一种抗体偶联药物及其药物组合物与应用
文献发布时间:2023-06-19 09:26:02
技术领域
本发明涉及生物医学技术领域,具体涉及一种抗体偶联药物及其药物组合物与应用。
背景技术
抗体偶联药物(ADC,Antibody-Drug Conjugates)是新一代的抗体靶向治疗药物,主要用于癌症和肿瘤的治疗,目前已经成为一个开发热点,多个ADC正处于不同的临床开发阶段。ADC主要是通过在大分子抗体上偶联多个高毒性的小分子化合物,在发挥抗体高靶向特异性优势的同时,将小分子毒物也带入到靶组织周围,甚至靶细胞内,从而发挥更强的杀伤效应。
从结构上,ADC由“弹头”药物(细胞毒药物)、抗体以及偶联抗体和药物的连接子3部分组成,通过化学偶联的方法将细胞毒性药物连接到抗体蛋白上。理想情况下,ADC中的抗体部分起到靶向运输“弹头”的作用,首先在肿瘤组织处富集,后内吞裂解,定点释放细胞毒药物,最终实现对肿瘤的遏制和杀伤。由于ADC能够确切地将药物携带至细胞内,而在外周血和细胞表面很少降解,使得ADC可以实现降低传统剧毒化疗药物最小有效剂量的同时,并增加其最大耐药剂量,降低了化疗药物对正常组织的毒副作用。
然而,在针对肿瘤的治疗中,传统ADC需要高度依赖肿瘤细胞的抗原高表达性,以Genentech的Kadcyla(Ado-trastuzumab-emtansine;T-DM1)为例,其抗体部分为一种针对人类表皮生长因子受体2(Her2)的人源化IgG1单克隆抗体(trastuzumab,曲妥珠单抗),在Her2高表达的肿瘤区域中治疗效果表现甚佳,但是在Her2低表达乃至不表达的恶性癌变区域治疗效果骤然下降。
这种针对抗原受体表现出来的异质性,在临床应用上极大限制了ADC的进一步应用,发展新型的药物以不受抗原表达异质性的制约成为ADC领域亟待解决的一个问题。
发明内容
为了解决上述技术问题,本发明的目的在于提供一种抗体偶联药物及其药物组合物与应用。
为此,本发明的第一方面,提供一种抗体偶联药物,
所述抗体偶联药物具有如式I所示的结构,
其中,
-CH
-L-位于-OSiR
L为杂原子、取代或未取代的烃基;
R
CTD为细胞毒性药物;
Ab选自下组:抗体或可特异性识别抗原表位的多肽;
m为1-10的整数。
本发明中,L作为抗体与细胞毒性药物的连接桥的一部分,可以采用多种化学基团,可以是杂原子、取代或未取代的烃基,例如肽连接、二硫键连接、腙连接、硫醚连接等,本发明并不限定以上连接方式。另外,所述L为连接体,所述L使得本发明所述抗体偶联药物在水溶液中稳定,可用于静脉注射;而且,所述L使得本发明所述抗体偶联药物在血液循环中保持稳定。
进一步,所述L为取代或未取代的C
在具体的实施方式中,所述-L-具有如式III所示的结构,
其中,*示出了与Ab相连的键,#示出了与苯环相连的键;
即,所述抗体偶联药物具有如式I-1所示的结构,
其中,n为1-10的整数,例如1、2、3、4、5、6、7、8、9或10等,在一个优选的实施例中为4;
R
进一步,R
进一步,所述SiR
进一步,所述Ab选自下组:单克隆抗体、Fab、F(ab)
进一步,所述Ab是能够与选自下组的肿瘤相关抗原结合的抗体或多肽:HER2、HER3、CD19、CD20、CD22、CD30、CD33、CD37、CD45、CD56、CD66e、CD70、CD74、CD79b、CD138、CD147、CD223、EpCAM、Mucin 1、STEAP、GPNMB、FGF2、FOLR1、EGFR、EGFRvIII、c-MET、FGFR、Nectin 4、AGS-16、Guanylyl cyclase C、Mesothelin、SLC44A4、PSMA、EphA2、AGS-5、c-KIT、RoR1、PD-L1、CD27L、5T4、Mucin16、NaPi2b、SLITRK6、ETBR、BCMA、Trop-2、SC-16、SLC39A6中的至少一种。
