用于介导EPS的组合物和方法
文献发布时间:2023-06-19 10:36:57
相关申请的交叉引用
本申请根据35 U.S.C. § 119(e)要求2018年6月29日提交的美国临时申请62/692,581号的优先权,其全部内容通过引用合并于此。
技术领域
本公开总体上涉及移除或抑制细菌细胞外聚合物(extracellular polymericsubstance,EPS)的方法和组合物。
背景技术
生物膜在医学、农业和工业环境中发挥重大作用。生物膜对动物和植物的大部分疾病以及工业设备的结垢负有责任,因此,生物膜是大量研究工作的重点。由于多种因素,根除或处理生物膜特别困难,这些因素包括形成对抗菌效应物的物理屏障的细胞外基质的产生、不易受到环境压力的影响的生理变化、以及生物膜成分之间的协同相互作用。生物膜基质可变地由多糖、蛋白质以及可能普遍地由细胞外DNA(eDNA)组成。微生物生物膜的eDNA是提供保护的细胞外基质的重要组成部分。通过DNA降解或去除稳定该结构的DNA结合蛋白来破坏生物膜eDNA的结构,会导致生物膜的灾难性倒塌,并使常驻细菌释放到更易受伤害的状态。
细菌在自然界中以两种不同的状态存在;浮游细菌是自由生活的,而发展成群落结构的细菌则被称为生物膜(在表面上或作为聚集体)。CDC和NIH估计,所有细菌感染中约有80%涉及必要的生物膜状态。Dongari-Bagtzoglou et al. (2008) Expert Rev AntiInfect Ther. 6(2):201-8。这些包括中耳炎(OM)、慢性鼻窦炎(CRS)、慢性肺部感染、慢性伤口感染、牙周炎、膀胱炎、以及医疗植入物和留置导管的感染,等等。确实,寻求儿科医疗服务的最常见原因之一是OM(由不可分型的流感嗜血杆菌(Nontypeable
因此,需要突破生物膜的保护性屏障以治疗或杀死相关的细菌感染,并从表面和水系统中清除它们。
概述
保护生物膜内存在的细菌免受免疫清除和抗微生物作用的自我产生的细胞外基质(或细胞外聚合物,EPS)对于致病性生物膜引起慢性和复发性感染至关重要,因为生物膜可作为这些引起疾病的细菌的顽固性储存库。EPS成分是特定于单个细菌物种的,但普遍包含源自生物膜内存在的细菌的细胞外DNA(eDNA)。实际上,不同属的细菌通常会进入多物种生物膜的共享群落结构,这要求EPS不仅在结构上有利于所有组成物种,而且还必须包含源自所有常驻细菌或可用于所有常驻细菌的EPS成分。在这方面,单个和多个物种生物膜的EPS包含支架eDNA,这似乎是潜在的通用EPS的常见结构。如本文公开的,申请人发现该eDNA依赖性结构被细菌DNA结合蛋白的普遍存在的DNABII家族稳定。虽然申请人已经表明,外源性DNA和DNABII蛋白可以将自由生活(浮游生物)的细菌带入生物膜的群落结构,但这两个组分不足以概括出具有特征性的eDNA支架。
申请人在本文中公开了多胺是通用eDNA-DNABII依赖性EPS的第三个关键组分。多胺是在细胞内和细胞外普遍存在的短的带正电的有机分子,当与DNA结合时,它会中和核苷酸磷酸的聚阴离子电荷,并使DNA分子凝聚/聚集。重要的是,申请人在本文中公开了多胺可以将DNA从最常见的右手B型驱动成抗核酸酶的左手Z型DNA。确实,尽管核酸酶可以
本文描述了用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的试剂接触,其中所述试剂不是HMGB1蛋白、其片段或其每一个的等效物。一方面,用于抑制生物膜的稳定性的方法以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂接触。本公开还涉及用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与干扰生物膜中多胺与DNA结合的试剂在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的消耗阳离子的试剂包被表面,其中该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一方面,用于抑制生物膜的稳定性的方法可以包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的试剂在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的一种或多种消耗阳离子的试剂包被表面。接触可以是体外或体内的。
在一个实施方案中,所述试剂干扰生物膜或其局部环境中B-DNA向Z-DNA转化。在第二实施方案中,所述试剂包括抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在第三实施方案中,所述试剂包括核黄素、溴化乙锭、双(甲锭)精胺(bis(methidium)spermine)、柔红霉素(daunorubicin)、TMPyP4、季苯并[
本文进一步描述了用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物包被表面。本公开内容还涉及用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的氯喹和抗B-DNA抗体或其片段或衍生物在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的氯喹和抗-B-DNA抗体或其片段或衍生物包被表面。接触可以是体外或体内的。
本文还提供了用于治疗对象中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的试剂,其中该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一方面,用于治疗对象中的生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂。
本公开内容还涉及用于防止在易于发展生物膜的对象中形成生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的试剂,其中该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一方面,用于防止在易于发展生物膜的对象中形成生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂。
本公开进一步涉及用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的试剂和抑制生物体复制的试剂,其中该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一方面,用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂。
对于上述任何方法,多胺可以选自:腐胺、精胺、尸胺、1,3-二氨基丙烷或亚精胺。在一个实施方案中,对于上述方法,干扰多胺与生物膜中DNA结合的试剂是tRNA。在另一个实施方案中,该试剂是多胺合成的抑制剂或抑制多胺与DNA结合的试剂。在第二实施方案中,该试剂包括多胺类似物二氟甲基鸟氨酸、反式-4-甲基环己胺、沙多齐特(sardomozide)、甲基乙二醛双[脒基腙](methylglyoxal-bis[guanylhydrazone],MGBG)、1-氨基氧基-3-氨基丙烷、奥沙利铂(oxaliplatin)、顺铂、二环己胺、其任何衍生物、或其盐,或基本上由其组成,或由其组成。在第三实施方案中,该试剂包括从生物膜中消耗阳离子的试剂,任选地阳离子交换树脂、氨基多羧酸、冠醚、氮杂冠(azacrown)、或穴醚(cryptand),或基本上由其组成,或由其组成。在第四实施方案中,从生物膜中消耗阳离子的试剂选自:磺酸盐,磺丙基,磷酸纤维素,P11磷酸纤维素,硫酸肝素,或其衍生物或类似物。在第五实施方案中,该试剂干扰生物膜或其局部环境中B-DNA向Z-DNA转化。在第六实施方案中,所述试剂包含抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在第八实施方案中,所述试剂包含核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[
本文还提供了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)的患者中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂,其中该试剂不是HMGB1蛋白、其片段或其每一个的等效物。本文公开了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)和/或TB的患者中的生物膜的方法,所述方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的一种或多种干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂,其中该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一方面,在不存在DNA酶(DNAse)的情况下施用所述试剂。在一个实施方案中,所述试剂在生物膜或其局部环境中干扰B-DNA向Z-DNA转化。在第二实施方案中,所述试剂包含抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在第三实施方案中,所述试剂包含核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[
