一种突变的人端粒酶RNA基因及其在预防和治疗泛癌种中的应用
文献发布时间:2023-06-19 18:29:06
相关申请的交叉参考
本申请引用申请人于同一申请日提交的、发明名称为“一种泛癌种超早期筛查方法”,其公开内容全部并入本文作为参考。
技术领域
本发明涉及一种突变的人端粒酶RNA基因(hTERC),包含所述突变的人端粒酶基因的重组构建体和/或表达载体,以及相应的突变的人端粒酶RNA,及其用于预防或治疗泛癌种的应用。
发明背景
大多数癌症是一种衰老性疾病,是人体组织中少数衰老细胞发生遗传变异,逃避了程序性凋亡,异常重新分裂,形成的异常增生性疾病。这种异常增生或细胞癌变可能也是人体细胞对衰老和凋亡的一种逃避方式,研究发现衰老细胞还更具有这种能力(Nishiguchi,M.A.et al.2018,Cell Reports,24,3383-3392),这可归因于在基因组进化上的一种遗传与适应。研究证明随着人年龄增加,机体逐渐衰老,表现在基因组DNA末端的端粒长度也在不断地缩短,表现出“生命时钟”的作用(Greider,C.W.,1996,Annu RevBiochem 65,337-365;Muezzinler,A.等,2013,Ageing Res Rev 12:509-519)。
深入研究发现,在衰老机体内仅有一部分端粒变短,另一部分端粒长度可保持相对恒定,且这部分逐渐变短的端粒与细胞衰老密切相关(Muezzinler,A.等,2013,AgeingRes Rev 12:509-519)。体内一些非衰老的正常细胞,如生殖细胞、干细胞等胚性细胞能通过端粒酶的催化作用将端粒延伸,维系端粒的正常长度和细胞可持续分裂,但是一般体细胞几乎没有端粒酶活性。
大量研究数据表明,在90%以上的癌变细胞中,端粒酶活性都异常增高,但其端粒却显著短于拥有端粒酶活性的正常胚性细胞,甚至比无端粒酶活性的正常体细胞更短(Blasco,M.A.,2005,Nat.Rev.Genet.6:611–622;Barthel,F.P.等,2017,Nat Genet 49:349-357),这或许是由于癌细胞恶性分裂导致的端粒长度快速缩减所致。当癌细胞端粒缩短到某一个关键节点之后又不再继续缩短,保持着相对恒定,推测这可能是因端粒酶催化端粒延伸的效率与恶性分裂伴随的缩短速率达到了一个平衡点。然而这一推断也不尽合理,因为端粒酶催化延伸端粒的效率数倍于细胞分裂缩短的速率。据报道,端粒可通过端粒结合蛋白Pot1和Tin2调节端粒酶活性(Lim,C.J.et al.2017,Nat Commun 8:1075);在端粒损伤和细胞衰老信号传递过程中,TRF2和P53之间进行正反馈信号调控(Fujita,K.etal.2010,Nat Cell Biol,12,1205-1212);当蛋白基因Pot1与P53伴随突变时细胞就会发生癌变(Pinzaru,A.M.et al 2016,Cell Rep,15,2170-2184)。又一研究也揭示了端粒与端粒酶共同作用,可通过加强和稳定P53的活性来调控细胞的分裂(Akbay,E.A.,et al.,2013,Oncogene 32:2211-2219);端粒酶除了催化延伸端粒以外,端粒酶还拥有一种前所未见的非典型功能,即促进含有hsp70-1和apollo的端粒保护复合体的形成,对端粒酶阳性的癌细胞持续增殖至关重要(Perera,O.N.et al 2019,Sci.Adv.5:eaav4409)。这些科学论据清晰阐明了细胞衰老、癌变与端粒、端粒酶之间存在极其密切的内在联系,2009年将诺贝尔医学或生理学奖授予了三位端粒和端粒酶的发现者。
基于癌变细胞端粒极度缩短以及端粒酶异常升高的发现,发明人推测癌细胞内端粒和端粒酶二者之间可能存在着一种关键的矛盾的统一体。一旦确定了这种矛盾统一体,并寻找到打破这种矛盾统一体的崭新方法和技术,将研发出前所未有颠覆性的创新抗癌药物,使得对大多数癌症的预防、治疗、甚至治愈成为可能。
在本发明中,发明人出乎意料地发现,癌细胞内端粒3’端末尾重复序列发生了显著变异,导致了其恶性分裂,通过采用相应的突变的人端粒酶RNA基因(hTERC)来“修复”这种在端粒3’端末尾发生了显著缺陷的端粒,可以有效地减弱癌细胞的生长,延缓癌细胞发展进程,甚至使异常分裂的癌细胞进入细胞程序性凋亡。
本发明由此提供了可用于有效地抑制癌细胞的分裂、治疗以及治愈和/或预防癌症疾病、延缓癌细胞发展进程的组合物和方法。
发明内容
在一个方面,本发明涉及一种突变的人端粒酶RNA基因,其在野生型人端粒酶RNA基因(SEQ ID NO.1)的模板域CR1(5'
在另一个方面,本发明涉及一种核酸重组构建体,其中包含有所述突变的人端粒酶RNA基因,以及与之可操作连接的真核生物基因表达元件,包括启动子、增强子、终止子和/或绝缘子。所述启动子可以是组成型启动子,或者是具有组织或细胞特异性的启动子,或者是诱导型启动子,包括人TK基因启动子、人HPRT基因启动子、人TERT基因启动子(hTERT启动子),或者可以是癌细胞特异性启动子。优选地,所述启动子是人TERT基因启动子。
在又一个方面,本发明涉及一种表达载体,其在载体骨架中包含所述突变的人端粒酶RNA基因,以及与之可操作连接的启动子。启动子的选择如上文针对重组构建体所述。所述载体可以是病毒表达载体,例如逆转录病毒载体(Retrovirus)、腺病毒载体(Adenvirus)、腺相关病毒载体(AAV)、疱疹病毒载体(Herpesvirus)等,优选地复制缺陷型病毒载体,例如人类低血清阳性率的新型黑猩猩复制缺陷型腺病毒载体(ChimpanzeeAdenovirus)或Ad5复制缺陷型腺病毒载体。
在又一个方面,本发明涉及一种突变的人端粒酶RNA,其具有与所述突变的人端粒酶RNA基因转录后相同的核糖核酸序列。
在进一步的方面,本发明涉及一种组合物,其中包含有所述突变的人端粒酶RNA基因、核酸构建体、表达载体和/或突变的人端粒酶RNA,以及任选地运载体。
本发明另外涉及一种药物组合物,其中包含所述突变的人端粒酶RNA基因、核酸构建体和/或表达载体,所述突变的人端粒酶RNA,以及药学上可接受的运载体和/或赋型剂。
本发明另外涉及一种在有此需要的受试者中预防和/或治疗肿瘤,和/或延缓肿瘤进展的方法,其中包括对所述受试者施用有效量的本发明所述突变的人端粒酶RNA基因、核酸构建体、表达载体、突变的人端粒酶RNA和/或药物组合物。
本发明还涉及所述突变的人端粒酶RNA基因、核酸构建体、表达载体、突变的人端粒酶RNA和/或组合物在制备用于预防和/或治疗肿瘤,和/或延缓肿瘤进展的药物中的用途。
不局限于具体理论,推测本发明的突变的人端粒酶RNA基因、核酸构建体或表达载体的重组体在被引入具有异常端粒酶活性的癌细胞和正常端粒酶活性的细胞中后将转录出突变的hTERC组分,或者体外合成的本发明突变的人端粒酶RNA在被递送入细胞内后,能够与细胞内的hTERT蛋白结合而形成具有催化活性的端粒酶,从而对相应的人染色体端粒3’末尾两个碱基为TC和/或TG的遗传缺陷端粒进行修复,使之变成TT结尾,进而在内源端粒酶作用下将端粒有效延伸,经Pot1和Tin2信号通路而降低端粒酶的异常表达,引导某(些)端粒已缩短至遗传缺陷区域的衰老细胞、潜在癌变细胞或已癌变细胞启动PCD凋亡,降低或消除癌变细胞的逃逸。对于体内正常分裂增殖的胚性细胞和组织,因其中不存在遗传缺陷的端粒,因此上述过程对它们没有影响。
本发明的突变hTERC基因、核酸构建体、表达载体和/或突变的人端粒酶RNA能够有效用于预防和治疗泛癌种,具有多种独特优势。它们通过对癌细胞遗传缺陷端粒进行修复,引导癌细胞启动程序性凋亡,实现癌细胞“自灭”的凋亡效果。本发明的方法彻底摒弃了“杀死”癌细胞的药物研发理念,极大地减少了癌细胞在现有药物作用下的遗传变异性逃逸,并且对胚性细胞无毒,对组织和器官无副作用,有效打破了现有抗癌药物的耐药性、易复发、毒性强、副作用大等瓶颈问题。本发明的突变hTERC基因、核酸构建体和/或表达载体的重组体,或体外合成的突变的端粒酶RNA同时具有预防和治疗的双重功效,适用于端粒酶异常增高的肿瘤患者,可覆盖约90%的癌症种类。利用本发明的人端粒酶RNA基因的突变体、核酸构建体、表达载体的重组体或体外合成的人端粒酶RNA,以及组合物能够开发出具有预防衰老、阻止癌变,治疗癌症及相关增生性疾病的无毒、无副作用的开创性靶向基因药物。
附图说明
图1A-图1F分别是乳腺癌、直肠癌、结肠癌、肝癌、肺癌和胃癌六种不同癌症患者的癌组织、癌旁组织和正常组织的端粒3’端末尾重复序列的测试结果,显示癌组织的端粒3’端末尾重复序列5’AGGGTT3’所占比例均比它的癌旁组织、正常组织显著下降。
图2显示HELA细胞端粒酶催化延伸端粒的特性。(上图展示端粒酶对六种3’末尾两个碱基差异的前导寡核苷酸被催化延伸之后的3’末尾重复序列的变化;下表展示催化延伸的端粒重复序列偏好,AGGGTT达到176.8,其次是TAGGGT为110.2,和TTAGGG为89.7。同时,也显示了端粒酶对3’末尾两个碱基结合位点的偏好是TT>GT>GG。
图3显示了多种癌症患者的细胞以及癌细胞株内,TC遗传缺陷端粒的存在水平显著高于正常受试者(上图展示PCR扩增产物电泳结果,下图展示TC遗传缺陷端粒的统计结果)。
图4显示了hTERC真核表达载体构建的示意图。
图5显示了携带hTERT启动子的hTERC腺病毒穿梭载体构建示意图。
图6显示了hTERC野生型真核表达的重组体在HeLa细胞中的表达。
图7显示了突变的hTERC基因对癌细胞系--HeLa细胞株的生长作用。
图8显示了Hoechst 33342染色观察HeLa细胞的凋亡特征。
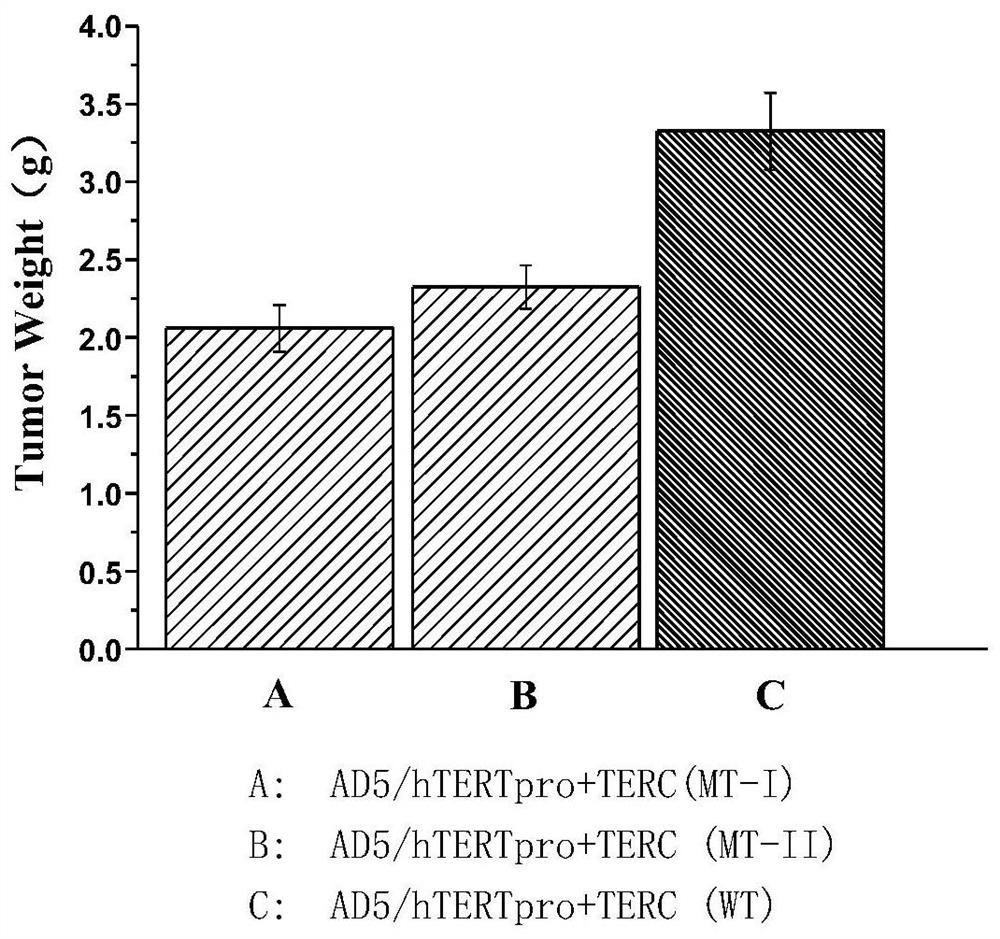