进一步,所述能够与HER2结合的抗体选自下组:曲妥珠单抗、帕妥珠单抗中的至少一种。
进一步,所述CTD选自下组:微管蛋白抑制剂、拓扑异构酶抑制剂、DNA作用剂中的至少一种。
进一步,所述的微管蛋白抑制剂选自下组:美登素衍生物、Monomethylauristatin E(MMAE)、Monomethyl auristatin F(MMAF)、Monomethyl Dolastatin 10、Tubulysin类衍生物、Cryptophycin衍生物、Taltobulin中的至少一种。
进一步,所述DNA作用剂选自下组:PBD类衍生物、duocarmycin类衍生物中的至少一种。
进一步,所述拓扑异构酶抑制剂选自下组:阿霉素代谢产物PNU-159682衍生物、依沙替康中的至少一种。
优选地,-CH
本发明的第二方面,提供一种药物组合物,包括,
激活剂和本发明所述的抗体偶联药物;
所述激活剂具有如式II所示的结构,
其中,R
进一步,所述R
进一步,所述激活剂为:氨基酸(Amino acids,AA)中的至少一个-COO
在具体的实施方式中,所述氨基酸选自下组,甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、苯丙氨酸、半胱氨酸、苏氨酸、谷氨酰胺、天冬酰胺、甲硫氨酸、丝氨酸、脯氨酸、酪氨酸、色氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸、组氨酸。
进一步,所述氨基酸选自下组:亮氨酸、苯丙氨酸、丙氨酸、脯氨酸。
在一个优选的实施例中,所述氨基酸为苯丙氨酸(Phe),所述激活剂命名为Phe-BF
进一步,所述抗体偶联药物与所述激活剂的重量份数比为1-3:10-200,优选1-3:10-100。
另外,本发明的组合物中还可选择性地包括至少一种药用辅助剂,例如抑菌性注射用水(BWFI)、pH缓冲溶液(例如磷酸缓冲盐水)、无菌盐水溶液、葡萄糖溶液等。
本发明组合物形成的制剂类型优选注射剂。
本发明的第三方面,提供本发明所述的抗体偶联药物的制备方法,当所述抗体偶联物的结构式为I-1时,包括(1)制备连接子-细胞毒性药物;(2)将抗体的二硫键还原产生巯基,通过所述巯基连接所述连接子-细胞毒性药物。
在一个具体的实施方式中,当所述抗体偶联药物的结构式为式I-1,n=4,SiR
以上合成路线可以达到具有工业推广价值的、良好的合成效率、收率和纯度等效果。
本发明的第四方面,提供本发明所述的抗体偶联药物或本发明所述的药物组合物在用于制备治疗肿瘤的药物的用途。
与现有技术相比,本发明的技术方案有以下优点:
(1)本发明提供的药物组合物在杀伤肿瘤细胞时具有双靶向作用,在肿瘤细胞识别过程中不受抗原表达异质性的制约,即使癌变区域不表达相应抗原表位,本发明提供的药物组合物也能进行高效特异性的杀伤;
(2)由于本发明提供的药物组合物可高效特异性杀伤肿瘤细胞,在外周血或正常细胞表面很少发生降解,对正常组织或细胞几乎无毒副作用,可显著降低剧毒化疗药物的最小有效剂量;
(3)本发明提供的药物组合物中的所述抗体偶联药物和所述激活剂均已验证过体内安全性,具有良好的临床应用前景。
附图说明
通过阅读下文优选实施方式的详细描述,各种其他的优点和益处对于本领域普通技术人员将变得清楚明了。附图仅用于示出优选实施方式的目的,而并不认为是对本发明的限制。在附图中:
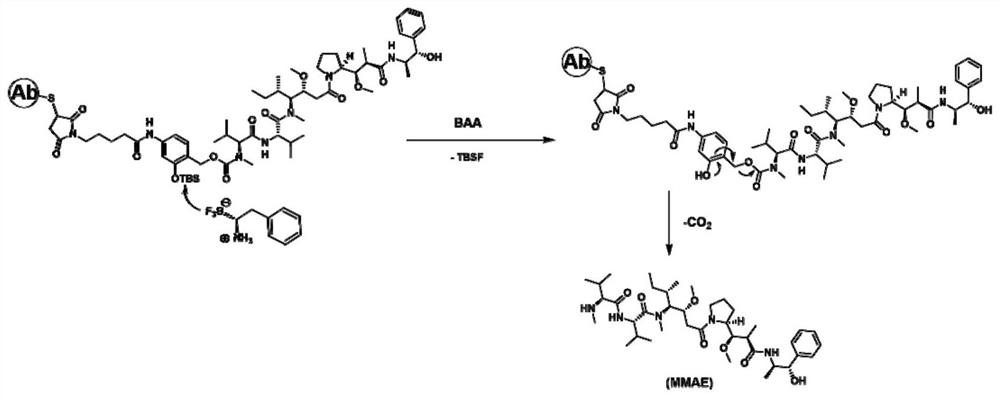