本文还提供了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)的患者中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物。在一方面,本文还提供了用于治疗在患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)和/或肺结核(TB)的患者中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的氯喹和抗B-DNA抗体或其片段或衍生物。本公开还涉及用于治疗在正在接受或已经接受化学疗法的患者中的因施用基于铂的化学疗法而发生的产生生物膜的感染的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂,其中所述试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一方面,所述方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的一种或多种干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在一方面,在不存在DNA酶的情况下所述试剂。在另一方面,所述试剂包含氯喹或其衍生物,或基本上由其组成,或由其组成。在一个特定的方面,氯喹衍生物保留了插入DNA碱基之间的能力。在另一方面,所述试剂包含抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在一个实施方案中,所述试剂包含核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[
本公开进一步涉及用于治疗在正在接受或已经接受化学疗法的患者中的因施用基于铂的化学疗法而发生的产生生物膜的感染的方法,所述方包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的 HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物。本文还提供了用于治疗在正在接受或已经接受化学疗法的患者中的因施用基于铂的化学疗法而发生的产生生物膜的感染的方法,所述方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的氯喹和抗B-DNA抗体或其片段或衍生物。
上述方法可以进一步包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰eDNA与DNA结合蛋白结合的试剂和/或抗菌剂接触,或替代地将所述试剂施用至对象,其中所述试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一方面,干扰eDNA与DNA结合蛋白结合的试剂包含抗DNABII抗体、抗IHF抗体和/或抗HU抗体、或其每一个的片段中的一种或多种,或基本上由其组成,或由其组成。在一个实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净负电荷。在第二实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净中性电荷。在第三实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净正电荷。在一方面,在不存在DNA酶的情况下施用所述试剂。上述方法可以在不施用DNA酶的情况下进行。
附图说明
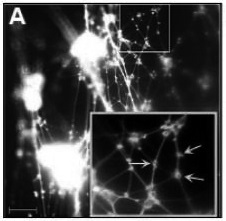
图1A-1C:多胺调节DNA结构。(图1A)在具有由于NTHI引起的急性OM的毛丝鼠(chinchilla)中耳中发现的粘膜生物膜EPS的免疫荧光显微图像(改编自Goodman et al.(2011) Mucosal Immunol. 4(6):625-37)。探测(probe)DNABII蛋白(灰色球)和eDNA(白色区域)。 DNABII在体内定位于生物膜中的eDNA链顶点。(图1B)普通多胺的化学结构(改编自Di Martino et al. (2013) Int J Med Microbiol. 303(8):484-91)。(图1C)单独的DNA或与多胺一起孵育后的DNA的原子力显微镜图像(改编自Iacomino et al. (2011)Biomacromolecules. 12(4):1178-86)。多胺在eDNA中引起粗纤维形成和结构复杂性。
图2A-2B:多胺诱导eDNA支架结构。(图2A)在由于NTHI引起的急性OM的毛丝鼠中耳中发现的粘膜生物膜EPS的免疫荧光CLSM图像。探测多胺(白点)(腐胺Put)、尸胺(Cad)、亚精胺(Spd))和eDNA(白色),并用DAPI(灰色区域)复染。多胺位于体内形成的生物膜中的eDNA链上。亚精胺是生物膜EPS中最普遍的多胺。(图2B)DNA(5 μM)和亚精胺(700 μM)(顶部)和DNA(5 μM)、亚精胺(700 μM)和HU(50 nM)(底部)的复合物的透射电子显微镜图像(改编自Sarkar et al. (2007) Nucleic Acids Res. 35(3):951 61)。DNA-聚胺-IHF复合物形成相似的结构。Sarkar et al. (2009) Biochemistry. 48(4):667-75。多胺诱导DNA缩合,并与DNABII蛋白结合形成粗纤维。
图3A-3C:多胺合成抑制剂减少生物膜形成。(图3A)在双环己胺(DCHA,50 μM)(一种亚精胺(Spd)合酶抑制剂)、Spd(1 mM)或两者同时存在的情况下生长的LIVE/DEAD®染色NTHI生物膜的COMSTAT定量。条形代表SEM。通过未配对t检验评估与对照组相比的统计学显著性,* P <0.05。DCHA降低了生物膜的平均厚度,而同时添加外源Spd可以恢复生物膜的形成。(图3B)在DCHA(50 μM)存在下生长3h的NTHI体外生物膜中eDNA支架结构的免疫荧光显微图像。探测dsDNA(白色区域)。通过抑制多胺的合成,大大减少了eDNA支架结构的产生。(图3C)在DCHA存在下生长40h的NTHI体外生物膜的免疫荧光CLSM图像。探测Spd(图3C下部小图中的白点)并用DAPI复染(灰色区域)。DCHA抑制多胺掺入生物膜EPS中。
图4A-4B:抗DNABII破坏DNABII-多胺(PA)依赖性结构。(图4A)通过将亚精胺(300μM)和HU(1 μM)在含有基因组DNA(gDNA;2μg/ml)的缓冲液中孵育40h来形成DNA结构。DNABII-多胺依赖性DNA结构的免疫荧光CLSM图像。探测DNABII蛋白(白色;图像右侧),并用DAPI复染(白色;图像左侧)。DNABII多胺依赖性DNA结构整合了DNABII蛋白。(图4B)如(图4A)中那样形成EPS结构24小时,并用1:50稀释的DNABII抗血清稀释处理16小时(在图像下方示出)。用DAPI(白色)染色的荧光CLSM图像。DNABII-多胺依赖性DNA结构需要DNABII蛋白。
图 5:阳离子交换剂磷酸纤维素(P11)破坏NTHI生物膜的形成。在将P11(1%)加入心尖室的同时,在Transwell平板系统的基底外侧室开始NTHI生物膜的生长。在播种时添加亚精胺(1 mM)和HU(1 μM),并保持16h。生物膜通过CLSM可视化,并通过COMSTAT分析。平均厚度(未显示)显示出相同的趋势。条形代表SEM。通过未配对t检验评估与对照组相比的统计学显著性,* P <0.05;** P <0.01。P11防止生物膜形成。外源性亚精胺和HU共同恢复了生物膜的发育,但不能单独恢复(未显示),这表明P11抗生物膜活性是来自生物膜EPS的滴定结构成分(多胺和DNABII蛋白)的结果。
图 6:成熟的生物膜对DNA酶的破坏具有抵抗力。在播种(预防)或24h(破坏)时,将DNA酶(Pulmozyme;5个单位)添加到各自的体外预先形成的NTHI和UPEC生物膜中。总共40小时后,生物膜用LIVE/DEAD®染色,通过CLSM可视化,并通过COMSTAT分析。条形代表SEM。通过未配对t检验评估与对照组相比的统计学显著性,* P <0.05;** P <0.01。DNA酶可以预防但不破坏现存的生物膜。
图7A-7D:DNABII蛋白和多胺(PA)协同相互作用以诱导DNA酶抗性。(图7A)生长40小时的NTHI体外生物膜的免疫荧光CLSM图像。探测亚精胺(深灰色球)、HU(浅灰色球),并用DAPI(灰色区域)复染。聚胺和DNABII蛋白在体外生物膜EPS中共定位(白球)。(图7B)在具有由于NTHI引起的急性OM的毛丝鼠中耳中发现的粘膜生物膜EPS的免疫荧光CLSM图像。探测腐胺(白色球)和HU(浅灰色球),并用DAPI(深灰色区域)复染。体内多胺和DNABII蛋白共定位在生物膜的eDNA链上(白色区域)。(图7C)将递增水平的亚精胺(Spd)和HU分别或一起与基因组DNA(2 μg/ml)在37℃温育1.5h,然后用Pulmozyme®处理20分钟。使用琼脂糖凝胶电泳测定DNA降解。Spd和HU协同保护基因组DNA免受DNA酶消化。(图7D)将Spd(300 μM)和HU(1μM)与基因组DNA(2 μg/ml)孵育40h,然后用Pulmozyme®处理20分钟。结构用LIVE/DEAD®染色并用CLSM成像(白色)。HU-Spd依赖性DNA结构对DNA酶处理具有抗性。