图9显示了突变的hTERC基因对人乳腺癌实体瘤生长的抑制作用。
图10显示了突变的hTERC基因对人乳腺癌细胞内端粒酶的抑制作用。
图11显示了切片观察hTERC基因单碱基突变对人乳腺癌实体瘤细胞的凋亡作用。
图12显示了突变的hTERC基因对人乳腺癌细胞的程序性凋亡作用。
发明详述
端粒结构和端粒酶催化
端粒是位于人染色体末端的DNA/蛋白复合体。其中,DNA部分是由(TTAGGG)
端粒3’末尾重复序列在端粒酶的催化过程中也起着至关重要的作用。端粒酶由人端粒酶蛋白亚基(hTERT)和端粒酶RNA亚基(hTERC)两部分组成,起着类似逆转录酶的功能。当端粒酶催化合成端粒时,端粒酶需要将hTERC自身携带的模板域CR1(5’CTAACCCTAAC3’,SEQ ID NO.2)与端粒的3’末尾重复序列配对结合,然后连续转位(Translocation)、合成、延伸端粒。端粒3’末尾重复序列不仅是端粒酶的底物(结合位点),也是端粒酶的产物(端粒重复序列)。发明人认为,这不仅说明了端粒酶是一个特殊的逆转录酶,催化反应的底物和产物完全一致,并且CR1与端粒的结合位点还决定着端粒合成的转位效率,从而影响延伸效率(Xiaodong Qi et al.,2012,EMBO Journal,31:150–161)。进一步地,端粒3’末尾重复序列的不同还可能暗示了端粒的延伸速率和长度并非完全取决于端粒酶自身,而且还决定于端粒3’末尾重复序列。
发明人利用一种端粒3’末尾重复序列检测方法对17例不同年龄的正常人全血基因组中的15个端粒进行了检测,结果显示,它们的3’末尾重复序列存在显著偏好,以5’AGGGTT3’结尾占比在26±7.7%,且随年龄的增加而逐渐减少。核酸动力学分析也显示5’AGGGTT3’结尾的重复序列明显优于其它的5种结尾方式,也佐证了体内端粒3’末尾重复序列偏好的存在。虽然在教科书中通常将人端粒重复序列写为(TTAGGG)
发明人利用Trap方法和端粒3’末端重复序列检测方法相结合,将六种3’末尾2个碱基差异的寡核苷酸作为前导序列对HELA细胞端粒酶的体外催化合成端粒的效率和特性进行了深入研究,出乎意料地发现端粒酶与端粒3’末尾的2个碱基配对形成有效结合位点,每次转位合成6个碱基,且不同的结合位点直接决定了端粒酶的转位效率和合成效率,从而影响端粒的延伸,其作用效率从高到低的结合位点依次是TT>GT>GG,催化合成的AGGGTT,TAGGGT和TTAGGG重复序列分别达到177、110和89.7个相对单位显著高于其它重复序列,详细结果显示于图2中。同时,HELA细胞端粒酶体外延伸的端粒也偏好5’AGGGTT3’结尾,占比达到35.29%,这说明癌细胞端粒酶催化的偏好特性并没有改变。根据10000例癌症患者大数据研究的报道,癌变细胞端粒酶的结构基因编码没有差异或突变,癌细胞端粒酶基因变异主要发生在hTERT结构基因的上游启动子区域,启动子区域呈现无规律变异(Zehir,A.,et al.,2017,Nat.Med.23,703–713)。这些都佐证了癌变细胞端粒3’末尾重复序列5’AGGGTT3’的占比下降与端粒酶自身催化特性无关。显然,导致癌变细胞端粒3’末尾重复序列5’AGGGTT3’的占比下降的原因很可能是端粒3’末尾重复序列本身的问题,特别是末尾2个碱基。因为3’末尾2个碱基不仅关系到Loop形成和稳定,也影响端粒酶的正常催化作用,还与细胞癌变密切关联,它无疑是至关重要的。
根据NCBI(https://www.ncbi.nlm.nih.gov/)提供的人类全基因组数据库信息,发明人对已定位的27个端粒序列进行了详细分析,包括已完成全染色体结构的X染色体和第8条染色体(Miga,K.H.,et al.,2020,Nature,585:79-84;Logsdon,G.A.,et al.,2021,Nature,DOI10.1038/s41586-021-03420-7;)。发明人惊奇地发现,其中有18个端粒在距染色体大约100~1000bp的区域内存在着(5’AGGGTC3’)n、(5’GGGTTC3’)n,(5’AGGGTG3’)n和/或(5’GGGTTG3’)n等n次异常重复序列,它们伴随人类基因组的遗传。各染色体端粒上的遗传缺陷重复序列情况总结于下表1中:
表1
发明人推测,由于细胞分裂的DNA半保留复制缺陷,导致细胞每分裂一次,端粒DNA重复序列将缩短约60-180bp。在细胞经过数十次分裂之后,某(些)端粒就会缩短至该遗传缺陷区域,即含有(5’AGGGTC3’)n、(5’GGGTTC3’)n,(5’AGGGTG3’)n和/或(5’GGGTTG3’)n的端粒区域,就可能会出现端粒3’末尾2个碱基为TC或TG的结尾。在本发明中,发明人将这种结尾的端粒分别定义为TC或TG遗传缺陷端粒。这种TC或TG遗传缺陷端粒一旦出现或存在,无疑将导致端粒酶异常,将在组织或细胞内出现一系列严重问题。
为了进一步验证在癌细胞中TC和TG遗传缺陷端粒的存在,发明人对16例癌症患者(包含了肺癌、结直肠癌、宫颈癌、卵巢癌、牙龈癌和贲门癌)的外周血基因组DNA和4个癌细胞系(结肠癌、肺癌、肝癌和宫颈癌)的DNA进行了TC遗传缺陷端粒的检测,显示所有癌症患者和癌细胞系都有不同数量的TC遗传缺陷端粒,而对照(5非癌症患者和1个正常的肝细胞系)则几乎不存在。结果示于图3。这个结果与预期完全一致,推测它普遍发生和存在于多种癌细胞中。显然,这一重大发现就能清晰阐明细胞衰老、癌变与端粒、端粒酶之间存在极其密切的内在关系,以及癌变细胞总是端粒酶异常高表达,端粒却很短等关键问题。
不局限于具体理论,发明人推测,随着年龄的增加,一部分细胞(例如上皮细胞)端粒逐渐缩短,某(些)关键染色体端粒缩短进入TC和TG遗传缺陷区域,出现了TC或TG遗传缺陷端粒。由于这种缺陷端粒的存在,端粒hTERC基因的CR1模板不能与这种遗传缺陷端粒3’末尾的2个碱基正常配对,严重影响端粒酶正常催化作用和催化效率。一方面扰乱了端粒酶以5’AGGGTT3’为主要方式的转位控制,导致延伸的端粒3’结尾紊乱,干扰loop的形成。另一方面,由于端粒酶催化效率严重降低,合成端粒3’凸出单链长度受限,通过Pot1和Tin2蛋白传递一个“端粒酶缺乏”的错误信号,引发端粒酶基因重启,并超高表达(Loayza,et al.,2003,Nature 423,1013-1018)。这种端粒酶异常高表达的结果,既能达到有限延伸端粒长度,维系细胞持续分裂的最低长度要求,又能促进含有hsp70-1和apollo的端粒保护复合体的形成,驱使细胞持续分裂,呈现出端粒酶活性高,端粒却很短,又能稳定地进行快速分裂,从而形成了癌变细胞典型的“矛盾”统一特征,其矛盾统一的关键就在于端粒3’结尾TC和TG遗传缺陷端粒出现和存在。所以,TC和TG遗传缺陷端粒是大多数细胞癌变初始驱动因子以及细胞初始癌变的重要生物标志物,只要能修复或打破端粒3’结尾TC和TG遗传缺陷端粒的方法、技术和药物都能达到对大多数癌症的预防,治疗,甚至治愈的目的。
突变的人端粒酶RNA基因
本发明由此提供了一种突变的人端粒酶RNA基因,与相应的野生型人端粒酶RNA组分(hTERC)基因的序列相比,其在模板域CR1(SEQ ID NO.2)中具有碱基突变,从而能与癌变细胞的遗传缺陷端粒的3’末尾的两个碱基形成互补的序列,并将遗传缺陷端粒修复成正常的TT结尾。在一个实施方案中,所述突变的人端粒酶RNA基因中第-54位的碱基“A”被突变成“G”(SEQ ID NO.15)或被突变成“C”(SEQ ID NO.16)。优选地,所述多核苷酸与相应的野生型人端粒酶RNA基因相比具有单碱基突变,称为“单碱基突变体”。
在本发明的上下文中,野生型hTERC基因指的是人基因组中编码端粒酶RNA组分的基因。人端粒酶RNA组分(hTERC)的编码基因可以从相应人血液或其它组织的细胞基因组DNA中容易地克隆,例如通过PCR克隆法,或者基因组文库筛选法。现有技术中也已经公开了人端粒酶RNA组分基因的核苷酸序列,其以例如Gene ID:7012登记在NCBI数据库中,它位于人第三染色体NC_000003.12(169764610bp~169765060bp),hTERC基因全长为451bp,无内含子,核苷酸序列如SEQ ID NO.1所示。
因此,在一个实施方案中,本发明涉及突变的hTERC基因,其对应的野生型hTERC基因具有SEQ ID NO.1所示的核苷酸序列,并且在其模板域CR1(5'-
本领域技术人员知晓如何计算两种核酸之间的序列同一性。例如,可以比对两种核酸的核苷酸序列一起获得最大配对,通过将比对序列之间相同的位置数除以位置总数,再乘以100%,即可以获得两种核酸之间的序列同一性百分比。
在本发明中,序列比对、确定序列同一性百分比和对应序列位置可以根据本领域中已知的一些软件进行,例如BLAST程序、CLUSTALW(http://www.ebi.ac.uk/clustalw/)、MULTALIN(http://prodes.toulouse.inra.fr/multalin/cgi-bin/multalin.pl)或MUSCLE(Multiple Sequence Alignment),以这些网站指示的缺省参数来探查(例如比对)获得的序列。如本文使用的,当关于比对核苷酸序列的特定对使用时,术语“同一性”、“同源性”或“百分比同一性”指核苷酸序列同一性百分比,其通过计数在比对中的相同匹配数目,并且将这样的相同匹配数目除以比对序列的长度来获得。
在本发明中,所述突变的hTERC基因可以是通过核酸载体扩增,重组的而来。或者,本发明的突变的hTERC基因可以通过PCR体外扩增,定点单碱基突变得到。在又一个实施方案中,本发明的突变的hTERC基因可以通过化学合成得到。
