mg空腹的受试者中在给药后1.5小时至16小时以及在120mg进食的受试者中在给药后2小时至6小时,平均给药前活性降低至10%或更低。
来自1期MAD研究中的四个多剂量队列的结果,化合物1在化合物1施用后表现出对AP溶血活性的快速和剂量依赖性抑制。40 mg BID (第1组)和80 mg BID (第2组)时的化合物1实现了部分至几乎完全的AP抑制,但在每个12小时的给药期结束前显著恢复。120 mgBID (第3组)和200 mg BID (第4组)时的化合物1实现了在12小时的给药期的大部分时间内维持的基本上完全的AP抑制,其中AP活性在第7天和第14天(代表稳态PK条件)在12小时的给药期中的10至12小时内维持低于10%。为本研究开发的两个计算的PD参数(持续时间
评价了多剂量队列间的血浆化合物1的浓度(PK)与血清AP溶血抑制(PD)之间的PK-PD关系。使用简单的E
已参考本发明的实施方案描述了本说明书。然而,本领域的一般技术人员应理解,在不脱离随附权利要求书中所阐明的本发明的范围的情况下,可以作出各种修改和改变。因此,本说明书应被视为说明性的而非限制性的,并且所有此类修改都旨在包括在本发明的范围内。
- 用于治疗补体介导的病症的靶向给药
- 补体介导的病症的治疗