图1为激活剂激活ADC的药物释放原理示意图;
图2为实施例1中连接子-细胞毒性药物(linker-CTD)的合成路线;
图3为ADC和曲妥珠单抗的UV-Vis谱图;
图4为ADC的HIC-HPLC和MALDI-TOF表征图;
图5为通过HPLC检测ADC的药物释放效量;
图6为通过MC检测ADC的药物释放效量;
图7为通过MC检测结果所计算的ADC的药物释放效率;
图8为本发明的药物组合物对不同细胞的细胞活性影响;
图9为通过PET成像检测[
图10为通过PET成像检测ADC药物在荷瘤小鼠体内的循环性示意图;
图11为NCI-N87荷瘤小鼠的肿瘤体积变化检测结果;
图12为BGC823荷瘤小鼠的肿瘤体积变化检测结果;
图13为三阴乳腺癌MDA-MB-231荷瘤小鼠的肿瘤体积变化检测结果。
具体实施方式
下面将参照附图更详细地描述本公开的示例性实施方式。虽然附图中显示了本公开的示例性实施方式,然而应当理解,可以以各种形式实现本公开而不应被这里阐述的实施方式所限制。相反,提供这些实施方式是为了能够更透彻地理解本公开,并且能够将本公开的范围完整的传达给本领域的技术人员。
本发明所述的药物组合物包括所述抗体偶联药物(结构如式I所示)和所述激活剂(结构如式II所示),
本发明提供的药物组合物在杀伤肿瘤细胞时具有双靶向功能,并可定点释放细胞毒性药物,原理说明如下:
药物释放原理
在本发明的技术方案中,药物组合物中的所述抗体偶联药物的连接子具有SiR
有机硅烷保护剂是最常用的活泼氢保护剂,分子式可表示为X-SiR
硅烷保护剂的一般作用是用硅烷保护基取代化合物中不参与主反应的活泼氢,生成稳定的中间体硅醚或硅酯;主反应结束后,通过水解反应,脱除硅烷保护基,使原先被硅烷基保护的基团再生,从而合成出特定的化合物。
在本发明的技术方案中,药物组合物中的所述抗体偶联药物的连接子具有硅烷保护基,当所述药物组合物进入体内环境后,所述激活剂上的BF
双靶向功能
本发明提供的药物组合物包括所述抗体偶联药物(ADC)和所述激活剂,该药物组合物在杀伤肿瘤细胞时具有双靶向功能。
所述激活剂可特异性被肿瘤细胞所摄取,且不依赖于免疫识别。根据本发明的发明人的前期工作(专利文献WO 2016/176572A1和学术期刊文献Liu Z,Chen H,Chen K,etal.Boramino acid as a marker for amino acid transporters.Sci Adv.2015;1(8):e1500694.Published 2015 Sep 11.doi:10.1126/sciadv.1500694),氨基酸转运蛋白(Amino acid transporters,AATs)是一系列位于细胞上的完整通道,用于细胞摄取营养物质和传递神经递质。相对于正常细胞,肿瘤细胞上的AATs表达异常,这种表达异常造成了所述激活剂可特异性被肿瘤细胞所摄取。本发明的发明人已在前期工作中通过影像学方法证实了,所述激活剂在体内可高度特异性地被肿瘤细胞摄取,而且可以从正常器官和组织中快速清除。即所述激活剂对肿瘤细胞具有高度特异的靶向性,且这种靶向性不依赖于肿瘤细胞的抗原表位。此处,需要指出的是,在图1及本发明公开的实施例中,所述激活剂优选为Phe-BF
所述ADC中的抗体部分可特异性识别肿瘤细胞上的抗原表位,从而具有靶向功能。
因此,本发明提供的药物组合物在体内可发挥双靶向功能,一方面,通过所述ADC中的抗体部分靶向具有相应抗原表位的肿瘤细胞,通过内吞作用或所述激活剂的激活作用,定点释放细胞毒性药物;另一方面,所述ADC在外周血和细胞表面很少降解,对于不表达相应抗原表位的肿瘤细胞,ADC不会发生内吞作用,但由于所述激活剂富集在肿瘤区域,可对肿瘤区域的ADC产生激活作用,使其释放细胞毒性药物,从而特异性杀伤肿瘤区域的癌细胞。