核酸构建体
本发明还涉及一种核酸构建体,其中包含本发明的突变的hTERC基因。在一个实施方案中,所述突变的hTERC基因与启动子可操作地连接。在本发明中,“可操作地”是指核酸构建体中各组分的连接方式使得它们能够发挥各自的生物学功能,例如启动子启动所连接的核酸序列的转录。启动子可以是组成型的,或者可以是细胞、组织或器官特异性的。在另一个实施方案中,启动子可以是诱导型的,响应于适当的刺激而启动所连接的核酸序列的转录。优选地,所述核酸构建体中还包含增强子、终止子和/或绝缘子等其它表达调控元件。适用于本发明的各种表达调控元件是本领域技术人员熟知的。通过选择合适的表达调控元件,可以精确地调节本发明多核苷酸的表达,具体地是转录。适合于在真核细胞、例如哺乳动物细胞、特别是人细胞中控制本发明的突变的人端粒酶RNA基因的转录/表达。此类诱导型表达系统包括例如由以下各项进行的调节:蜕皮激素、雌激素、黄体酮、四环素、二聚作用的化学诱导物以及异丙基-β-D-硫代吡喃半乳糖苷(IPTG)。本领域技术人员将能够根据需要选择适宜的调控元件/启动子序列。
表达载体
本发明还涉及一种表达载体,在本发明中,表达载体可以是质粒载体或病毒载体。与真核细胞相容的表达载体、优选地与脊椎动物细胞相容的那些,可以用来产生用于表达如本文所述的突变的hTERC基因的重组构建体。真核细胞表达载体是本领域中熟知的并且从许多商业来源可获得。
在一个实施方案中,所述表达载体是病毒表达载体系统。优选地,所述病毒表达载体选自下组中:(a)腺病毒载体;(b)逆转录病毒载体,包括但不局限于慢病毒载体、莫洛尼鼠白血病病毒等;(c)腺伴随病毒载体;(d)单纯疱疹病毒载体;(e)SV40载体;(f)多瘤病毒载体;(g)乳头瘤病毒载体;(h)微小核糖核酸病毒载体;(i)痘病毒载体,如正痘病毒,例如痘苗病毒载体,或禽痘病毒,例如金丝雀痘病毒或鸡痘病毒;以及(j)辅助病毒依赖性腺病毒或无肠腺病毒。所述病毒表达载体还可以是复制缺陷型病毒。本领域技术人员知晓各种表达载体的结构、制备、引入和表达。还可以通过用包膜蛋白或来自其他病毒的其他表面抗原来对这些载体假型化而进行修饰,或者适当时通过取代不同的病毒衣壳蛋白而修饰病毒载体的偏嗜性。例如慢病毒载体可以是用来自水泡性口炎病毒(VSV)、狂犬病病毒、埃博拉病毒、Mokola等的表面蛋白进行假病毒化。可以通过将载体工程化以表达不同衣壳蛋白血清型,使得载体靶向不同的细胞。
在本发明中,可以通过多种方法将本发明的表达载体引入目标细胞中,包括例如感染法、转染法、显微注射、微粒轰击等。一旦被引入细胞内,本发明的表达载体可以整合或不整合到细胞的基因组中。通过合适的调节元件,例如启动子、增强子等,可以确保本发明的多核苷酸在靶细胞中的转录。
突变的人端粒酶RNA
本发明还涉及一种突变的人端粒酶RNA,其具有对应于上述本发明的突变的hTERC基因的核酸序列。更具体地说,本发明的突变的人端粒酶RNA与所述突变的人端粒酶RNA基因的转录产物基本相同的核糖核酸。本发明的突变的人端粒酶RNA可以是在体外转录或者直接化学合成而来。所以,本发明的突变的人端粒酶RNA可以是所述突变的hTERC基因的“转录”的RNA序列。
在一个实施方案中,所述突变的人端粒酶RNA具有SEQ ID NO.23或SEQ ID NO.24所示的核糖核苷酸序列。为了改善RNA的活性和/或提高其稳定性,优选地在所述RNA的5’端具有修饰,例如采用“加帽”修饰。在一个实施方案中,可以在5’端第一个鸟嘌呤(
在又一个实施方案中,本发明的突变的人端粒酶RNA可以缀合有合适的聚合物,如PEG、PVP等。优选地,本发明所述突变的人端粒酶RNA在5’端缀合了合适的聚合物。本领域技术人员清楚如何提高RNA稳定性的方法,这些内容也包括在本发明范围内。
药物组合物
本发明还包括一种药物组合物,其中包含治疗或预防有效量的本发明的突变的人端粒酶RNA基因、重组构建体、表达载体、突变的人端粒酶RNA和/或组合物,以及药学上可接受的运载体/赋型剂。
如本文所用的,“治疗有效量”是指在将本发明的重组体,体外合成的人端粒酶RNA组合物和/或药物组合物施用于受试者以治疗泛癌种时足以实现泛癌种的治疗(例如通过削弱、改善或维持该现有疾病或一种或多种疾病症状)的量。“治疗有效量”将取决于突变的人端粒酶RNA基因、重组构建体、表达载体、突变的人端粒酶RNA本身的性质、给药方式、被治疗疾病的类型及其严重度、以及受试者的病史、年龄、体重、家族史、遗传组成、患病阶段、先前或伴随治疗(如果有的话)的类型、以及待治疗的患者的其他个体特征而变化。
如本文所用的,“预防有效量”是指在将本发明的重组体、体外合成的人端粒酶RNA、组合物和/或药物组合物施用于有患癌风险的受试者时足以降低患癌的风险或改善患癌后的一种或多种症状的量。改善包括减缓患癌的进程或减少后发疾病的严重度。“预防有效量”将取决于该重组体、体外合成的人端粒酶RNA、组合物和/或药物组合物本身的性质、给药方式、被治疗疾病的类型及其严重度、以及受试者的病史、年龄、体重、家族史、遗传组成、患病阶段、先前或伴随治疗(如果有的话)的类型、以及待治疗的患者的其他个体特征而变化。
如本文中所使用的,术语“泛癌种”是指细胞端粒酶异常升高的癌症种类,这类癌症大约占人类癌症种类的90%以上,大多数来源于上皮组织中细胞的癌变,也包括肉瘤,母细胞瘤等。
“运载体”或“赋形剂”是药学上可接受的溶剂、悬浮剂或用于将一种或多种核酸递送至动物的任何其他药理学上惰性的媒介物。该赋形剂可以是液体或固体,并且当与核酸和特定药物组合物的其他组分组合时,参考意欲的给予方式,对赋形剂进行选择以提供希望的容积、稠度等。典型的运载体包括但不限于结合剂(例如,糯性玉米淀粉、聚乙烯吡咯烷酮或羟丙基甲基纤维素等);填充剂(例如,乳糖和其他糖、微晶纤维素、果胶、明胶、硫酸钙、乙基纤维素、聚丙烯酸酯或磷酸氢钙等);润滑剂(例如,硬脂酸镁、滑石、二氧化硅、胶态二氧化硅、硬脂酸、金属硬脂酸盐、氢化植物油、玉米淀粉、聚乙二醇、苯甲酸钠、乙酸钠等);崩解剂(例如,淀粉、淀粉乙醇酸钠等);以及润湿剂(例如,月桂基硫酸钠)。
适合于非肠胃外给予的、不与核酸发生有毒反应的、药学上可接受的有机或无机赋形剂也可以用来配制本发明的组合物。适当的药学上可接受载体包括但不限于:水、盐溶液、醇、聚乙二醇、明胶、乳糖、直链淀粉、硬脂酸镁、滑石、硅酸、粘性石蜡、羟甲基纤维素、聚乙烯吡咯烷酮等。
用于局部给予核酸的配制品可以包括在普通溶剂如醇中的无菌或非无菌的水溶液、非水溶液,或在液体或固体油基质中的核酸溶液。这些溶液还可以包括缓冲液、稀释液和其他合适的添加剂。可以使用适合于非肠胃外给予的、且不与核酸发生有毒反应的、药学上可接受的有机或无机赋形剂。
适当的药学上可接受赋形剂包括但不限于:水、盐溶液、醇、聚乙二醇、明胶、乳糖、直链淀粉、硬脂酸镁、滑石、硅酸、粘性石蜡、羟甲基纤维素、聚乙烯吡咯烷酮等。
合适的运载体/赋型剂是本领域普通技术人员可以常规确定的,并且记载在例如Remington’s Pharmaceuticals中。
本发明的药物组合物可以是水性或非水性溶液、混悬液、脂质体配制品、胶束配制品、乳剂、结晶组合物等的形式。
在一个实施方案中,本发明的药物组合物可以被配制在水性缓冲溶液中,例如乙酸盐、柠檬酸盐、醇溶谷蛋白、碳酸盐或磷酸盐,或其任何组合。优选地,所述缓冲溶液是磷酸盐缓冲盐水(PBS)。在一些实施方案中,该缓冲溶液进一步包含一种或多种用于控制该溶液的克分子渗透压浓度的试剂,以使得克分子渗透压浓度被保持在一个所希望的值,例如在人血浆的生理值。可以加入到该缓冲溶液以控制克分子渗透压浓度的溶质包括但不限于蛋白质、肽、氨基酸、非代谢聚合物、维生素、离子、糖、代谢物、有机酸、脂质或盐。在一些实施方案中,用于控制溶液的克分子渗透压浓度的所述试剂是盐,优选地是氯化钠和/或氯化钾。
在另一个实施方案中,本发明的药物组合物被配制成透皮贴剂、软膏、洗剂、乳膏、凝胶剂、滴剂、栓剂、喷雾剂、液体以及粉剂。常规的药物载体、水、粉末或油基、增稠剂等可以是。适合的局部配制品包括其中在本发明中的药物活性成分与局部用递送剂如脂质、脂质体、脂肪酸、脂肪酸酯、类固醇、螯合剂和表面活性剂混合的那些。适合的脂质和脂质体包括中性(例如二油酰磷脂酰DOPE乙醇胺、二肉豆蔻酰磷脂酰胆碱DMPC、二硬脂酰磷脂酰胆碱)、阴性(例如二肉豆蔻酰磷脂酰甘油DMPG)和阳离子脂质和脂质体(例如二油酰四甲基氨基丙基DOTAP和二油酰磷脂酰乙醇胺DOTMA)。本发明中表征的药物组合物可以封装于脂质体内部或可以与阳离子脂质体、尤其是与阳离子脂质体形成复合物。可替代地,本发明的核酸可以与脂质、具体地与阳离子脂质复合。合适的脂肪酸与酯包括但不限于花生四烯酸、油酸、花生酸、月桂酸、羊脂酸、羊蜡酸、肉豆蔻酸、棕榈酸、硬脂酸、亚油酸、亚麻酸、二癸酸酯、三癸酸酯、甘油单油酸酯、甘油二月桂酸酯、1-单癸酸甘油酯或其药学上可接受的盐。局部配制品被详细地描述于美国专利号6,747,014中,该美国专利通过引用方式结合在此。
本发明的药物组合物中还可以另外地含有本领域熟知用量的在药物组合物中常用的其他辅助组分。在本发明的药物组合物中可以包括另外的、可相容的药学上有活性的物质,例如收敛剂、局部麻醉剂或抗炎剂等,或者可以包括对本发明的药物组合物的各种剂型的配制有用的其他物质,如染料、芳香剂、防腐剂、抗氧化剂、遮光剂、增稠剂和/或稳定剂。然而,当加入此类物质时,它们不应当过度干扰本发明的组合物的成份的生物活性。可以将这些配制品进行灭菌并且如果希望的话与助剂如润滑剂、防腐剂、稳定剂、湿润剂、乳化剂、盐混合,用于影响渗透压的盐、缓冲液、着色物质、芳香物质和/或芬芳物质等进行混合,这些助剂不与该配制品中的一种或多种核酸发生有害的相互作用。对于水性悬浮液而言,可以包括增加该悬浮液的粘度的物质,例如羧甲基纤维素钠、山梨糖醇和/或葡聚糖等。混悬液或者还可以含有稳定剂。