图8A-8B:DNABII和多胺结合来将B-DNA转化为ZDNA形式。(图8A)通过将基因组DNA(2 μg/ ml)与HU(1 μM)和亚精胺(300 μM)温育16h来形成DNABII-多胺依赖性DNA结构。DNABII-多胺依赖性DNA结构的免疫荧光CLSM图像。探测Z-DNA(白色)并用DAPI染色(深灰色)。多胺和DNABII蛋白协同作用以诱导B-DNA到Z-DNA的转化。(图8B)上图:通过增加Z-DNA催化剂的浓度将B-DNA底物的圆二色性光谱转换为Z-DNA(改编自Jang et al. (2015) SciRep. 5:9943)。注意250 nm附近的负峰和280 nm附近的正峰反转。底部:将聚(dGdC)DNA(20μg/ml)与HU(15 μM)孵育2h,并收集CD光谱。HU将聚(dGdC)的CD光谱移向Z-DNA标记。
图 9:Z-DNA存在于多种人类病原体的生物膜EPS中。上图:所示细菌40h生物膜的免疫荧光CLSM图像,用无一抗(No 1º)或用Z-DNA抗体(白色)进行了探测。Z-DNA是处于不同稳态水平的多个细菌生物膜EPS的组成部分。下图:用HU(白色)和亚精胺(深灰色)抗体探测的所示生物膜的免疫荧光CLSM图像,共定位为(白色)。DNABII和多胺组分在稳态细菌水平上共定位在多个细菌生物膜的EPS中,这些水平与Z-DNA丰度相关。
图10:随着UPEC和NTHI生物膜的成熟,Z-DNA和多胺的含量增加。UTI89和NTHI体外生物膜在形成的各个阶段的免疫荧光CLSM图像,分别用抗Z-DNA(白色)或抗亚精胺(深灰色)探测。随着时间的推移,成熟的生物膜在生物膜EPS中掺入了越来越多的Z-DNA(白色)和亚精胺(深灰色)。
图11:B- DNA向Z-DNA转化以及将多胺掺入生物膜EPS中需要HU。40h NTHI野生型和ΔHU突变体体外生物膜的免疫荧光CLSM图像,用亚精胺(深灰色)或抗Z-DNA(白色)探测。在没有HU的情况下,生物膜EPS中的多胺和Z-DNA的丰度降低。
图12:不可分型的流感嗜血杆菌生物膜在补充的BHI培养基中在8孔室盖玻片中,于37℃和5% CO
图13:不可分型的流感嗜血杆菌在含有上述添加剂的BHI培养基中,于37℃和5%CO
图14:不可分型的流感嗜血杆菌生物膜在含有如下面每个条形所示添加剂的补充的BHI培养基中,在37℃和5% CO
图15:不可分型的流感嗜血杆菌生物膜在含有上述添加剂的BHI培养基中在8孔室盖玻片中,于37℃和5% CO
图16:将表面附着的不可分型的流感嗜血杆菌在含有上述添加剂的BHI培养基中于37℃和5% CO
图17A-17D:亚精胺存在于多种人类病原体形成的生物膜EPS中。(图17A)用HU(浅灰色)和亚精胺(深灰色)抗体探测的所示生物膜的免疫荧光CLSM图像,共定位为白色。DNABII和多胺组分在稳态水平下共定位于多个细菌生物膜的EPS中。(图17B)二环己胺(DCHA)对亚精胺生物合成的抑制降低了IFHI信号降低所指示的NTHI和UPEC生物膜中的亚精胺水平,并且与sBHI对照相比导致平均厚度显著降低(图17C)。UPEC表示为与LB对照相比的平均厚度变化百分比(图17D)。
图18A-18B:磷酸纤维素在体外对生物膜形成和预先形成的NTHI生物膜稳定性具有剂量依赖性的负面影响。(图18A)开始生物膜生长,然后维持24小时,然后用0(SBHI对照)、0.1%、1%和5%(w/v)的磷酸纤维素(P11)处理16小时。(图18B)开始生物膜生长,然后在0(sBHI对照)、0.1%、1%和5%(w/v)(P11)的存在下维持40小时。生物膜用盐水洗涤,并用LIVE/DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量。使用63倍物镜捕获所有图像。
图19:肝素琼脂糖凝胶对体外NTHI生物膜形成有负面影响。开始生物膜生长,然后在0(sBHI对照)或5%(w/v)肝素琼脂糖树脂存在下维持40小时。生物膜用盐水洗涤,并用LIVE /DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量。使用63倍物镜捕获所有图像。
图20:外源加入HU可拯救磷酸纤维素对NTHI生物膜稳定性的负面影响。启动生物膜生长并维持24小时,然后如所示处理16小时。生物膜用盐水洗涤,并用LIVE /DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量,并与sBHI对照进行比较。使用63倍物镜捕获所有图像。条形代表SEM。通过未配对t检验评估与对照组相比的统计学显著性,* P <0.05;** P <0.01。
图21:外源添加MgCl
图22:外源添加亚精胺可拯救磷酸纤维素对体外NTHI生物膜稳定性的负面影响。开始生物膜生长,然后维持24小时,然后如所示处理16小时。生物膜用盐水洗涤,并用LIVE/DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量。使用63倍物镜捕获所有图像。
图23:P11磷酸纤维素的阳离子耗竭效应不需要与生物膜直接接触。在通孔板系统的基底外侧室开始生物膜的生长,同时将0、0.5、1或1.5%(w/v)的P11磷酸纤维素添加到顶端室中,并保持16小时。生物膜用盐水洗涤,并用LIVE/DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量,并将其与sBHI对照进行比较。使用63倍物镜捕获所有图像。条形代表SEM。通过未配对t检验评估与对照组相比的统计学显著性,* P <0.05;** P<0.01。
图24:外源添加亚精胺可减少P11磷酸纤维素的阳离子耗竭作用,而无需直接与生物膜接触。在transwell板系统的基底腔中开始生物膜的生长,同时将0或1.5%(w/v)P11磷酸纤维素添加到顶端腔中,并在存在100、500或1000uM亚精胺的情况下洗涤16小时。生物膜用盐水洗涤,并用LIVE/DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量,并将其与sBHI对照进行比较。使用63倍物镜捕获所有图像。条形代表SEM。通过未配对t检验评估与对照组相比的统计学显著性,* P <0.05;** P <0.01。方括号表示条件之间的统计比较。
图25:外源添加亚精胺和DNABII可减少P11磷酸纤维素的阳离子耗竭作用,而无需直接与生物膜接触。在transwell板系统的基底外侧腔室中开始生物膜生长,同时将0或1.5%(w/v)P11磷酸纤维素添加到顶端腔室中,并在100uM亚精胺或500nM HU或组合存在的情况下保持16小时。生物膜用盐水洗涤,并用LIVE /DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量,并将其与sBHI对照进行比较。使用63倍物镜捕获所有图像。条形代表SEM。通过未配对t检验评估与对照组相比的统计学显著性,* P <0.05;** P <0.01。方括号表示条件之间的统计比较。
图26A-26B:用阳离子交换树脂包被非生物表面可防止生物膜形成,且呈剂量依赖方式。如图所示,用P11磷酸纤维素(图26A)或肝素琼脂糖凝胶(图26B)溶液包被腔室载玻片。生物膜生长开始并在包被的载玻片上保持40小时。生物膜用盐水洗涤,并用LIVE /DEAD®染色剂染色。通过COMSTAT分析图像以计算平均厚度和生物量。使用63倍物镜捕获所有图像。条形代表SEM。
图27:成熟的生物膜对DNA酶的破坏具有抵抗力。将DNA酶(Pulmozyme;5个单位)在播种(预防)或24 h(破坏)时分别添加到各自体外预先形成的NTHI或UPEC生物膜中。总共40小时后,生物膜用LIVE /DEAD®染色,通过CLSM可视化,并通过COMSTAT分析。条形代表SEM。对于生物量观察到相似的趋势。通过未配对t检验评估与对照组相比的统计学显著性,* P<0.05;** P <0.01。脱氧核糖核酸酶可以预防但不破坏现存的生物膜。
图28:随着UPEC和NTHI生物膜的成熟,Z-DNA和多胺的含量增加。UPEC和NTHI生物膜在不同成熟阶段体外形成的免疫荧光CLSM图像,分别用抗Z-DNA(浅灰色)或抗亚精胺(深灰色)探测。随着时间的推移,成熟的生物膜在生物膜EPS中掺入了越来越多的Z-DNA(浅灰色)和亚精胺(深灰色)。
图29:Z-DNA存在于成熟的病原真菌生物膜中。体外由白色念珠菌形成的生物膜的免疫荧光CLSM图像,用DAPI复染色,并用抗B-DNA、抗Z-DNA或无一抗(浅灰色)探测。成熟的真菌生物膜在生物膜EPS中掺入了Z-DNA(浅灰色)。
图30:抗Z-DNA抗体刺激生物膜的生物发生。在播种时,将抗Z-DNA抗体(1 mg)添加到NTHI体外生物膜中。16小时后,生物膜用LIVE /DEAD®染色,通过CLSM可视化,并通过COMSTAT分析。条形代表SEM。对于生物量观察到相似的趋势。通过配对t检验评估与对照组相比的统计学显著性。抗Z-DNA抗体可稳定生物膜的细胞外基质,刺激生物膜的生物发生,而抗B-DNA的抗体(例如抗dsDNA)不会刺激生物膜的生物发生。
图 31:DNA酶降解生物膜细胞外基质内的B-DNA,但不降解Z-DNA。在生物膜播种后24小时,将DNA酶(Pulmozyme;5个单位)添加到体外预先形成的NTHI生物膜中。总共40小时后,用抗Z-DNA、抗B-DNA或无一抗探测生物膜,这些抗体通过使用相应的荧光二抗显露并通过CLSM可视化。DNA酶处理可降解B-DNA形式的eDNA结构,但揭示了生物膜细胞外基质内大量Z-DNA形式的eDNA。