除非另外定义,本公开中使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同意义。实施例中未注明具体技术或条件者,按照本领域内的常规技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
实施例1制备连接子-细胞毒性药物(linker-MMAE)
步骤1制备式b化合物
将底物a(1g,5mmol)溶于无水THF(20ml)中,冷却至0℃,然后向溶液中加入LiAlH
白色固体的表征结果:
步骤2制备式c化合物
将步骤1制备得到的底物b(1.5g,3.8mmol)溶解在MeOH(15ml)中,在N
棕色固体表征结果:
步骤3制备式d化合物
将2,5-二氧代吡咯烷-1-基5-(2,5-二氧代-2,5-二氢-1H-吡咯-1-基)戊酸酯(100mg,0.34mmol)溶于无水THF(5ml)中,在N
该步得到的棕色油状物表征结果:
步骤4制备式e化合物
将底物d(80mg,0.15mmol)溶解在MeOH(1ml)中,然后加入TsOH(10%mol)。将反应在惰性气氛下搅拌2小时直至起始原料耗尽。减压除去MeOH,并通过柱色谱纯化残余物,得棕色油状物(56mg,收率:90%)。
该步得到的棕色油状物表征结果:
步骤5制备式f化合物
将底物e(50mg,0.11mmol)溶于无水THF(0.5ml)中,在惰性气氛下加入DIPEA(2当量0.22mol)。在0度下将4-硝基苯基氯甲酸酯(4-nitrophenylcarbonochloridate,2当量0.22mol)缓慢加入溶液中。反应在55℃加热3小时。然后减压除去THF,并通过色谱法纯化残余物。将中间体溶于DMF(0.5ml)中,随后加入DIPEA(1当量0.11mmol)和HOBT(1当量0.11mmol)。缓慢加入MMAE(0.5当量,0.05mmol)。将溶液在37℃加热过夜。以DCM和MeOH作为溶剂,通过色谱法纯化混合物。得白色固体(27mg,收率:47%)。
该步得到的白色固体的表征结果:
实施例2制备抗体偶联药物(ADC)
本实施例以曲妥珠单抗作为抗体,偶联实施例1制备得到的连接子-细胞毒性药物,具体步骤如下:
(1)制备浓度为1mM的TCEP的PBS溶液,备用;
(2)将市售曲妥珠单抗浓缩成40-50mg/ml的PBS储备液,4℃保存备用;
(3)制备反应用的缓冲液,该缓冲液由25mM的硼酸钠,25mM的NaCl,1mM的DTPA溶液组成;
(4)在EP管内取40μL所述缓冲液,20μL曲妥珠单抗储备液,根据储备液浓度加入3.25当量的1mM的TCEP(PBS溶液,15μL左右),充分振荡,37℃反应90-120min;
(5)将步骤(4)制备得到的混合溶液冷却至常温,加入实施例1制备得到的连接子-细胞毒性药物的DMSO溶液(3.5mM)10μL,轻轻振荡摇匀,于室温条件反应30min;
(6)将步骤(5)制备得到的混合溶液通过PD10(已经过预处理)除去多余小分子,接收2.5ml-4ml的部分,即制备得到目标产物抗体偶联药物,将其用于下文中的实施例中,简称为ADC。
实施例3测定ADC的药物抗体比
本实施例对实施例2制备得到的ADC的药物抗体比进行测定。
(1)通过A260吸光度和A280吸光度测定药物抗体比
药物抗体比(DAR)计算公式如下:
通过NanoDrop测定实施例2制备得到的ADC的A260吸光度和A280吸光度之比恒为0.75,即R=A260/A280=0.75;