当以化学合成或者体外转录生成的形式对细胞或者受试者施用本发明的突变的人端粒酶RNA时,优选地对所述RNA进行配制,以保持其活性和/或提高其稳定性。因此,在一个实施方案中,本发明提供了一种药物组合物,其中包含本发明的突变的人端粒酶RNA以及合适的递送载体。优选地,所述递送载体是鱼精蛋白载体、纳米颗粒脂质粒载体、多聚载体。所述突变的人端粒酶RNA与递送载体可以是直接混合的、缔合的或者是化学偶联的。例如,本发明的RNA可以与GalNAc(N-乙酰化半乳糖胺)偶联。这些递送载体同时能很好保护RNA不被降解。可替代地或者额外地,可以纳米颗粒形式施用本发明的突变的人端粒酶RNA。在一个实施方案中,在纳米颗粒中还包含胆固醇分子,以增加纳米颗粒的流动性和/或提高纳米颗粒的穿膜能力。在另一个实施方案中,所述药物组合物中包含PEG作为佐剂,躲避宿主免疫系统,使其更具水溶性,并减缓其被降解的速度。
本发明的药物组合物能够以足够治疗和/或预防泛癌种疾病类型的剂量施用于有此需要的受试者,例如人。施用剂量是医师根据本领域中的一般性知识通过实验能够常规确定的。例如,施用剂量是每天约1μg/kg体重至500mg/kg体重,例如10μg/kg、50μg/kg、100μg/kg、500μg/kg体重至1mg/kg、5mg/kg、10mg/kg、50mg/kg、100mg/kg或500mg/kg体重。本发明也涵盖这些范围中的任何中间范围。
本发明的药物组合物可以以合适的给药频率施用。合适的施用频率是本领域技术人员通过常规方法可以确定的。例如,可以每日给予一次,或者可以在一天内以适当的间隔给予两次、三次或更多次亚剂量,或者甚至可以通过控释配制品使用连续输注或递送而给予。或者,可以隔天、每三天、每周、每半个月、每个月、每两个月、每季度、每半年或每年施用一次本发明的组合物。本发明的药物组合物也可以制成常规的持续释放配制品。持续释放配制品是本领域中熟知的并且对于在特定位点递送试剂是特别有用的。
本领域技术人员将理解,某些因素可以影响有效治疗受试者所要求的剂量和时间安排,这些因素包括但不限于疾病或病症的严重性、先前的治疗、该受试者的总体健康和/或年龄以及其他存在的疾病。此外,用治疗有效剂量的组合物治疗受试者可以包括单次治疗或一系列治疗。如在此的其他地方所描述,使用常规方法或基于使用适当动物模型的体内测试,可以评估本发明涵盖的突变的人端粒酶RNA基因、核酸构建体、表达载体、突变的人端粒酶RNA、组合物和/或药物组合物的有效剂量和体内半衰期。
施用途径
本发明的突变的人端粒酶RNA基因、核酸构建体、表达载体、突变的人端粒酶RNA在通过化学合成、体外转录或重组方法制备之后,可以有效量通过合适的施用方式施用于有此需要的受试者体内或细胞内,特别是被靶向的器官、组织或细胞内。这样的施用技术包括例如静脉、皮下、腹膜内、经皮、透皮、口服等。或者,本发明的突变的人端粒酶RNA基因可以通过基因枪、脂转染法、脂质体法、基因注射等进行给药。或者,还可以以上述核酸构建体或表达载体方式递送到有此需要的受试者体内。
因此,本发明的药物组合物可以通过各种合适的施用途径施用于有此需要的受试者,这取决于待治疗的区域以及期望获得的效果。例如,可以局部施用本发明的药物组合物,例如通过透皮贴剂;可以通过吸入或吹入粉剂或气雾剂,包括通过喷雾器而施用于肺,或者可以通过气管内、鼻内、表皮、透皮、口服或肠胃外等途径施用。肠胃外给予包括静脉内、皮下、腹膜内或肌肉内注射或输注;皮下的,例如通过植入性器械的给予;或颅内的,例如通过实质内或鞘内等途径进行施用。
疾病治疗和预防方法
本发明还涉及一种治疗或预防泛癌种的方法,其中包括对有此需要的受试者施用治疗或预防有效量的本发明的药物组合物。有关“治疗有效量”和“预防有效量”的含义、对受试者的施用方式、施用频率、药物组合物的形式,参考上文针对药物组合物所述。
可以通过直接以合成的RNA形式、体外转录本发明的突变的人端粒酶RNA基因而获得的突变的人端粒酶RNA(SEQ ID NO.23或SEQ ID NO.24),通过脂质体或其它纳米方式进入细胞,或者突变的人hTERC基因I或突变的人hTERC基因II通过表达载体进入细胞后,转录的RNA与hTERT组分结合,形成携带hTERC突变的端粒酶,对目标细胞的遗传缺陷端粒进行修正。携带hTERC突变的端粒酶能明显抑制HELA细胞和人乳腺癌实体瘤的生长,显著降低癌组织异常端粒酶活性,有效地引导癌细胞进入PCD自然凋亡。由于正常胚性细胞是分生性细胞,非衰老细胞,端粒长度较长,不存在TC和TG遗传缺陷的端粒,无需修复,所以重组体不影响正常分裂的胚性细胞。而癌变细胞是衰老细胞,又疯狂分裂,端粒异常变短,存在不同数量的TC和TG遗传缺陷端粒,直接引入的或者在细胞内重组表达的突变的hTERC组分能够与这些TC或TG遗传缺陷端粒有效结合,将它们修复成正常的端粒5’AGGGTT3’,并借助于内源的hTERC将端粒继续延伸,引导它们进入程序性凋亡,从而达到预防和治疗癌症以及延缓衰老的目的。重组体针对的是端粒酶异常升高的癌变细胞,所以它适用于90%以上的癌种类型,具有治疗和预防泛癌种的特性。
试剂盒
本发明还提供了一种试剂盒,其中包含本发明的突变的人端粒酶RNA基因、核酸构建体、表达载体、突变的人端粒酶RNA、组合物中的一种或多种,和/或用于实施本发明的方法的使用说明书。试剂盒可以任选地包含使目标细胞与本发明的突变的人端粒酶RNA基因、核酸构建体、表达载体、突变的人端粒酶RNA和/或组合物接触的工具(例如,注射装置)。
制备方法
本发明的突变的人hTERC基因可以与前述的各种基因载体重组、包装制备。对于本发明的突变的人端粒酶RNA可以直接化学合成和/或通过体外转录获得。这些生产制备方法是本领域技术人员熟知的。
在一个实施方案中,本发明还提供了一种制备本发明的突变的人hTERC基因的方法,其中包括:在质粒或病毒载体内插入本发明的突变的人端粒酶RNA基因。优选地,在所述载体中,本发明的突变的人端粒酶RNA基因与指导其转录的启动子可操作地连接。例如,可以以AD5复制缺陷型腺病毒为基因载体,在其中插入hTERC基因突变体I(SEQ ID NO.15)或hTERC基因突变体II(SEQ ID NO.16),并使之与人端粒酶启动子hTERTpro可操作连接,由此制备重组体I和重组体II。由本发明的方法制备的该重组体在具有端粒酶活性的细胞中表达hTERC突变体基因,分别对TC和TG遗传缺陷端粒进行修复。在本发明中,所述基因载体不限于AD5复制缺陷型腺病毒,也可用前述的表达载体或类似的基因表达载体替代。
除非另外限定,否则本文中所用的全部技术与科学术语具有如本发明所属领域的普通技术人员通常理解的相同含义。本文描述了用于实施本发明的一些合适的方法和材料,但与本文所述的那些方法和材料相似或等同的方法和材料同样可以用于实施本发明。本文所提及的全部出版物、专利申请、专利和其他参考文献均通过引用方式整体并入本文中。在有冲突的情况下,以本发明的说明书(包括定义)为准。此外,这些材料、方法和实施例仅是说明性的,并非意图限定本发明的范围。
本发明将通过下述实施例进一步进行描述。本领域普通技术人员十分清楚,这些实施例是非限制性的,仅仅以举例的方式说明了本发明。本领域技术人员可以在不背离本发明的精神和范围的情况下对本发明作出各种修饰。这些修饰同样在本发明范围内。本发明的保护范围由所附的权利要求书进行限定。
实施例
下述实施例分别涉及以下方面:
1、癌症患者外周血和癌细胞系的基因组DNA中TC遗传缺陷端粒的检测;
2、创建hTERC基因单碱基突变体;
3、构建hTERC基因突变体的重组真核表达载体;
4、构建人端粒酶启动子转移载体;
5、构建hTERC基因突变体的AD5复制缺陷型腺病毒穿梭载体;
6、用AdMAX系统包装AD5复制缺陷型重组腺病毒;
7、用hTERC野生型真核表达的重组体,测试重组体的基因表达盒;
8、测试重组hTERC基因突变体对HeLa细胞株的生长作用;
9、测试重组hTERC基因突变体对人乳腺癌实体瘤的生长作用;
10、测试重组hTERC基因突变体对人乳腺癌端粒酶的抑制作用;
11、观察重组hTERC基因突变体对人乳腺癌细胞的结构变化;
12、检测重组hTERC基因突变体对人乳腺癌细胞的程序性凋亡作用;
实施例1癌症患者外周血基因组DNA和癌细胞系基因组DNA中TC遗传缺陷端粒的检测
获取16例癌症患者(包含了肺癌、结直肠癌、宫颈癌、卵巢癌、牙龈癌和贲门癌)的外周血和4个癌细胞系(结肠癌、肺癌、肝癌和宫颈癌)的样品,采用下述方法测定TC遗传缺陷端粒的存在情况。
1)基因组DNA样品制备:
用宝生物工程(大连)有限公司(TAKARA)提供的MiniBEST Whole Blood GenomicDNA Extraction Kit试剂盒,根据试剂盒说明书分别提取、制备人血液基因组DNA样品。最后,用1xTE,pH7.6缓冲液替代试剂盒提供的“Elution”洗脱液。
2)寡核苷酸合成及接头制作:
a)上海生物工程公司合成序列表中所列的寡核苷酸,并按要求进行化学修饰。
b)采用双蒸水(ddH
3)内切酶处理基因组DNA:
用1xTE pH7.6缓冲液将制备的基因组DNA稀释成20ng/ul,取25ul(500ng DNA)与25ul酶切混合液[5ul 10x EcoR V buffer,17ul ddH2O和3.0ul 15U/ul EcoR V(Takara)]混合后,37℃保温2小时,并标记为:M1。
4)酶切样品纯化:
每份酶切样品补入50ul ddH2O,再加入40ul磁珠混合液(Hieff NGSTM SmarterDNA Clean Beads试剂盒),并按试剂盒要求进行样品纯化。用28.0ul 0.1xTE,pH7.6洗脱,吸取26.5ulDNA纯化样品,并标记为:MC1。
5)纯化的酶切DNA片段与接头连接:
取12.0ul MC1 DNA片段,加入15ul 2x T7 DNA Ligase Buffer,1.5ul 5.0uMLK1,1.5ul T7 DNA Ligase(Biolab NEB)混合、离心,25℃保温2小时,并标记为:MCL1。
6)连接样品纯化:
每份MCL样品补入70ul ddH2O,再按第4步方法进行样品纯化,并用34ul 0.1xTEpH7.6洗脱,吸取32.5ul DNA纯化样品,并依次标记为:MCLC1。
7)PCR扩增纯化的样品:
取出8ul MCLC1,加入2ul ddH
8)PCR产物电泳分离、染色、成像:
将PCR产物样品和60ng(4ul)DNA标样(DL2000 DNA Marker,TAKARA),在1.5%琼脂糖凝胶上分离;然后,用0.1%溴化乙锭进行标准化染色,并用GeneGenius Bio-imagingSystem(SYNGENE,Synoptics Ltd,Cambridge,UK),记录成像。
9)定量分析,取得结果:
利用Genetool(SYNGENE,Synoptics Ltd,Cambridge,UK)图像定量分析工具。根据DNA标样说明书,对泳道M的DNA条带分子量2000bp、1000bp、750bp、500bp、250bp和100bp进行定义,其中750bp条带定义60ng dsDNA。然后,分别对PCR产物各目标条带的分子量和扩增产物量进行测定。结果总结于以下表2中:
表2 不同癌种5’AGGGTC3’结尾端粒检测结果
结果清晰地显示,无论是肺癌、结肠癌、直肠癌、卵巢癌、宫颈癌还是不常见牙龈癌,其受试者的全血基因组DNA中都能扩增出不同数量的清晰DNA目标条带,显著多于非癌症受试者,而且部分非癌症受试者没有检测到遗传缺陷端粒。同时,癌症受试者的DNA条带的含量均显著更高。在癌细胞系中也检测到不同数量的TC遗传缺陷端粒,而作为对照的正常肝细胞系中则几乎不存在。
实施例2创建hTERC基因单碱基突变体
基于NCBI数据库中hTERC基因全长为451bp,无内含子,直接从基因组DNA中获得人源hTERC序列,作为野生型基因(WT),其序列如SEQ ID NO.1所示。用重叠PCR方法将hTERC序列中第-54位脱氧腺嘌呤核苷酸(A)突变成脱氧鸟嘌呤核苷酸(G),定义为hTERC基因突变体I(TI)。将第-54位脱氧腺嘌呤核苷酸(A)突变成脱氧胞嘧啶核苷酸(C),定义为hTERC基因突变体II(TII)。并将它们克隆到T-载体中。具体实施如下:
A:人全基因组DNA制备。采用TaKaRa MiniBEST Whole Blood Genomic DNAExtraction Kit试剂盒,制备全基因组DNA,4℃保存,待用。
B:重叠延伸PCR方法,定点突变所用引物为:
1)TF:5’-TTCCATTTTTAAGGTAGTCGAGG-3’(SEQ ID NO.NO.7);
2)TR:5’-TAAAAGGCAACAAAAAGCGGAAG-3’(SEQ ID NO.8);
3)MF-I:5’-GTCTAACCCTGACTGAGAAGG-3’(SEQ ID NO.9);
4)MR-I:5’-CCTTCTCAGTCAGGGTTAGAC-3’(SEQ ID NO.10)。
第一轮PCR:反应体系20ul(2.0ul 10x PCR Buffer with Mg2+,1.6ul 10mMdNTP,2.0ul 2.0uM Primer TF,2.0ul 2.0uM Primer MR-I,5.0ul 10ng/ul Genomic DNA,0.2ul 5U/ul Pfu polymerase和7.2ul ddH2O)。反应程序:95℃/5min,(95℃/30sec,58℃/30sec,72℃/60sec)x30个循环,72℃/10min,8℃停止。用1%琼脂糖凝胶电泳PCR产物。然后,用Takara MiniBEST Agarose Gel DNA Extraction Kit回收376bp目标片段,并标记为PD1,稀释定量成10ng/ul。同时,用Primer TR替换Primer TF,PrimerMF-I替换Primer MR-I,其余完全相同,回收639bp目标片段,并标记为PD2,稀释定量成10ng/ul。第二轮PCR:反应体系20ul(2.0ul 10x Pfu Buffer with Mg2+,1.6ul 10mM dNTP,2.0ul 2.0uM PrimerTF,2.0ul 2.0uM Primer TR,0.5ul 10ng/ul PD1,0.5ul 10ng/ul PD2,0.2ul 5U/ul Pfupolymerase和11.2ul ddH2O)。反应程序:(95℃/30sec,62℃/30sec,72℃/60sec)x 30个循环,72℃/10min,8℃停止。用1%琼脂糖凝胶电泳PCR产物。然后,用Takara MiniBESTAgarose Gel DNA Extraction Kit回收967bp目标片段,并标记为TD-I。同时,用1.0ul50ng/ul Genomic DNA替换0.5ul 10ng/ul PD1和0.5ul 10ng/ul PD2,获得野生型目标基因片段,并标记为WD。用pMD18-T分别克隆TD-I和WD。将目标片段
TD-I或WD进行3’末端加A反应,反应体积10ul(2.0ul 10x PCR Buffer with Mg2+,1.0 1mM dATP,7.8ul TD-I或WD目标片段,0.2ul 5U/ul rTaq,),72℃保温15分钟。然后,根据Takara pMDTM18-T Vector Cloning Kit的指导书进行连接、克隆,用TakaraMiniBESTPlasmid Purification Kit提取DNA样品,并进行测序确认无误后,分别标记为:pMD18/+TD-I和pMD18/+WD。
C:构建pMD18/+hTERC(MT-I)转移载体。所用引物为:
1)EcoRTER:
5’-GCGTGAATTCTCCCTTTATAAGCCGAC-3’(SEQ ID NO.11);
2)BamHNoSP:
5’-CAGGATCCAAAAAAAAATGGGGGCTCACAAGCC-3’(SEQ ID NO.12)。
PCR反应体系20ul(10ul 2x GC Buffer,3.2ul 10mM dNTP,2.0ul 2.0uMEcoRTER,2.0ul 2.0uM BamHNoSP,1ul DNA from pMD18/TD-I,0.2ul 5U/ul Pfupolymerase,1.6ul ddH2O);PCR反应程序:95℃/2min,(95℃/30sec,60℃/30sec,72℃/60sec)x 30个循环,72℃/10min,8℃停止。用1%琼脂糖凝胶电泳PCR产物,用TakaraMiniBEST Agarose Gel DNA Extraction Kit回收扩增片段。再将回收的扩增片段和pMDTM18-T Vector DNA分别用BamHI,EcoRI进行双酶切。酶切反应体系25ul(2.5ul 10xBuffer T,10ul 60ng/ul DNA,0.6ul 5U/ul BamHI,1.2ul 5U/ul EcoRI,3.25ddH2O)。反应条件:37℃保温2小时。用TakaraMiniBEST DNA Fragment Purification Kit进行纯化。然后,根据Takara pMDTM18-T Vector Cloning Kit的指导书进行连接、克隆,再用TakaraMiniBEST Plasmid Purification Kit提取DNA样品,并进行测序确认,获得hTERC基因的单碱基突变I的转移载体,标记为:pMD18/+hTERC(MT-I)。
D:用引物MF-II:5’-GTCTAACCCTCACTGAGAAGG-3’(SEQ ID NO.13)替代MF-I:5’-GTCTAACCCTGACTGAGAAGG-3’(SEQ ID NO.9);MR-II 5’-CCTTCTCAGTGAGGGTTAGAC-3’(SEQID NO.14)替代引物MR-I,5’-CCTTCTCAGTCAGGGTTAGAC-3’(SEQ ID NO.10),其余重复步骤B和步骤C,获得hTERC基因的单碱基突变II的转移载体,并标记为:pMD18/+hTERC(MT-II)。
E:用步骤B中构建的pMD18/+WD替代pMD18/+TD-I,其余重复步骤C,获得hTERC基因的野生型的转移载体,并标记为:pMD18/+hTERC(WT)。
实施例3构建hTERC基因突变体的重组真核表达载体
利用真核表达载体pcDNA3.1(-)和实施例2中获得的MT-I、MT-II和WT转移载体,通过BamHI和EcoRI双酶切,将hTERC目标基因插入pcDNA3.1(-)真核表达载体,具体实施如下:
用Takara MiniBEST Plasmid Purification Kit制备pcDNA3.1(-)(Invitrogen,Life Technology)和pMD18/+hTERC(MT-I)的质粒DNA。双酶切pMD18/+hTERC(MT-I)DNA反应体系:(5ul 10x Buffer K,12ul 180ng/ul pMD18/+hTERC(MT-I)DNA,2.5ul 5U/ul BamHI,2.5ul 5U/ul EcoRI,28ul ddH2O);反应液37℃保温2小时后,用1%琼脂糖凝胶电泳酶切产物。然后,用Takara MiniBEST Agarose Gel DNA Extraction Kit回收hTERC(MT-I)目标片段(577bp)。同时,双酶切pcDNA3.1(-)DNA反应体系:(5ul 10x Buffer K,40ul 50ng/ulpcDNA3.1(-)DNA,2.5ul 5U/ul BamHI,2.5ul 5U/ul EcoRI);反应液37℃保温2小时后,直接用Takara MiniBEST DNA Fragment Purification Kit纯化酶切样品,获得线性pcDNA3.1(-)DNA。然后,将hTERC(MT-I)目标片段与线性pcDNA3.1(-)DNA样品,用T4连接酶连接。连接体系10ul(1ul 10x T4 Ligase buffer,5ul 30ng/ul回收的线性pcDNA3.1(-)DNA片段,1ul 40ng/ul回收的hTERC(MT-I)目标片段,1ul 5U/ul T4 DNA Ligase),16℃保温12小时后,按常规方法进行连接片段的转化、筛选、酶切鉴定以及测序鉴定,分别获得携带目标基因的真核表达载体:pcDNA3.1(-)/+hTERC(MT-I)。
用pMD18/+hTERC(MT-II)或pMD18/+hTERC(WT)分别替代pMD18/+hTERC(MT-I),其余完全相同,重复上述方法。分别获得携带目标基因的真核表达载体:pcDNA3.1(-)/+hTERC(MT-II)和pcDNA3.1(-)/+hTERC(WT)。(图4)
实施例4构建人端粒酶启动子转移载体
克隆hTERTpro所用引物:1)hTERTpro-F:5’-ACATACGTACACGCACCTGTTCCCAG-3’(SEQ ID NO.17);2)hTERTpro–R:5’-TCCTGAATTCGCGGGGGTGGCCGGGGCCA-3’(SEQ IDNO.18);PCR反应体系20ul(10ul 2x GC Buffer,3.2ul 10mM dNTP,2.0ul+2.0uM+hTERTpro-F,2.0ul+2.0uM+hTERTpro-R,0.5ul+50ng/ul+genomic+DNA,0.2ul+5U/ul+rTaq+Polymerase,5.7ul+ddH2O);反应条件:95℃/2min,(95℃/30sec,61℃/30sec,72℃/60sec)x30个循环,72℃/10min,8℃停止。用1%琼脂糖凝胶电泳PCR产物,用Takara+MiniBEST+Agarose+Gel+DNA+Extraction+Kit回收505bp扩增片段。再根据Takara+pMDTM18-T+VectorαCloning Kit的指导书,进行连接、克隆,并进行测序确认无误后,将人端粒酶启动子(hTERTpro)转移载体标记为:pMD18/+hTERTpro。
实施例5构建hTERC基因突变体的AD5复制缺陷型腺病毒穿梭载体
A:用Takara MiniBEST Plasmid Purification Kit制备删去HCMV腺病毒载体pCA13/-HCMV和质粒pMD18/+hTERTpro的DNA样品,用HindIII和EcoRI双酶切方法将hTERTpro片段转入pCA13/-HCMV质粒。双酶切反应体系1:(2.5ul 10x Buffer R,8ulpMD18/+hTERTpro DNA,0.8ul 5U/ul HindIII,1.25ul 5U/ul EcoRI,12.45ul ddH2O);反应液37℃保温2小时后,用1%琼脂糖凝胶电泳酶切产物。然后,用Takara MiniBESTAgarose Gel DNA Extraction Kit回收519bp目标片段。同时,双酶切反应体系2:(2.5ul10x Buffer R,8ul pCA13/-HCMV DNA,0.8ul 5U/ul HindIII,1.25ul 5U/ul EcoRI,12.45ul ddH2O);反应液37℃保温2小时后,直接用Takara MiniBEST DNA FragmentPurification Kit纯化酶切样品。最后,将519bp片段与双酶切、纯化的pCA13/-HCMV DNA样品,用T4连接酶连接。连接体系10ul(1ul 10x T4 Ligase buffer,1.5ul回收的载体pCA13/-HCMV片段,6.5ul回收的hTERTpro目标片段,1ul5U/ul T4DNA连接酶),16℃保温2小时后,按常规方法进行连接片段的转化、筛选、酶切以及测序鉴定,获得目标重组质粒:pCA13/+hTERTpro。
B:用Takara MiniBEST Plasmid Purification Kit制备pCA13/+hTERTpro和pMD18/+hTERC(MT-I)质粒的DNA,用BamHI和EcoRI双酶切方法将hTERC(MT-I)片段转入pCA13/+hTERTpro载体。双酶切pMD18/+hTERC(MT-I)DNA反应体系:(5ul 10x Buffer K,12ul 180ng/ul pMD18/+hTERC(MT-I)DNA,2.5ul 5U/ul BamHI,2.5ul 5U/ul EcoRI,28ulddH2O);反应液37℃保温2小时后,用1%琼脂糖凝胶电泳酶切产物。然后,用TakaraMiniBEST Agarose Gel DNA Extraction Kit回收hTERC(MT-I)目标片段(577bp)。同时,双酶切pCA13/+hTERTpro DNA反应体系:(5ul 10x Buffer K,40ul 50ng/ul pCA13/+hTERTpro DNA,2.5ul 5U/ul BamHI,2.5ul 5U/ul EcoRI);反应液37℃保温2小时后,直接用Takara MiniBEST DNA Fragment Purification Kit纯化酶切样品,获得线性pCA13/+hTERTpro DNA。最后,将hTERC(MT-I)目标片段与酶切、线性pCA13/+hTERTpro DNA样品,用T4连接酶连接。连接体系10ul(1ul 10x T4 Ligase buffer,5ul 30ng/ul回收的线性pCA13/+hTERTpro DNA片段,1ul 40ng/ul回收的hTERC(MT-I)目标片段,1ul 5U/ul T4 DNA连接酶),16℃保温2小时后,按常规方法进行连接片段的转化、筛选、酶切以及测序鉴定,获得目标基因穿梭载体:pCA13/+hTERTpro+hTERC(MT-I)。
用pMD18/+hTERC(MT-II)或pMD18/+hTERC(WT)替代pMD18/+hTERC(MT-I),其余完全相同,重复上述方法。分别获得目标基因穿梭载体:pCA13/+hTERTpro+hTERC(MT-II)和pCA13/+hTERTpro+hTERC(WT)。(图5)
实施例6用AdMAX系统包装AD5复制缺陷型重组腺病毒
A:通过脂质体介导将目标基因转移载体pCA13/+hTERTpro+hTERC(MT-I)与人类腺病毒血清5型(E1/E3缺失)骨架载体pBHG11共转染至HEK293细胞,获得目标重组腺病毒。具体方法:在转染前一天,在60mm平皿中以7.5×105个人胚肾293细胞铺板,用5%DMEM,置于37℃、5%CO2细胞培养箱内培养。转染前3-4小时(细胞回合率最好小于70%),更换成新鲜培养液,加入5ml无血清培养基,确保细胞快速生长。然后,将目标基因转移载体pCA13/+hTERTpro+hTERC(MT-I),病毒骨架载体pBHG11和脂质体Lipofectamine 2000(购自Invitrogen公司)加入293细胞(具体操作省略),共培养7-14天后出现病毒空斑,经过3次病毒空斑纯化后收集细胞,于-80℃和37℃之间反复冻融3次,然后4℃、5000rpm离心10分钟,去除细胞碎片,将包装好的重组腺病毒颗粒分装后-80℃,保存备用。
B:重组腺病毒的鉴定
应用QIAGEN DNA Blood Mini Kit,根据产品说明书,提取重组腺病毒DNA。
再通过PCR扩增目标基因表达盒(hTERTpro+hTERC,1002bp),上游引物Target-F:5’-ATTACATACGTACACGCACCT-3’(SEQ ID NO.19);下游引物Target-R:5’-AACGGGAAAGCGAACTGCAT-3’(SEQID NO.20)。反应条件:95℃/3min,(95℃/30sec,60℃/30sec,72℃/60sec)x30个循环,72℃/10min,8℃停止。扩增产物用1%琼脂糖凝胶电泳。用Takara MiniBEST Agarose Gel DNA Extraction Kit回收1002bp目标片段,经常规T-克隆后,送生工生物工程(上海)有限公司测序,确认测序序列中hTERC基因第-54号位点为鸟嘌呤脱氧核糖核苷酸“G”。并将重组腺病毒定名为:AD5+hTERC(MT-I)。
用pCA13/+hTERTpro+hTERC(MT-II)替代pCA13/+hTERTpro+hTERC(MT-I),其余完全重复上述步骤A和步骤B,确认测序序列中hTERC基因第-54号位点为胞嘧啶脱氧核糖核苷酸“C”,并将重组腺病毒定名为:AD5+hTERC(MT-II)。
用pCA13/+hTERTpro+hTERC(WT)替代pCA13/+hTERTpro+hTERC(MT-I),其余完全重复上述步骤A和步骤B,确认测序序列中hTERC基因第-54号位点为腺嘌呤脱氧核糖核苷酸“A”,并将重组腺病毒定名为:AD5+hTERC(WT)。