图 32:Z-DNA的形成保护DNA免受核酸酶降解。如上所述,将聚(dG-dC)底物与盐浓度的核酸酶(SAN)或DNA酶 I一起以不断增加的浓度的NaCl、亚精胺或亚精胺温育。降解产物通过凝胶电泳可视化。高盐和多胺可防止DNA通过转化为Z-DNA形式而降解。
图33:DNABII蛋白和多胺共定位在生物膜细胞外基质内。不可分型的流感嗜血杆菌生物膜在补充的BHI培养基中于37℃和5% CO
图34A-34B:DNABII蛋白通过转变为Z-DNA形式来保护DNA。(图34A)如上所述,将聚(dG-dC)底物与DNA酶 I一起以浓度增加的NTHi HU孵育。降解产物通过凝胶电泳可视化。HU保护DNA免受降解。(图34B)上图:通过增加Z-DNA催化剂的浓度将B-DNA底物转换为Z-DNA的圆二色性光谱(改编自Rahmouni (1992) Mol Microbiol. 6(5):569-72)。注意250 nm附近的负峰和280 nm附近的正峰反转。下图:将聚(dGdC)DNA(5 µg)与HU(15 µM)孵育2 h,并收集CD光谱。HU将聚(dGdC)的CD光谱移向Z-DNA标记。
图35A-35D:DNABII蛋白和多胺(PA)协同相互作用以诱导DNA酶抗性。(图35A)将递增水平的亚精胺(Spd)和HU分别或一起与基因组DNA(gDNA;2 μg/ml)一起在37℃下孵育1.5小时,然后用Pulmozyme®处理20分钟。使用琼脂糖凝胶电泳测定DNA降解。Spd和HU协同保护基因组DNA免受DNA酶消化。(图35B)将Spd(300 µM)和HU(1 µM)与基因组DNA(2 µg/ml)孵育40小时,然后处理20分钟。结构用LIVE/DEAD®染色并用CLSM成像(白色)。(图35C)在具有由于NTHI引起的实验性OM的毛丝鼠中耳中发现的粘膜生物膜EPS的免疫荧光CLSM图像。探测腐胺(深灰色)和HU(浅灰色),并用DAPI(灰色)复染。 HU-Spd依赖性DNA结构对DNA酶处理具有抗性。体内多胺和DNABII蛋白共定位在生物膜中的eDNA链上(白色)。(图35D):在播种(预防)或24小时(破坏)时,向NTHI或UPEC生物膜中添加浓度增加的DNA酶 16小时。对生物膜进行染色,固定,通过CLSM进行可视化,并通过COMSTAT进行分析。条形代表SEM。通过未配对t检验评估与对照组(无DNA酶)相比的统计学显著性,** P <0.01。脱氧核糖核酸酶可以防止但不破坏现存的生物膜。
图36:Z-DNA存在于多种人类病原体的生物膜EPS中。上图:由所示细菌形成的40 h生物膜的免疫荧光CLSM图像,用无一抗(No1º)或用Z-DNA特异性抗体(浅灰色)探测。 Z-DNA是处于不同稳态水平的多个细菌生物膜EPS的组成部分。下图:用HU(浅灰色)和亚精胺(深灰色)抗体探测的所示生物膜的免疫荧光CLSM图像,共定位为(白色)。DNABII和多胺组分在稳态细菌水平上共定位在多个细菌生物膜的EPS中,这些水平与Z-DNA丰度相关。
图37:HMGB1使生物膜细胞外基质中的Z-DNA结构不稳定。不可分型的流感嗜血杆菌生物膜在补充的BHI培养基中,在37℃和5%CO
图 38A-38C:HMGB1取代DNABII蛋白从而破坏NTHI生物膜。不可分型的流感嗜血杆菌生物膜在补充的BHI培养基中,在37℃和5%CO
图39A-39B:HMGB1在OM的实验模型(毛丝鼠宿主)中促进生物膜分辨率。在感染NTHI后第4天和第5天,将稀释剂或5μg rHMGB1或mHMGB1直接递送至毛丝鼠的中耳。24小时后处死动物,并根据(图39A)底部描述的标准对它们的中耳成像(图39A)并盲目评分(图39B)。条形代表SEM。*** P <0.001。图像和评分显示HMGB1促进了原位NTHI生物膜的清除。
图40A-40C:HMGB1破坏新洋葱伯克霍尔德菌(
图41:HMGB1将多胺诱导的Z-DNA还原为核酸酶敏感的B-DNA状态。将聚(dG-dC)底物与亚精胺和HMGB1一起温育。降解产物通过凝胶电泳可视化。 HMGB1处理可恢复核酸酶对亚精胺诱导的Z-DNA底物的敏感性。
图42A-42D:需要DNABII蛋白和多胺来挽救阳离子交换剂(P11)介导的生物膜预防。(图42A)NTHI生物膜的生长在Transwell板系统的基底外侧腔中于37℃ 5%CO
图43A-43B:B-DNA和Z-DNA抗体的特异性。(图43A)将溴化的基因组DNA(2 μg/mL)和聚dGdC在缓冲液中温育,测量260nm和295nm的吸光度值,并计算A260/295比。
图44:随着时间的推移,DNABII蛋白、多胺和eDNA(B-DNA和Z-DNA)稳定地积累在NTHI生物膜的EPS内。NTHI生物膜在不同形成阶段的免疫荧光CLSM图像,分别用抗B-DNA抗体(浅灰色;左起第一个)、抗抗亚精胺(浅灰色;左起第二个)、抗DNABII(灰色)探测;左起第三个)、或抗Z-DNA(白色;左起第四个)。随着时间的推移,成熟的生物膜在EPS内包含越来越多的每种TEDS成分。
图45:多胺和DNABII蛋白刺激Z-DNA,它对DNA酶具有抗性。通过将DNABII蛋白(HU
图46:TEDS和Z-DNA存在于由多种人类病原体形成的生物膜EPS中。(A)上图:用幼稚(naive)或抗HU(灰色)和抗亚精胺(Spd,浅灰色)抗体探测的所示生物膜的免疫荧光CLSM图像,共定位为白色。下图:使用幼稚或抗B-DNA(深灰色)和抗Z-DNA抗体(白色)探测的所示细菌形成40小时的生物膜的免疫荧光CLSM图像。将一种特征明确的Z-DNA特异性单克隆抗体(克隆Heydorn et al. (2002) COMSTAT. Microbiology; 146; Yang et al. (2017)Paediatr Respir Rev. 21:65-7; Xu et al. (2016) Molecules; 21(8))用于检测Z-DNA。DNABII、多胺、Z-DNA和B-DNA成分都存在于多种细菌生物膜的EPS中。Z-DNA是处于不同稳态水平的多个细菌生物膜EPS的组成部分。
图47A-47B:Z-DNA存在于体内和离体样品中。左:在(图47A)实验性NTHI诱导的OM中的NTHI生物膜和(图47B)囊性纤维化患者的痰液内的Z-DNA(白色)和B-DNA(深灰色)。插图,阴性对照。比例尺10μm。右:在(图47A)实验性NTHI诱导的OM中的NTHI生物膜和(图47B)囊性纤维化患者的痰液内的DNABII蛋白(灰色)和亚精胺(Spd,浅灰色)。插图,阴性对照。比例尺10μm。DNABII、多胺、Z-DNA和B-DNA组分都存在于被NTHI生物膜感染的毛丝鼠中耳部分。Z-DNA是处于不同稳态水平的多个细菌生物膜EPS的组成部分。
图48A-48B:生物膜中的Z-DNA形成耐核酸酶的支架。(图48A)将由所示细菌(Kp =肺炎克雷伯氏菌)形成的生物膜24小时与DNA酶(Pulmozyme®; 40 U/ml)一起温育另外16小时。然后用抗B-DNA(深灰色)和抗Z-DNA(白色)抗体探测生物膜,并用细菌细胞膜染色剂FM4-64
图49:Z-DNA稳定刺激生物膜形成。在幼稚或抗Z-DNA抗体(5 mg/ml)存在的情况下,启动NTHI和UPEC生物膜生长16小时。对生物膜进行染色,固定,通过CLSM进行可视化,并通过COMSTAT进行分析。条形代表SEM。通过配对t检验评估与对照组(无抗体)相比的统计学显著性,* P <0.05,** P <0.01。抗Z-DNA抗体以剂量依赖性方式刺激生物膜的形成,而幼稚型抗体则不会。
图50A-50B:B-DNA向Z-DNA转化的平衡转变改变了生物膜的形成。将形成24小时的NTHI生物膜与氯化铈(CeCl
图51A-51B:B-DNA向Z-DNA转化的平衡转变改变了生物膜的形成。将形成24小时的NTHI生物膜与氯化铈(CeCl
图52A-52B:RNA稳态调节细菌生物膜的发育。(图52A)在RNase A存在下开始NTHI生物膜生长16小时。对生物膜进行染色,固定,通过CLSM进行可视化,并通过COMSTAT进行分析。RNase A的添加以剂量依赖性方式刺激了生物膜的形成,可能是通过释放多胺而实现的,多胺是B-DNA向Z-DNA细胞外转化的关键催化剂。 (图52B)将形成16小时的NTHI生物膜与tRNA一起温育并按照(图52A)进行分析。鸟苷一磷酸(GMP)用作阴性对照。条形代表SEM。通过配对t检验评估与对照组(无RNase A或tRNA/GMP)相比的统计学显著性,* P <0.05,**P <0.01。tRNA(而非GMP)的添加以剂量依赖性方式破坏了较早的NTHI生物膜,这可能是由于从生物膜EPS中螯合了多胺。
图53:DNABII和多胺协同作用以将B-DNA转换为Z-DNA。在一方面,在不存在DNA酶的情况下施用试剂。左:将基因组DNA(2µg/mL)与亚精胺(spd:300 µM)、HU(500 nM)、spd和HU的混合物在37℃孵育2小时。将gDNA与3.6M NaCl一起孵育用作Z-DNA阳性对照。中:将gDNA(2 µg/mL)与增加浓度的氯化铈(CeCl
详细说明
除非另有定义,否则本文中使用的所有技术和科学的术语具有与本发明所属领域的普通技术人员通常所理解的相同含义。本文提供的所有核苷酸序列均以5'至3'方向呈现。尽管类似于或等同于本文描述的方法和材料的任何方法和材料都可以用于本公开的实践或测试中,但是现在描述特定的、非限制性的示例性方法、装置和材料。本文引用的所有技术和专利出版物均通过引用全文并入本文。本文中的任何内容均不得解释为承认本公开无权先于现有公开而早于该公开。
除非另有说明,否则本公开的实践将采用组织培养、免疫学、分子生物学、微生物学、细胞生物学和重组DNA的常规技术,它们在本领域技术范围内。参见例如Sambrook andRussell eds, (2001) Molecular Cloning: A Laboratory Manual, 3
所有数字值(包括范围),例如pH、温度、时间、浓度、以及分子质量,都是变化(+)或(-)0.1的增值的近似值,适当地或以+/− 15%、或10%或5%或2%变化。应当理解的是,尽管并非总是明确地说明,但所有数字值前面都有术语“约”。还应当理解的是,尽管并非总是明确说明,但本文所述的试剂仅是示例性的,并且其等效物在本领域中是已知的。
如说明书和权利要求书中所使用的,单数形式“一”、“一个”和“该/所述”包括复数引用,除非上下文另外明确指出。 例如,术语“多肽”包括多个多肽,包括其混合物。
如本文所用,术语“包含”或“包括”旨在表示包括所列举的要素但不排除其他要素的组合物和方法。当用于定义组合物和方法时,“基本上由……组成”应表示排除对于所述目的而言对组合具有任何实质意义的其他元素。因此,基本上由本文所定义的元素组成的组合物将不会从分离和纯化方法以及药学上可接受的载体(例如磷酸盐缓冲盐水、防腐剂等)中排除痕量污染物。“由……组成”是指排除其他成分和用于施用本文公开的组合物的实质性方法步骤的痕量元素。由这些过渡术语中的每一个限定的实施方案在本公开的范围内。
“生物膜”意指有时附着于可以是有机或无机的结构表面的微生物的有组织的群落,与它们分泌和/或释放的诸如DNA的聚合物一起。生物膜对微生物和抗菌剂具有很高的抵抗力。它们生活在牙龈组织、牙齿和修复体上,引起龋齿和牙周疾病(也称为牙周斑块病)。它们还会引起慢性中耳感染。生物膜也可以在牙科植入物、支架、导管和隐形眼镜的表面形成。它们在起搏器、心脏瓣膜替代物、人工关节和其他外科植入物上生长。疾病控制中心估计,超过65%的医院(医院获得性)感染是由生物膜引起的。它们会导致慢性阴道感染,并导致免疫系统低下的人危及生命的全身感染。生物膜也参与多种疾病。例如,囊性纤维化患者患有假单胞菌感染,通常导致抗生素耐药性生物膜形成。
术语“抑制、竞争或滴定”旨在减少作为微生物生物膜成分的DNA/蛋白质基质的形成。
“DNABII多肽或蛋白质”意指由DNA结合结构域组成并因此对微生物DNA具有特异性或一般性亲和力的DNA结合蛋白质或多肽。在一方面,它们在小沟中结合DNA。DNABII蛋白的非限制性实例是整合宿主因子(IHF)蛋白和来自大肠杆菌菌株U93(HU)的组蛋白样蛋白。可能与生物膜相关的其他DNA结合蛋白包括DPS(Genbank登录号:CAA49169)、H-NS(Genbank登录号:CAA47740)、Hfq(Genbank登录号:ACE63256)、CbpA(Genbank登录号:BAA03950)和CbpB(Genbank登录号:NP
“HF”蛋白的“整合宿主因子”是这样的细菌蛋白,其被噬菌体用于将其DNA掺入宿主细菌中。它们还结合细胞外微生物DNA。编码大肠杆菌中IHF蛋白亚基的基因是himA(Genbank登录号:POA6X7.1)和himD(POA6Y1.1)基因。这些基因的同源物在其他生物中发现,并且与来自其他生物的这些基因相对应的肽在本领域中公开,例如在美国专利号8,999,291的表10中。
“HMGB1”是高迁移率族盒(high mobility group box,HMGB)1蛋白,其据报道可结合并扭曲DNA的小沟,是试剂的一个例子。重组或分离的蛋白质和多肽可从Atgenglobal、ProSpecBio、Protein1和Abnova商购。HMGB1是由215个氨基酸组成的小蛋白(约30 Kda),由3个域组成:两个带正电荷的域A和B盒(box),每个由80个氨基酸组成;以及带负电荷的羰基末端,酸性C尾巴,由大约30个连续的天冬氨酸和谷氨酸残基组成。以下提供的是野生型HMGB1的多肽序列的非限制性实例:
MGKGDPKKPRRKMSSYAFFVQTCREEHKKKHPDASVNFSEFSKKCSERWKTMSAKEKGKFEDMAKADKARYEREMKTYI_PPKGETKKKF_
粗体氨基酸(氨基酸1-70)描述了A Box结构域。
斜体氨基酸(氨基酸88-164)描述了B Box结构域。
带下划线的氨基酸(氨基酸186-215)描述了C-尾结构域。这些是片段的非限制性实例,例如A Box结构域、B Box结构域、A和B box结构域(AB box结构域)、C尾结构域和N结构域(氨基酸1-185) 。在一方面,片段基本上由C末端结构域或包含B Box结构域的多肽组成。
“HU”或“来自大肠杆菌菌株U93的组蛋白样蛋白”是指通常与大肠杆菌结合的一类异二聚体蛋白。已知HU蛋白会结合DNA汇合点。相关蛋白已从其他微生物中分离出来。Laineet al. (1980) Eur. J. Biochem 103(3)447-481报道了大肠杆菌HU的完整氨基酸序列。HU蛋白的抗体可从Abeam商购。
术语“表面抗原”或“表面蛋白质”是指细胞(如细菌细胞)表面上的蛋白质或肽。表面抗原的实例是外膜蛋白,例如OMP P5(Genbank登录号:YP
术语“流感嗜血杆菌”是指可以引起许多不同感染(例如耳部感染、眼部感染和鼻窦炎)的病原细菌。已分离出许多不同的流感嗜血杆菌菌株,并具有IhfA基因或蛋白质。不同流感嗜血杆菌菌株的一些非限制性实例包括Rd KW20、86-028NP、R2866、PittGG、PittEE、R2846和2019。
“微生物DNA”意指来自产生生物膜的微生物的单链或双链DNA。
“抑制、预防或破坏生物膜”旨在预防或治疗性减少生物膜的结构。
“弯曲的多核苷酸”意指这样的双链多核苷酸,其在一个链上包含小环,该环不与另一条链配对。在一些实施方案中,所述环的长度为1个碱基至约20个碱基,或替代地为2个碱基至约15个碱基长,或替代地为约3个碱基至约12个碱基长,或替代地为约4个碱基至约10个碱基长、或替代地具有约4、5、或6、或7、或8、9、或10个碱基。
诊断或治疗的“对象”是细胞或动物,例如哺乳动物或人类。接受诊断或治疗的非人类动物是经受感染或动物模型的动物,例如猿猴、鼠类(例如大鼠、小鼠、毛丝鼠、犬类(例如狗)、类脂动物(例如兔子)、家畜、运动动物和宠物。术语“对象”、“宿主”、“个体”和“患者”在本文中可互换使用,是指动物,通常是哺乳动物。哺乳动物的非限制性示例包括人类、非人类灵长类动物(例如猿、长臂猿、黑猩猩、猩猩、猴子、猕猴等)、家畜(例如狗和猫)、农场动物(例如马、牛、山羊、绵羊、猪)和实验动物(例如小鼠、大鼠、兔子、豚鼠)。在一些实施方案中,哺乳动物是人。哺乳动物可以是任何年龄或处于任何发育阶段(例如成年、青少年、儿童、婴儿或子宫内的哺乳动物)。哺乳动物可以是雄性或雌性。在一些实施方案中,对象是人。
术语“蛋白质”、“肽”和“多肽”可互换使用,并且在其最广义上是指两个或更多个亚基氨基酸、氨基酸类似物或拟肽的化合物。亚基可以通过肽键连接。在另一个实施方案中,亚基可以通过其他键连接,例如酯、醚等。蛋白质或肽必须包含至少两个氨基酸,并且对可以构成蛋白质或肽序列的最大氨基酸数没有限制。如本文所用,术语“氨基酸”是指天然和/或非天然或合成的氨基酸,包括甘氨酸以及D和L旋光异构体、氨基酸类似物和拟肽。
术语“多核苷酸”和“寡核苷酸”可互换使用,并且是指任何长度的核苷酸的聚合形式,所述核苷酸是脱氧核糖核苷酸或核糖核苷酸或其类似物。多核苷酸可以具有任何三维结构,并且可以执行任何已知或未知的功能。以下是多核苷酸的非限制性示例:基因或基因片段(例如探针、引物、EST或SAGE标签)、外显子、内含子、信使RNA(mRNA)、转移RNA、核糖体RNA、RNAi、核酶、cDNA、重组多核苷酸、分支多核苷酸、质粒、载体、任何序列的分离DNA、任何序列的分离RNA、核酸探针和引物。多核苷酸可以包含修饰的核苷酸,例如甲基化的核苷酸和核苷酸类似物。如果存在,则可以在多核苷酸组装之前或之后赋予核苷酸结构修饰。核苷酸的序列可以被非核苷酸成分打断。多核苷酸可在聚合后进一步修饰,例如通过与标记组分缀合。该术语还指双链和单链分子。除非另有说明或要求,否则本文公开的为多核苷酸的任何实施方案既包括双链形式,也包括已知或预测组成双链形式的两个互补单链形式的每一个。
多核苷酸由四个核苷酸碱基的特定序列组成:腺嘌呤(A);胞嘧啶(C);鸟嘌呤(G);胸腺嘧啶(T);当多核苷酸为RNA时,胸腺嘧啶为尿嘧啶(U)。因此,术语“多核苷酸序列”是多核苷酸分子的字母表示。可以将该字母表示形式输入到具有中央处理单元的计算机中的数据库中,并用于生物信息学应用,例如功能基因组学和同源性搜索。
如本文关于核酸(例如DNA或RNA)所用,术语“分离的”或“重组的”是指分别与其他DNA或RNA分离的分子,其存在于大分子以及多肽的天然来源中。术语“分离的或重组的核酸”是指包括并非天然存在为片段并且不会以天然状态发现的核酸片段。术语“分离的”在本文中也用于指从其他细胞蛋白分离的多核苷酸、多肽和蛋白,并且意在包括纯化的和重组的多肽。在其他实施方案中,术语“分离的或重组的”是指与成分、细胞或其他分离,其中细胞、组织、多核苷酸、肽、多肽、蛋白质、抗体或其片段通常在自然界中是相关联的。例如,分离的细胞是与具有不同表型或基因型的组织或细胞分离的细胞。分离的多核苷酸与3'和5'连续核苷酸分离,在其天然或天然环境(例如在染色体上)通常与其是相关联的。对本领域技术人员显而易见的是,非天然存在的多核苷酸、肽、多肽、蛋白质、抗体或其片段不需要“分离”即可将其与天然存在的对应物区分开。
可以推断出,没有明确的叙述,除非另有说明,否则当本公开内容涉及多肽、蛋白质、多核苷酸或抗体时,其等效物或生物学等效物应在本公开内容的范围内。如本文所用,术语“其生物等效物”在指代参考蛋白、抗体、片段、多肽或核酸时旨在与“其等效物”同义,意指具有最小同源性而仍保持所需结构或功能的那些。除非本文具体叙述,否则预期本文提及的任何多核苷酸、多肽或蛋白质也包括其等效物。在一方面,等效的多核苷酸是在严格条件下与本文所述的多核苷酸或多核苷酸的互补序列杂交以用于所述方法的核苷酸。在另一方面,等效的抗体或抗原结合多肽是指以至少70%、或至少75%、或至少80%、或至少85%、或至少90%、或至少为95%亲和力或更高的亲和力与参照抗体或抗原结合片段结合的那些。在另一方面,其等效物在竞争性ELISA分析中与抗体或抗原结合片段与其抗原的结合竞争。在另一方面,等效物意指至少约80%的同源性或同一性,并且替代地至少约85%,或替代地至少约90%,或替代地至少约95%,或98%的百分比同源性或同一性并表现出与参照蛋白、多肽或核酸基本相等的生物学活性。
多核苷酸或多核苷酸区域(或多肽或多肽区域)与另一序列具有一定百分比(例如80%、85%、90%或95%)的“序列同一性”是指,当比对时,在比较两个序列时该百分比的碱基(或氨基酸)是相同的。可以使用本领域已知的软件程序来确定比对和同源性或序列同一性百分数,例如在Current Protocols in Molecular Biology (Ausubel et al., eds.1987) Supplement 30, section 7.7.18, Table 7.7.1中描述的那些程序。在某些实施例中,使用默认参数进行比对。非限制性示例性比对程序是BLAST,使用默认参数。特别是,示例性的程序包括BLASTN和BLASTP,使用以下默认参数:遗传密码=标准;筛选=无;链=两个;截点=60;预期=10;矩阵=BLOSUM62;描述=50个序列;排序= HIGH SCORE;数据库=不重复的,GenBank+EMBL+DDBJ+PDB+GenBank CDS translations+SwissProtein+SPupdate+PIR。这些程序的详情可以在以下网址找到:ncbi.nlm.nih.gov/cgi-bin/BLAST。序列同一性和同一性百分比是通过将它们合并到clustalW(可在网址:align.genome.jp获取,上次访问于2011年3月7日)中确定的。
“同源性”或“同一性”或“相似性”是指两个肽之间或两个核酸分子之间的序列相似性。同源性可以通过比较每个序列中的位置来确定,所述位置可以为了比较的目的而被比对。当比较序列中的一个位置被相同的碱基或氨基酸占据时,则分子在该位置是同源的。序列之间的同源性程度是序列共享的匹配或同源位置数目的函数。“无关”或“非同源”序列与本公开的序列之一共享小于40%的同一性、或替代地小于25%的同一性。
“同源性”或“同一性”或“相似性”还可以指在严格条件下杂交的两个核酸分子。
“杂交”是指一个或多个多核苷酸反应形成复合物的反应,该复合物通过核苷酸残基的碱基之间的氢键稳定。氢键可通过Watson-Crick碱基配对、Hoogstein结合或以任何其他序列特异性方式发生。该复合物可以包括形成双链体结构的两条链、形成多链复合物的三条或更多条链、单一的自杂交链、或它们的任何组合。杂交反应可以构成更广泛过程中的一个步骤,例如PCR反应的启动、或核酶对多核苷酸的酶促切割。
严格杂交条件的例子包括:约25℃至约37℃的孵育温度;约6×SSC至约10×SSC的杂交缓冲液浓度;约0%至约25%的甲酰胺浓度;以及约4×SSC至约8×SSC的洗涤溶液。中度杂交条件的例子包括:约40℃至约50℃的孵育温度;约9×SSC至约2×SSC的缓冲液浓度;约30%至约50%的甲酰胺浓度;以及约5×SSC至约2×SSC的洗涤溶液。高严格杂交条件的例子包括:约55℃至约68℃的孵育温度;约1×SSC至约0.1×SSC的缓冲液浓度;约55%至约75%的甲酰胺浓度;以及约1×SSC至约0.1×SSC的洗涤溶液、或去离子水。一般来说,杂交孵育时间为5分钟至24小时,具有1、2或更多个洗涤步骤,并且洗涤孵育时间为约1、2或15分钟。SSC是0.15 M NaCl和15 mM柠檬酸缓冲液。应该理解的是,可以采用使用其他缓冲系统的SSC的等效物。
如本文所用,“表达”是指多核苷酸被转录成mRNA的过程和/或转录的mRNA随后被翻译成肽、多肽或蛋白质的过程。如果多核苷酸衍生自基因组DNA,则表达可包括在真核细胞中剪接mRNA。
当应用于多核苷酸时,术语“编码”是指被说成“编码”多肽的多核苷酸在其天然状态或通过本领域技术人员公知的方法操作时,可以被转录和/或翻译以产生多肽和/或其片段的mRNA。反义链是这种核酸的互补物,并且可以从其推导编码序列。
如本文所用,术语“治疗”等在本文中用于意指获得期望的药理和/或生理作用。就部分或完全治愈疾病和/或归因于该疾病的不良影响而言,该作用可以是治疗性的。如本文所用,对象中疾病的“治疗”还可以指(1)防止症状或疾病在易患或尚未表现出疾病症状的对象中发生;(2)抑制疾病或阻止其发展;或(3)改善或导致疾病或疾病症状的消退。如本领域中所理解的,“治疗”是用于获得有益的或期望的结果(包括临床结果)的方法。为了本技术的目的,有益的或期望的结果可以包括以下中的一种或多种但不限于:减轻或改善一种或多种症状,减轻病症(包括疾病)的程度,稳定(即不恶化)病症(包括疾病)的状态,病症(包括疾病)的延缓或减慢,病症(包括疾病)的进展、改善或减轻,状态和缓解(无论是部分还是全部),无论是可检测的还是不可检测的。在一方面,治疗排除了预防。当疾病是SLE(系统性红斑狼疮)和/或囊性纤维化(CF)时,治疗的证据包括炎症的证据和/或自身免疫活性或症状的水平减少。
预防旨在预防易患疾病或效应的系统或对象体内或体外的疾病或效应。这样的一个例子是防止在被已知会产生生物膜的微生物感染的系统中形成生物膜。
“组合物”意指活性剂与另一种化合物或组合物的组合,该另一种化合物或组合物是惰性的,例如可检测的试剂或标记,或是活性的,例如佐剂、稀释剂、粘合剂、稳定剂、缓冲剂、盐、亲脂性溶剂、防腐剂、佐剂等,并包括药学上可接受的载体。载体还包括药物赋形剂和添加剂蛋白质、肽、氨基酸、脂质和碳水化合物(例如糖,包括单糖、二寡糖、三寡糖、四寡糖和寡糖;衍生糖,例如醛醇、醛糖酸、酯化糖等;以及多糖或糖聚合物),其可以单独地或组合地存在,以重量或体积计单独地或组合地 1-99.99%。示例性蛋白质赋形剂包括血清白蛋白(例如人血清白蛋白(HSA)、重组人白蛋白(rHA))、明胶、酪蛋白等。还以缓冲能力起作用的代表性的氨基酸/抗体组分包括丙氨酸、精氨酸、甘氨酸、精氨酸、甜菜碱、组氨酸、谷氨酸、天冬氨酸、半胱氨酸、赖氨酸、亮氨酸、异亮氨酸、缬氨酸、甲硫氨酸、苯丙氨酸、阿斯巴甜等。碳水化合物赋形剂也旨在位于在本技术的范围之内,其例子包括但不限于:单糖,例如果糖、麦芽糖、半乳糖、葡萄糖、D-甘露糖、山梨糖等;二糖,例如乳糖、蔗糖、海藻糖、纤维二糖等;多糖,例如棉子糖、松三糖、麦芽糖糊精、葡聚糖、淀粉等;以及糖醇,例如甘露醇、木糖醇、麦芽糖醇、乳糖醇、木糖醇山梨醇(葡萄糖醇)和肌醇。
“药物组合物”旨在包括活性剂与惰性或活性载体的组合,使得该组合物适合于体外、体内或离体的诊断或治疗用途。
“药学上可接受的载体”是指可以在本文公开的组合物中使用的任何稀释剂、赋形剂或载体。药学上可接受的载体包括:离子交换剂、氧化铝、硬脂酸铝、卵磷脂、血清蛋白(例如人血清白蛋白)、缓冲物质(例如磷酸盐、甘氨酸、山梨酸、山梨酸钾)、饱和植物脂肪酸的部分甘油酯混合物、水、盐或电解质(例如硫酸鱼精蛋白)、磷酸氢二钠、磷酸氢钾、氯化钠、锌盐、胶体二氧化硅、三硅酸镁、聚乙烯吡咯烷酮,纤维素基物质、聚乙二醇、羧甲基纤维素钠、聚丙烯酸酯、蜡、聚乙烯-聚氧丙烯-嵌段聚合物、聚乙二醇和羊毛脂。合适的药物载体在该领域的标准参考书Remington's Pharmaceutical Sciences, Mack PublishingCompany中有描述。可以根据预期的给药形式,即口服片剂、胶囊剂、酏剂、糖浆剂等来选择它们,并且与常规药学实践一致。
根据本发明使用的组合物可以以剂量单位形式包装,以易于施用和剂量均匀。术语“单位剂量”或“剂量”是指适合在对象中使用的物理上离散的单位,每个单位包含预定量的组合物,其经计算以与其施用相关地产生期望的应答,即适当的途径和方案。根据治疗次数和单位剂量的给药量取决于结果和/或所需的保护。组合物的精确量还取决于从业者的判断,并且对于每个人都是独特的。影响剂量的因素包括对象的身体和临床状态、给药途径、预期的治疗目标(缓解症状与治愈)以及特定组合物的效力、稳定性和毒性。在配制后,以与剂量制剂相容的方式和以治疗或以预防有效量的量施用溶液。容易以各种剂型(例如本文所述的可注射溶液的类型)来施用制剂。
术语“接触”是指两个或更多个之间的直接或间接结合或相互作用。直接相互作用的一个特定示例是结合。间接相互作用的一个特定示例是一个实体作用于中间分子,而中间分子又作用于第二个所引用的实体。本文所用的接触包括在溶液中、在固相中、体外、离体、细胞和体内。体内接触可以称为给药或施用。
本文公开的“生物活性剂”或活性剂旨在表示以下中的一种或多种:分离的或重组的多肽、分离的或重组的多核苷酸、载体、分离的宿主细胞、或抗体、以及包含其一种或多种的组合物。
“施用/给药”可以在整个治疗过程中以一剂连续或间歇地进行。确定最有效的给药方式和剂量的方法是本领域技术人员已知的,并且将根据用于治疗的组合物、治疗的目的、所治疗的靶细胞和所治疗的对象而变化。可以通过治疗医师选择剂量水平和方式进行单次或多次给药。合适的剂型和施用试剂的方法是本领域已知的。也可以确定给药途径,并且确定最有效给药途径的方法是本领域技术人员已知的,并且将根据用于治疗的组合物、治疗目的、被治疗的对象的健康状况或疾病阶段、以及靶细胞或组织而变化。施用途径的非限制性实例包括口服施用、鼻腔施用、注射和局部施用。
本公开的试剂/药剂可以通过任何合适的施用途径施用以进行治疗。还应当理解,最佳途径将随着对象的状况和年龄以及所治疗的疾病而变化。
术语“有效量”是指足以实现期望效果的量。在治疗或预防应用的情况下,有效量将取决于所讨论病症的类型和严重性以及个体对象的特征,例如总体健康、年龄、性别、体重和对药物组合物的耐受性。在免疫原性组合物的情况下,在一些实施方案中,有效量是足以引起针对病原体的保护性应答的量。在其他实施方案中,免疫原性组合物的有效量是足以导致产生针对抗原的抗体的量。在一些实施方案中,有效量是赋予有需要的对象被动免疫所需的量。关于免疫原性组合物,在一些实施方案中,除了上述因素之外,有效量将取决于预期用途、特定抗原性化合物的免疫原性程度和对象免疫系统的健康/应答性。技术人员将能够根据这些和其他因素确定适当的量。
在体外应用的情况下,在一些实施方案中,有效量将取决于所讨论的应用的大小和性质。它还将取决于体外靶标的性质和敏感性以及所使用的方法。本领域技术人员将能够基于这些和其他考虑因素确定有效量。根据实施方案,有效量可以包括组合物的一次或多次施用。
“肽缀合物”是指通过一个或多个多肽与另一种化学或生物化合物的共价或非共价键结合。在非限制性实例中,多肽与化合物的“缀合”导致多肽出于其预期目的的改善的稳定性或功效。 在一个实施方案中,将肽缀合至载体,其中所述载体是脂质体、胶束或药学上可接受的聚合物。
“脂质体”是由同心脂质双层构成的微观囊泡。从结构上讲,脂质体的大小和形状范围从长管到球形,尺寸范围从几百埃到几分之一毫米。选择形成囊泡的脂质以实现最终复合物的指定程度的流动性或刚性,从而提供外层的脂质组合物。这些是中性的(胆固醇)或双极性的,包括磷脂,例如磷脂酰胆碱(PC)、磷脂酰乙醇胺(PE)、磷脂酰肌醇(PI)和鞘磷脂(SM)和其他类型的双极性脂质,包括但不限于二醇油酰磷脂酰乙醇胺(DOPE),其烃链长度在14-22范围内,且是饱和的或具有一个或多个双C═C键。能够单独或与其他脂质组分组合产生稳定脂质体的脂质的实例是磷脂,例如氢化大豆磷脂酰胆碱(HSPC)、卵磷脂、磷脂酰乙醇胺、溶血卵磷脂、溶血磷脂酰乙醇胺、磷脂酰丝氨酸、磷脂酰肌醇、鞘磷脂、脑磷脂、心磷脂、磷脂酸、脑苷脂、二硬脂酰磷脂酰胆碱(DSPE)、二油酰磷脂酰胆碱(DOPC)、二棕榈酰磷脂酰胆碱(DPPC)、棕榈酰脂酰磷脂酰胆碱(POPC)、棕榈酰油酰磷脂酰乙醇胺(POPE)和四(N-马来酰亚胺基-三乙基)环己烷-1-羧酸二油酰基磷脂酰乙醇胺(POPE-mal)。可以掺入脂质体的其他非磷脂质包括硬脂胺、十二烷基胺、十六烷基胺、肉豆蔻酸异丙酯、三乙醇胺-十二烷基硫酸盐、烷基芳基硫酸盐、乙酰棕榈酸酯、甘油蓖麻油酸酯、十六烷基硬脂酸酯、两性丙烯酸聚合物、聚乙氧基化脂肪酸酰胺、以及上面提到的阳离子脂质(DDAB、DODAC、DMRIE、DMTAP、DOGS、DOTAP(DOTMA)、DOSPA、DPTAP、DSTAP、DC-Chol)。带负电荷的脂质包括能够形成囊泡的磷脂酸(PA)、二棕榈酰磷脂酰甘油(DPPG)、二棕榈酰磷脂酰甘油和(DOPG)、鲸蜡二磷酸酯。通常,脂质体可根据其整体大小和层状结构的性质分为三类。 1977年12月,由纽约科学院科学会议制定的“脂质体及其在生物学和医学中的用途”中这三个分类是多层囊泡(MLV)、小型单层囊泡(SUV)和大型单层囊泡(LUV)。可以将生物活性剂封装在其中以根据本文所述的方法施用。
“胶束”是分散在液体胶体中的表面活性剂分子的聚集体。水溶液中的典型胶束形成与周围溶剂接触的亲水性“头部”区域的聚集体,将胶束中心的疏水性尾部区域隔离。这种胶束称为正相胶束(水包油胶束)。反胶束的头组在中心,而尾巴则伸出(油包水胶束)。胶束可用于附着本文所述的多核苷酸、多肽、抗体或组合物,以促进有效递送至靶细胞或组织。
短语“药学上可接受的聚合物”是指可以与本文所述的一种或多种多肽缀合的一组化合物。 预期聚合物与多肽的缀合能够在体内和体外延长多肽的半衰期。非限制性实例包括聚乙二醇、聚乙烯基吡咯烷酮、聚乙烯醇、纤维素衍生物、聚丙烯酸酯、聚甲基丙烯酸酯、糖、多元醇及其混合物。可以根据本文描述的方法将生物活性剂缀合至药学上可接受的聚合物以进行施用。
“基因递送载体”定义为可以将插入的多核苷酸携带到宿主细胞中的任何分子。基因传递载体的例子是:脂质体,胶束生物相容性聚合物,包括天然聚合物和合成聚合物;脂蛋白;多肽;多糖;脂多糖;人工病毒包膜;金属颗粒;以及细菌或病毒,例如杆状病毒、腺病毒和逆转录病毒、噬菌体、粘粒、质粒、真菌载体和本领域通常使用的其他重组载体,其已被描述用于在各种真核和原核宿主中表达,并可以用于基因治疗以及简单的蛋白质表达。
可使用基因递送载体将本文公开的多核苷酸递送至细胞或组织。如本文所用,“基因递送”、“基因转移”、“转导”等是指将外源多核苷酸(有时称为“转基因”)引入宿主细胞中的术语,而与引入的方法无关。此类方法包括多种众所周知的技术,例如载体介导的基因转移(例如,通过病毒感染/转染,或各种其他基于蛋白质或基于脂质的基因递送复合物),以及促进“裸”多核苷酸递送的技术(例如电穿孔,“基因枪”递送以及用于引入多核苷酸的各种其他技术)。所引入的多核苷酸可以在宿主细胞中稳定或瞬时保持。稳定的保持通常要求引入的多核苷酸包含与宿主细胞相容的复制起点或整合到宿主细胞的复制子中,例如染色体外复制子(例如质粒)或核或线粒体染色体。如本领域已知和本文所述,已知许多载体能够介导基因向哺乳动物细胞的转移。
如本文所用,术语“eDNA”是指被发现为病原性生物膜的组分的细胞外DNA。
“质粒”是与染色体DNA分离的染色体外DNA分子,其能够独立于染色体DNA复制。在许多情况下,它是圆形的和双链的。质粒提供了在微生物种群内水平基因转移的机制,并且通常在给定的环境状态下提供选择优势。质粒可以携带在竞争性环境中对天然存在的抗生素具有抗性的基因,或者在相似情况下,产生的蛋白质可以充当毒素。
基因工程中使用的“质粒”被称为“质粒载体”。许多质粒可用商业地于此类用途。将要复制的基因插入质粒的拷贝中,该质粒含有使细胞对特定抗生素具有抗性的基因和多个克隆位点(MCS或多接头),其是一个短区域,包含几个常用的限制性酶切位点,可轻松插入DNA 片段在此位置。质粒的另一主要用途是制备大量蛋白质。在这种情况下,研究人员可以使细菌生长,该细菌含有带有目的基因的质粒。正如细菌产生赋予其抗生素抗性的蛋白质一样,它也可以被诱导从插入的基因中产生大量蛋白质。这是大量生产一种基因或其随后编码的蛋白质的廉价而简便的方法。
“酵母人工染色体”或“ YAC”是指用于克隆大DNA片段(大于100 kb且最大3000kb)的载体。它是人工构建的染色体,包含在酵母细胞中复制和保存所需的端粒、着丝粒和复制起点序列。使用最初的环状质粒构建,然后使用限制酶将它们线性化,然后DNA连接酶可以通过使用粘性末端在线性分子内添加目标序列或基因。酵母表达载体,例如YAC、YIp(酵母整合质粒)和YEp(酵母游离质粒),非常有用,因为可以得到具有翻译后修饰的真核蛋白产物,因为酵母本身就是真核细胞,但是已经发现YAC是 比BAC更不稳定,产生嵌合效应。
“病毒载体”定义为重组产生的病毒或病毒颗粒,其包含待体内、离体或体外递送至宿主细胞的多核苷酸。病毒载体的实例包括逆转录病毒载体、腺病毒载体、腺相关病毒载体、α病毒载体等。基于传染性烟草花叶病毒(TMV)的载体可用于制造蛋白质,并且据报道在烟草叶中表达Griffithsin (O'Keefe et al. (2009) Proc. Nat. Acad. Sci. USA 106(15):6099-6104)。α病毒载体,例如基于Semliki Forest病毒的载体和基于Sindbis病毒的载体,也已被开发用于基因治疗和免疫治疗。参见Schlesinger & Dubensky (1999) Curr.Opin. Biotechnol. 5:434-439以及Ying et al. (1999) Nat. Med. 5(7):823-827。在逆转录病毒载体介导基因转移的方面,载体构建体是指包含逆转录病毒基因组或其一部分和治疗基因的多核苷酸。
如本文所用,“逆转录病毒介导的基因转移”或“逆转录病毒转导”具有相同的含义,并且是指借助于病毒进入细胞并将其基因组整合到宿主细胞基因组中而将基因或核酸序列稳定地转移到宿主细胞中的过程。病毒可以通过其正常的感染机制进入宿主细胞,也可以对其进行修饰,使其与不同的宿主细胞表面受体或配体结合以进入细胞。如本文所用,逆转录病毒载体是指能够通过病毒或类病毒进入机制将外源核酸引入细胞的病毒颗粒。
逆转录病毒以RNA形式携带其遗传信息;但是,一旦病毒感染细胞,RNA就会逆转录为DNA形式,其整合到被感染细胞的基因组DNA中。整合的DNA形式称为原病毒。
在通过DNA病毒载体(例如腺病毒(Ad)或腺相关病毒(AAV))介导基因转移的方面,载体构建体是指包含病毒基因组或其一部分和转基因的多核苷酸 。腺病毒(Ad)是表征相对较完善的同质病毒,包括超过50种血清型。参见例如PCT国际申请公开号WO 95/27071。Ad不需要整合到宿主细胞基因组中。还构建了重组Ad衍生载体,特别是减少重组可能性和产生野生型病毒的载体。参见PCT国际申请公开号WO 95/00655和WO 95/11984,野生型AAV具有整合到宿主细胞基因组中的高感染性和特异性。 参见Hermonat & Muzyczka (1984)Proc. Natl. Acad. Sci. USA 81:6466-6470 和Lebkowski et al. (1988) Mol. Cell.Biol. 8:3988-3996。
既包含启动子又包含可将多核苷酸可操作地连接至的克隆位点的载体是本领域众所周知的。这样的载体能够在体外或体内转录RNA,并且可以从诸如Stratagene (LaJolla, Calif.) 和Promega Biotech (Madison, Wis.)的来源商购获得。为了优化表达和/或体外转录,可能有必要去除、添加或改变克隆的5'和/或3'非翻译部分,以消除可能干扰的额外的、潜在的不适当的替代翻译起始密码子或在转录或翻译水平干扰或减少表达的其他序列。或者,可以在起始密码子的5'处紧接着插入共有核糖体结合位点以增强表达。
基因递送载体还包括DNA/脂质体复合物、胶束和靶向病毒蛋白-DNA复合物。同样包含靶向抗体或其片段的脂质体可用于本文公开的方法中。除了将多核苷酸递送至细胞或细胞群外,本文所述的蛋白质直接导入细胞或细胞群中还可以通过蛋白质转染的非限制性技术来完成,或者通过培养条件来增强表达和/或或促进本文公开的蛋白质的活性是其他非限制性技术。
如本文所用,术语“抗体”和“免疫球蛋白”包括完整的抗体及其任何抗原结合片段或其单链。因此,术语“抗体”包括包含至少一部分免疫球蛋白分子的任何蛋白质或肽分子。术语“抗体”和“免疫球蛋白”还包括任何同种型的免疫球蛋白、保留与抗原特异性结合的抗体片段,包括但不限于Fab、Fab'、F(ab)2、Fv、scFv、dsFv、Fd片段、dAb、VH、VL、VhH和V-NAR域;小体、双体、三体、四体和κ体;由抗体片段形成的一种或多种分离的多特异性抗体片段。这样的例子包括但不限于重链或轻链或其配体结合部分的互补决定区(CDR)、重链或轻链可变区、重链或轻链恒定区、框架(FR)区或其任何部分,结合蛋白、嵌合抗体、人源化抗体、单链抗体和包含抗体的抗原结合部分和非抗体蛋白的融合蛋白的至少一部分。免疫球蛋白分子的重链和轻链的可变区包含与抗原相互作用的结合域。抗体(Ab)的恒定区可以介导免疫球蛋白与宿主组织的结合。当在蛋白质名称(例如抗DNABII、抗IHF、抗HU、抗OMP P5)之前使用时,术语“抗”是指与特定蛋白质结合和/或具有亲和力的单克隆或多克隆抗体 。例如,“抗IHF”是指与IHF蛋白结合的抗体。特异性抗体可以与针对其产生抗性的蛋白质以外的蛋白质具有亲和力或结合。例如,抗IHF虽然针对IHF蛋白特异性产生,但也可以结合通过序列同源性或通过结构同源性相关的其他蛋白。
抗体可以是多克隆抗体、单克隆抗体、多特异性抗体(例如双特异性抗体)和抗体片段,只要它们表现出所需的生物学活性即可。可以从任何合适的生物学来源(例如鼠、大鼠、绵羊和犬)中分离出抗体。
如本文所用,“单克隆抗体”是指从基本上同质的抗体群体获得的抗体。单克隆抗体具有高度特异性,因为每种单克隆抗体都针对抗原上的单个决定簇。可以用例如放射性同位素、产生可检测产物的酶、荧光蛋白等可检测地标记抗体。抗体可以进一步与其他部分偶联,例如特异性结合对的成员,例如生物素(生物素-亲和素特异性结合对的成员)等。抗体也可以结合到固相支持物上,包括但不限于聚苯乙烯板或珠等。
单克隆抗体可以使用本领域已知的杂交瘤技术或重组DNA方法产生。杂交瘤是在实验室中由产生抗体的淋巴细胞与非抗体产生的癌细胞(通常是骨髓瘤或淋巴瘤)融合产生的细胞。杂交瘤增殖并产生特定单克隆抗体的连续样品。产生或选择抗体的替代技术包括将淋巴细胞体外暴露于目标抗原,以及在细胞、噬菌体或类似系统中筛选抗体展示文库。
如本文所用,术语“人抗体”旨在包括具有源自人种系免疫球蛋白序列的可变区和恒定区的抗体。本文公开的人抗体可包括未由人种系免疫球蛋白序列(例如,由体外随机或位点特异性诱变或体内体细胞突变引入的突变)编码的氨基酸残基。然而,本文所用的术语“人抗体”不意图包括其中衍生自另一哺乳动物物种(例如小鼠)的种系的CDR序列已嫁接到人框架序列上的抗体。因此,如本文所用,术语“人抗体”是指这样的蛋白质,其中基本上每个蛋白质部分(例如CDR、构架、C
如本文所用,如果抗体是从使用人免疫球蛋白序列的系统获得的,例如通过免疫携带人免疫球蛋白基因的转基因小鼠或通过筛选人免疫球蛋白基因文库,则人抗体是“衍生自”特定种系序列。可以通过将人抗体的氨基酸序列与人种系免疫球蛋白的氨基酸序列进行比较来鉴定“衍生自”人种系免疫球蛋白序列的人抗体。所选择的人抗体通常在氨基酸序列上与人种系免疫球蛋白基因编码的氨基酸序列具有至少90%的同一性,并且含有与其他物种(例如鼠种系序列)的种系免疫球蛋白的氨基酸序列相比鉴定该人抗体为人源的氨基酸残基。在某些情况下,人抗体与种系免疫球蛋白基因编码的氨基酸序列可以在氨基酸序列上至少95%、或甚至至少96%、97%、98%或99%相同。通常,衍生自特定人种系序列的人抗体与由人种系免疫球蛋白基因编码的氨基酸序列所显示的氨基酸差异不超过10个氨基酸。在某些情况下,人抗体与种系免疫球蛋白基因编码的氨基酸序列的氨基酸差异不超过5个、甚至不超过4、3、2或1个氨基酸。
“人单克隆抗体”是指显示单一结合特异性的抗体,其具有衍生自人种系免疫球蛋白序列的可变区和恒定区。该术语还意指重组人抗体。本文描述了制备这些抗体的方法。
如本文所用,术语“重组人抗体”包括通过重组方式制备、表达、产生或分离的所有人抗体,例如:从用于人免疫球蛋白基因的转基因或转染色体动物(例如小鼠)分离的抗体或由其制备的杂交瘤,从被转化以表达抗体(例如从转染瘤中表达)的宿主细胞分离的抗体,从重组的组合人抗体文库分离的抗体,以及通过涉及将人免疫球蛋白基因序列剪接至其他DNA序列的任何方法制备、表达、产生或分离的抗体。此类重组人抗体具有源自人种系免疫球蛋白序列的可变区和恒定区。然而,在某些实施方案中,可以对此类重组人抗体进行体外诱变(或,当使用针对人Ig序列转基因的动物时,进行体内体细胞诱变),并因此,重组抗体的VH和VL区的氨基酸序列是这样的序列,尽管这些序列衍生自人类种系VH和VL并与之相关,但在体内人类抗体种系库中可能不自然存在。本文描述了制备这些抗体的方法。
如本文所用,嵌合抗体是通常通过遗传工程从属于不同物种的抗体可变区和恒定区基因构建的轻链和重链基因的抗体。
如本文所用,术语“人源化抗体”或“人源化免疫球蛋白”是指含有源自非人免疫球蛋白的最小序列的人/非人嵌合抗体。在大多数情况下,人源化抗体是人免疫球蛋白(受体抗体),其中来自受体可变区的残基被来自非人类物种(供体抗体)(例如小鼠、大鼠、兔、或具有所需特异性、亲和力和能力的非人灵长类动物)的残基取代。人源化抗体可包含在受体抗体或供体抗体中找不到的残基。人源化抗体还可任选地包含免疫球蛋白恒定区(Fc)的至少一部分,通常是人免疫球蛋白、在构架区(恒定区或CDR)中包含已经被来自人抗体的相应定位的氨基酸取代的一个或多个氨基酸的非人抗体的至少一部分。通常,与相同抗体的非人源化形式相比,预期人源化抗体在人宿主中产生降低的免疫反应。人源化抗体可以具有保守的氨基酸取代,其对抗原结合或其他抗体功能基本上没有影响。保守取代组合包括:甘氨酸-丙氨酸,缬氨酸-亮氨酸-异亮氨酸,苯丙氨酸-酪氨酸,赖氨酸-精氨酸,丙氨酸-缬氨酸,丝氨酸-苏氨酸和天冬酰胺-谷氨酰胺。
如本文所用,术语“多克隆抗体”或“多克隆抗体组合物”是指衍生自不同B细胞系的抗体的制剂。它们是针对特定抗原分泌的免疫球蛋白分子的混合物,每个分子都识别不同的表位。
如本文所用,术语“抗体衍生物”包括全长抗体或抗体的片段,其中一个或多个氨基酸通过烷基化、聚乙二醇化、酰化、酯形成或酰胺形成等等(例如用于将抗体连接至第二分子)而被化学修饰。这包括但不限于聚乙二醇化抗体、半胱氨酸聚乙二醇化抗体及其变体。
如本文所用,术语“标记”意指直接或间接可检测的化合物或组合物直接或间接缀合至待检测的组合物,例如N-末端组氨酸标签(N-His)、磁活性同位素(例如
如本文所用,术语“免疫缀合物”包括与第二试剂结合或连接的抗体或抗体衍生物,所述第二试剂例如是细胞毒性试剂、可检测试剂、放射性试剂、靶向试剂、人抗体、人源化抗体、嵌合抗体、合成抗体、半合成抗体或多特异性抗体。
合适的荧光标记的实例包括但不限于荧光素、罗丹明、四甲基罗丹明、曙红、赤藓红、香豆素、甲基香豆素、芘、孔雀石绿、二苯乙烯、荧光素黄、级联蓝™和得克萨斯红。其他合适的光学染料在Haugland, Richard P. (1996) Handbook of Fluorescent Probesand Research Chemicals(第6版)中进行了描述。
在另一方面,对荧光标记进行功能化以促进共价附接到细胞或组织的表面中或表面上存在的细胞成分(例如细胞表面标记)。合适的官能团包括但不限于异硫氰酸酯基、氨基、卤代乙酰基、马来酰亚胺、琥珀酰亚胺酯和磺酰卤,所有这些均可以用于将荧光标记附着于第二分子。荧光标记的官能团的选择将取决于与接头、试剂、标记或第二标记试剂的连接位点。
“真核细胞”包括除原核生物外的所有生命。通过膜结合核可以轻松地区分它们。动物、植物、真菌和原生生物是真核生物或生物,其细胞通过内膜和细胞骨架组织成复杂的结构。最有特色的膜结合结构是细胞核。除非特别叙述,否则术语“宿主”包括真核宿主,包括例如酵母、高等植物、昆虫和哺乳动物细胞。真核细胞或宿主的非限制性实例包括猿猴、牛、猪、鼠、大鼠、禽、爬行动物和人。
“原核细胞”通常缺乏细胞核或任何其他膜结合的细胞器,并且分为两个域,细菌和古细菌。除了染色体DNA,这些细胞还可以在称为附加体的环状环中包含遗传信息。细菌细胞非常小,大约相当于动物线粒体的大小(直径约1-2 μm,长10 μm)。原核细胞具有三种主要形状:