经检测,曲妥珠单抗的A260吸光度和A280吸光度参数如下(ADC和曲妥珠单抗的UV-Vis谱图见图3所示):
药物MMAE的A260吸光度和A280吸光度参数如下:
将以上数值带入公式,计算得到DAR=3.56。
(2)通过HIC-HPLC测定药物抗体比
对曲妥珠单抗和实施例2制备得到的ADC进行HIC-HPLC和MALDI-TOF测定,结果如图4所示,与曲妥珠单抗的峰相比,制备得到的ADC表现为几乎完全均一的疏水性较强的峰。相应地,通过MALDI-TOF测定曲妥珠单抗的二价峰M/Z为72766,制备得到的ADC的相应主峰为二价的M/Z 75054,表明制备得到的是连上4个单体的ADC。此外,通过22nm处的积分分析表明,平均DAR=4×0.9+2×0.02=3.64,与Nanodrop测定结果相吻合。
以上检测结果均说明了实施例2制备得到的ADC具有良好的均一性。
实施例4体外条件下药物释放效率的测定
本实施例测定了在体外条件下,实施例2制备得到的ADC与Phe-BF
方式1:通过色谱测定MMAE的释放效率
(1)制备药物组合物:分别取实施例2制备得到的ADC和Phe-BF
(2)将所述药物组合物溶解于PBS中,使得以ADC的质量计,浓度为1.5mg/mL,于37℃孵育,随着孵育时间的进行,通过HIC柱子分析峰的位移变化。
检测结果见图5所示,由图5可知,随着反应的持续进行,30h后ADC的峰完全消失,说明连接抗体的药物完全脱落释放。
方式2:通过质谱测定MMAE的释放效率
通过双四极杆的3Q质谱仪对MMAE进行半定量释放研究。
(1)通过HPLC-MS分析测定MMAE浓度的标准曲线,根据该标准曲线计算以下MMAE的释放量;
(2)制备药物组合物:分别取实施例2制备得到的ADC和Phe-BF
(3)在试管中,将所述药物组合物溶解于PBS中,以ADC的质量计,浓度为50nM(该浓度是为了模拟ADC在体内的富集而设置的),于37℃孵育,随着孵育时间的进行,在不同时间点取出100μL反应液,加入冰的100μL乙腈沉淀蛋白,高速离心3min,取上清液,进行HPLC-MS分析,并计算MMAE的浓度。
HPLC-MC检测的结果见图6所示,根据MMAE峰面积计算MMAE的浓度。由于ADC的浓度为50nM,平均DAR为3.6,可计算得出MMAE的理论上限释放浓度为180nM,由于测试过程中加入等体积的乙腈,所以理论上检测得到的上限释放浓度为90nM。随着孵育时间的进行,以90nM为100%,反应体系中MMAE的释放量如图7所示。由图7可知,孵育至24h时,ADC上偶联的MMAE几乎已经释放完全。
实施例5细胞水平上肿瘤抑制作用的验证
本实施例所用的细胞系如下:6种人源细胞系:BT474(HER2高表达)、NCI-N87(HER2低表达)、BGC823(HER2低表达)、MDA-MB-231(三阴乳腺癌)、U87-MG、A549;3种鼠源细胞系:4T1、B16F10、MC38。以上细胞均购自ATCC。
首先,通过流式细胞仪检测上述人源细胞系的HER2表达量,根据检测结果,各细胞系的HER2表达量由高到低排序为:BT474、NCI-N87、BGC823、A549、U87-MG、MDA-MB-231,其中BGC823和A549的HER2表达水平相当。
在细胞水平上检测本发明的药物组合物对肿瘤细胞的抑制作用,具体步骤如下:
将以上细胞系分别进行复苏扩增,复苏扩增后用96孔板进行铺板,复孔数为5。按照以下分组进行实验:
MMAE组、ADC组、ADC+Phe-BF