C:重组腺病毒的扩增、纯化、及滴度测定
基于常用重组腺病毒扩增方法,将HEK293细胞接种于75cm2培养瓶,加入20ml10%FBS的DMEM,培养至80-90%的细胞融合度时,换成含15ml 2%FBS DMEM。取0.5ul经病毒空斑纯化、鉴定正确的病毒保存液,小心加入培养瓶,水平十字形慢慢晃动3次。在37℃5%CO2培养箱中培养48小时后收集细胞沉淀,加入5ml 5%DMEM悬浮沉淀,-20℃至37℃反复冻融3次,600g离心20分钟后去沉淀取上清。按上述步骤反复扩增至需要的病毒量,用HPLC离子交换柱纯化病毒颗粒,并测定纯化样品的物理滴度(vp/ml)和感染性滴度(TCID50/ml)。结果:AD5+hTERC(MT-I)为5.3×10
实施例7用hTERC野生型真核表达的重组体,测试重组体的基因表达盒
基于HeLa细胞基因组DNA存在端粒3’末尾含有和遗传缺陷端粒,以及很高的端粒酶活性,突变型重组载体对细胞生长有严重干扰(见:
实施例8)。所以,测试重组体的基因表达盒选用野生型的重组真核表达载体。具体实施如下:
A:确定G418最佳筛选浓度。取生长状态良好的HeLa细胞,用0.25%胰酶消化,制备成细胞悬浮液,按1000个/ml接种在24孔培养板中,培养6小时,吸去培养基,PBS洗涤一次,每孔中加入不同浓度的筛选培养基(按100ug/ml、200ug/ml、300ug/ml、400ug/ml、500ug/ml、600ug/ml、700ug/ml、800ug/ml、900ug/ml、1000ug/ml的梯度浓度用培养基稀释G418制成筛选培养基)。每3天更换一次筛选培养基。以10-14天内,将细胞全部杀死的浓度确定为最佳筛选浓度,即400ug/ml G418。
B:用重组载体pcDNA3.1(-)/+hTERC(WT)转染HeLa细胞,获得稳定转染的单细胞系。挑选生长状态良好的HeLa细胞,在转染前一天铺六孔板,待细胞汇合度达到90%-95%时进行转染(Effectene细胞转染按试剂盒说明书)。转染24小时后将细胞用胰酶消化,以1:10的比例传代,贴壁24小时候开始G418筛选,浓度为400ug/ml,十四天后绝大部分细胞死亡,极少数细胞形成克隆。在超净台内,弃去培养皿内的培养液,胰酶消化细胞,用200ul移液枪挑取单克隆,加入已预先加好培养基的24孔板内,待细胞贴壁后加入G418筛选培养基继续培养2-5天,细胞长满后,0.25%胰酶消化,转入6孔板内继续培养,获得稳定转染的HeLa细胞株。
C:用Trizol方法提取稳定转染的HeLa细胞株总RNA,通过RT-PCR方法检测目标基因的表达,确定重组基因的表达正确。(图6)
实施例8测试重组hTERC基因突变体对HeLa细胞株的生长作用
A:MTT方法测定HeLa细胞的生长。
用构建的重组真核表达载体pcDNA3.1(-)/+hTERC(MT-I),pcDNA3.1(-)/+hTERC(MT-II)和pcDNA3.1(-)/+hTERC(WT),另增加一个空载体pcDNA3.1(-)/(空)为阴性对照,分别转染HeLa细胞。(按Effectene试剂盒说明书操作)。首先,挑选生长状态良好的HeLa细胞,于转染前一天铺六孔板,待细胞汇合度达到90%-95%时进行转染(Effectene细胞转染按试剂盒说明书)。转染24小时后,用胰酶消化收集,调整单细胞悬液密度为10
(图7)
B:Hoechst 33342染色观察细胞凋亡特征
Hoechst 33342染色观察细胞的形态,突变的pcDNA3.1(-)/+hTERC(MT-I)和pcDNA3.1(-)/+hTERC(TII)转染细胞一部分核膨大,有部分细胞出现染色质浓缩,高荧光状态呈现出细胞凋亡的特征。但未突变的阳性对照pcDNA3.1(-)/+hTERC(WT)和阴性对照pcDNA3.1(-)均呈现正常细胞状态(图8)。
实施例9测试重组hTERC基因突变体对人乳腺癌实体瘤生长的抑制作用
取人乳腺癌Bcap-37经培养、传代细胞系,用3000离心3min,PBS悬浮至3x10
实施例10测试重组hTERC基因突变体对人乳腺癌端粒酶的抑制作用
A:用TRAP方法,检测将实施例9中获得的实体瘤样本。取实体瘤100mg,放入预冷的研钵中,用液氮捣碎,加入200ul裂解液(10mM Tris-HCl pH7.5,1mM EGTA,0.1mM AEBSF,5mMβ-巯基乙醇,1%NP-40,0.25mM NaDOC,150mM NaCl,1mM MgCl2,10%甘油),充分匀浆;将匀浆液移入1.5ml的Ep管中,冰浴25min;再12,000g,4℃离心20min,将上清移取至1.5ml的Ep管中,充分混匀,取5μl,用Bradford方法进行蛋白质浓度测定,其余均匀分装到8个100μl的Ep管中,置于-80℃保存,待用。
B:端粒酶反应与PCR扩增。TS引物:5’-GGATCCAATCCGTCGAGCAGAGTT-3’(SEQ IDNO.21);CX引物:5’-CCCTTA CCCTTACCCTTACCCTTA-3’(SEQ ID NO.22)。反应体系50ul(20mMTris-HCl pH8.5,2.0mM MgCl2,50mM KCl,0.01%Triton X-100,1mM EGTA,1mM DTT,5ngBSA,50uM dNTPs,2uM TS引物,2.5ul 100ng总蛋白/ul端粒酶提取液,DEPC H
C:用10%聚丙烯胺凝胶电泳分离PCR产物。
制备10%非变性聚丙烯酰胺凝胶20ml(5.46ml,37%Acr-Bis36:1,12.4ml ddH2O,2ml 10×TBE,125ul 10%APS,15ul TEMED),用0.5xTBE电泳缓冲液,电压17.5V/cm,垂直电泳预跑1小时,再取15ul PCR产物与1.5ul 10x上样缓冲液进行上样,继续电泳1小时。银染:将凝胶置10%乙酸固定30min,蒸馏水漂洗3次,每次5min;用0.2g/L硫代硫酸钠浸泡1min,蒸馏水漂洗3次,每次30s;将凝胶放入0.2%硝酸银染液浸泡30min,用蒸馏水漂洗15s;然后,显色液中显色10min左右,放入10%乙酸固定10min。用Genetool软件,进行相对定量分析。结果:hTERC基因单碱基突变实体瘤样本相对端粒酶活性仅占野生型样本的21.6%(MT-I)和28.3%(MT-II)呈现显著下降。(图10)
实施例11观察重组hTERC基因突变体对人乳腺癌细胞的结构变化
利用常用组织石蜡切片和Schiff试剂染色方法,观察实施例9获得的实体瘤组织样本,尤其是细胞核的变化。取实体瘤组织切成4x4mm的组织放入FAA固定剂中固定,经一系列脱水、浸蜡、包埋、切成2-3um薄片,固定在载玻片上,经脱蜡、Schiff试剂染色、封片。在10x100倍的普通显微镜下观察、拍照,记录结果:A:AD5+hTERC(MT-I);B:AD5+hTERC(MT-II)与C:对照AD5+hTERC(WT)样本相比,细胞核浓缩变小,大小不均一,有较多细胞核固缩导致的致密浓染的现象,细胞间接触变得松散,呈现出明显细胞凋亡的现象。(图11)
实施例12检测重组hTERC基因突变体对人乳腺癌细胞的程序性凋亡作用
细胞程序性细胞凋亡的基因组DNA片段由大约180bp的多聚体组成,在琼脂糖凝胶电泳分离上呈现出典型的“梯形”特征,这可明显区别于其它死亡细胞的DNA降解特征。具体实施如下:
采用Takara MiniBEST Universal Genomic DNA Extraction Kit制备从实施例9中获得的实体瘤样本的基因组DNA,并用Nanodrop 2000进行定量,用1%琼脂糖凝胶电泳分析实体瘤基因组DNA,结果呈现出几乎完全相同的特征。说明hTERC基因单碱基突变引发了癌细胞的程序性凋亡。(图12)
以上实施例表明,使用本发明的重组端粒酶RNA基因突变体,通过对或遗传缺陷端粒的修复,显著降低它们的高端粒酶活性,明显抑制衰老细胞、癌变细胞及增生组织的异常生长,并将它们逆转进入正常的细胞程序性凋亡,从而实现其延缓衰老、预防细胞癌变、治疗癌症及增生性疾病的目的。
最后,还需要特别注意的是,以上例举的仅是本发明的若干具体实施方式中的一部分。显然,本发明不限于以上实施方式。所有涉及TC和TG遗传缺陷端粒的修复,而达到相同目的的任何方式,变形技术、方法和途径,均应认为是本发明的保护范围。
序列表
<110> 刘小川
<120> 一种突变的人端粒酶RNA基因及其在预防和治疗泛癌症中的应用
<130> IDC206048
<160> 24
<170> PatentIn version 3.5
<210> 1
<211> 451
<212> DNA
<213> 人
<400> 1
gggttgcgga gggtgggcct gggaggggtg gtggccattt tttgtctaac cctaactgag 60
aagggcgtag gcgccgtgct tttgctcccc gcgcgctgtt tttctcgctg actttcagcg 120
ggcggaaaag cctcggcctg ccgccttcca ccgttcattc tagagcaaac aaaaaatgtc 180
agctgctggc ccgttcgccc ctcccgggga cctgcggcgg gtcgcctgcc cagcccccga 240
accccgcctg gaggccgcgg tcggcccggg gcttctccgg aggcacccac tgccaccgcg 300
aagagttggg ctctgtcagc cgcgggtctc tcgggggcga gggcgaggtt caggcctttc 360
aggccgcagg aagaggaacg gagcgagtcc ccgcgcgcgg cgcgattccc tgagctgtgg 420
gacgtgcacc caggactcgg ctcacacatg c 451
<210> 2
<211> 11
<212> DNA
<213> 人
<400> 2
ctaaccctaa c 11
<210> 3
<211> 17
<212> DNA
<213> 人工序列
<400> 3
gcagtctacg agaccct 17
<210> 4
<211> 18
<212> DNA
<213> 人工序列
<400> 4
tcgtagactg cgtaccga 18
<210> 5
<211> 23
<212> DNA
<213> 人工序列
<400> 5
gaatcctgcg caccgagatt ctc 23
<210> 6
<211> 20
<212> DNA
<213> 人工序列
<400> 6
cggtacgcag tctacgagac 20
<210> 7
<211> 23
<212> DNA
<213> 人工序列
<400> 7
ttccattttt aaggtagtcg agg 23
<210> 8
<211> 23
<212> DNA
<213> 人工序列
<400> 8
taaaaggcaa caaaaagcgg aag 23
<210> 9
<211> 21
<212> DNA
<213> 人工序列
<400> 9
gtctaaccct gactgagaag g 21
<210> 10
<211> 21
<212> DNA
<213> 人工序列
<400> 10
ccttctcagt cagggttaga c 21
<210> 11
<211> 27
<212> DNA
<213> 人工序列
<400> 11
gcgtgaattc tccctttata agccgac 27
<210> 12
<211> 33
<212> DNA
<213> 人工序列
<400> 12
caggatccaa aaaaaaatgg gggctcacaa gcc 33
<210> 13
<211> 21
<212> DNA
<213> 人工序列
<400> 13
gtctaaccct cactgagaag g 21
<210> 14
<211> 21
<212> DNA
<213> 人工序列
<400> 14
ccttctcagt gagggttaga c 21
<210> 15
<211> 451
<212> DNA
<213> 人
<400> 15
gggttgcgga gggtgggcct gggaggggtg gtggccattt tttgtctaac cctgactgag 60
aagggcgtag gcgccgtgct tttgctcccc gcgcgctgtt tttctcgctg actttcagcg 120
ggcggaaaag cctcggcctg ccgccttcca ccgttcattc tagagcaaac aaaaaatgtc 180
agctgctggc ccgttcgccc ctcccgggga cctgcggcgg gtcgcctgcc cagcccccga 240
accccgcctg gaggccgcgg tcggcccggg gcttctccgg aggcacccac tgccaccgcg 300
aagagttggg ctctgtcagc cgcgggtctc tcgggggcga gggcgaggtt caggcctttc 360
aggccgcagg aagaggaacg gagcgagtcc ccgcgcgcgg cgcgattccc tgagctgtgg 420
gacgtgcacc caggactcgg ctcacacatg c 451
<210> 16
<211> 451
<212> DNA
<213> 人
<400> 16
gggttgcgga gggtgggcct gggaggggtg gtggccattt tttgtctaac cctcactgag 60
aagggcgtag gcgccgtgct tttgctcccc gcgcgctgtt tttctcgctg actttcagcg 120
ggcggaaaag cctcggcctg ccgccttcca ccgttcattc tagagcaaac aaaaaatgtc 180
agctgctggc ccgttcgccc ctcccgggga cctgcggcgg gtcgcctgcc cagcccccga 240
accccgcctg gaggccgcgg tcggcccggg gcttctccgg aggcacccac tgccaccgcg 300
aagagttggg ctctgtcagc cgcgggtctc tcgggggcga gggcgaggtt caggcctttc 360
aggccgcagg aagaggaacg gagcgagtcc ccgcgcgcgg cgcgattccc tgagctgtgg 420
gacgtgcacc caggactcgg ctcacacatg c 451
<210> 17
<211> 26
<212> DNA
<213> 人工序列
<400> 17
acatacgtac acgcacctgt tcccag 26
<210> 18
<211> 29
<212> DNA
<213> 人工序列
<400> 18
tcctgaattc gcgggggtgg ccggggcca 29
<210> 19
<211> 21
<212> DNA
<213> 人工序列
<400> 19
attacatacg tacacgcacc t 21
<210> 20
<211> 20
<212> DNA
<213> 人工序列
<400> 20
aacgggaaag cgaactgcat 20
<210> 21
<211> 24
<212> DNA
<213> 人工序列
<400> 21
ggatccaatc cgtcgagcag agtt 24
<210> 22
<211> 24
<212> DNA
<213> 人工序列
<400> 22
cccttaccct tacccttacc ctta 24
<210> 23
<211> 451
<212> RNA
<213> 人工序列或突变的人hTERC基因I的转录物
<400> 23
ggguugcgga gggugggccu gggaggggug guggccauuu uuugucuaac ccugacugag 60
aagggcguag gcgccgugcu uuugcucccc gcgcgcuguu uuucucgcug acuuucagcg 120
ggcggaaaag ccucggccug ccgccuucca ccguucauuc uagagcaaac aaaaaauguc 180
agcugcuggc ccguucgccc cucccgggga ccugcggcgg gucgccugcc cagcccccga 240
accccgccug gaggccgcgg ucggcccggg gcuucuccgg aggcacccac ugccaccgcg 300
aagaguuggg cucugucagc cgcgggucuc ucgggggcga gggcgagguu caggccuuuc 360
aggccgcagg aagaggaacg gagcgagucc ccgcgcgcgg cgcgauuccc ugagcugugg 420
gacgugcacc caggacucgg cucacacaug c 451
<210> 24
<211> 451
<212> RNA
<213> 人工序列或突变的人hTERC基因II的转录物
<400> 24
ggguugcgga gggugggccu gggaggggug guggccauuu uuugucuaac ccucacugag 60
aagggcguag gcgccgugcu uuugcucccc gcgcgcuguu uuucucgcug acuuucagcg 120
ggcggaaaag ccucggccug ccgccuucca ccguucauuc uagagcaaac aaaaaauguc 180
agcugcuggc ccguucgccc cucccgggga ccugcggcgg gucgccugcc cagcccccga 240
accccgccug gaggccgcgg ucggcccggg gcuucuccgg aggcacccac ugccaccgcg 300
aagaguuggg cucugucagc cgcgggucuc ucgggggcga gggcgagguu caggccuuuc 360
aggccgcagg aagaggaacg gagcgagucc ccgcgcgcgg cgcgauuccc ugagcugugg 420
gacgugcacc caggacucgg cucacacaug c 451
- 一种长链非编码RNA及其组合物在诊断治疗胆管癌中的应用
- 一种食管癌检测、诊断或者预后评价制剂,治疗食管癌的药物及RND2基因的应用
- 一种水稻ACCase突变型基因及其在植物抗除草剂中的应用
- 选自由胰腺癌、肺癌、大肠癌、胆管癌及肝癌组成的组中的至少一种癌的预防或治疗剂、用于与该药剂组合的复方医药的前述癌的预防或治疗剂、包含该药剂的组合医药、以及对癌的预防或治疗剂进行筛选的方法
- PEG10基因的双链小干扰RNA在制备治疗或预防肝癌药物中的应用