分组给药后孵育48h,换液加入培养基稀释的CCK8,在细胞培养箱内孵育30m,用酶标仪测定吸光度,测定结果见图8所示。由图8可知,对于HER2高表达的BT474细胞系,曲妥珠单抗本身就展现出良好的治疗效果,ADC+Phe-BF
同时,通过比较ADC+Phe-BF
实施例8通过影像学验证药物靶向功能
本实施例通过影像学的方法,验证了本发明提供的药物组合物具有双靶向性功能,在体内不仅对具有对应抗原表位的肿瘤细胞具有靶向性,对不具有抗原表位的其他肿瘤细胞,也具有靶向性。具体步骤如下:
(1)标记Phe-BF
在eppendorf管中,将Phe-BF
(2)标记ADC
在eppendorf管中加入900μL实施例2制备得到的ADC溶液(2mg/ml,PBS),加入100μL NaHCO
将170μL 0.2M Na
(3)PET成像
取7.4MBq 200μL生理盐水中的[
进一步,以NCI-N87荷瘤小鼠为例,在注射[
由图9可知,[
由图10可知,[
实施例9体内水平上肿瘤抑制作用的验证
本实施例在活体小鼠层面验证药物组合物的肿瘤抑制作用。具体步骤如下:
(1)荷瘤小鼠造模
选用NCI-N87、BGC823细胞系分别进行扩增,扩增后进行计数和胰酶裂解,用PBS重悬细胞后于裸鼠右肩皮下注射(裸鼠品系:Nu/Nu female,购自维通利华),注射计量为每只裸鼠约10
(2)给药:
待瘤生长至50mm
(3)观测小鼠生长状态,从第0天起,每3天量取肿瘤大小,并进行称重(肿瘤生长至1000mm
结合实施例5对各细胞系的HER2表达量检测结果,对图11-12分析如下:
由实施例5对各细胞系的HER2表达量检测可知,NCI-N87细胞系和BGC823细胞系均不属于典型的HER2高表达细胞,但NCI-N87的HER2表达量较BGC823更高。
由图11可知,针对NCI-N87肿瘤细胞的治疗,单独施用ADC的效果较单独施用曲妥珠单抗的效果更好;而图12针对BGC823肿瘤细胞的治疗,单独施用ADC与单独施用曲妥珠单抗的效果相当,甚至与PBS对照组的效果相近。这与实施例5的分析结果相吻合,这也体现了现有技术中抗体偶联药物在实际应用中的瓶颈,即传统ADC的治疗高度依赖肿瘤细胞的抗原高表达性,对于抗原低表达或不表达的恶性癌变区域治疗效果骤然下降,甚至没有治疗效果。
而本发明提供的药物组合物可有效克服传统ADC的这一缺陷,由于本发明提供的药物组合物中ADC和激活剂具有双靶向性,即使肿瘤细胞低表达或不表达相应抗原,也可以实现对肿瘤细胞的靶向治疗。由图11-12的结果可知,即使针对HER2低表达或不表达的肿瘤细胞,通过向小鼠施用ADC+Phe-BF
实施例10体内水平上三阴乳腺癌肿瘤抑制作用的验证
本实施例在活体小鼠层面进一步验证了,本发明提供的药物组合物对的MDA-MB-231(三阴乳腺癌)肿瘤抑制作用。具体步骤如下:
(1)荷瘤小鼠造模
选用MDA-MB-231细胞系分别进行扩增,扩增后进行计数和胰酶裂解,用PBS重悬细胞后于裸鼠右肩皮下注射(裸鼠品系:Nu/Nu female,购自维通利华),注射计量为每只裸鼠约10
(2)给药:
待瘤生长至50mm
(3)观测小鼠生长状态,从第0天起,每3天量取肿瘤大小,并进行称重(肿瘤生长至1000mm
由图13可知,对于不表达HER2的三阴乳腺癌MDA-MB-231,强烈依赖靶向性以及内吞释放机制的T-DM1在MDA-MB-231荷瘤小鼠中的治疗效果不显著,即使增加给药疗程,其治疗效果也几乎没有改善。
而使用本发明提供的药物组合物ADC+Phe-BF
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
- 一种含二甲双胍和抗体药物偶联物的药物组合物及其应用
- 抗体药物偶联物、中间体、制备方法、药物组合物及应用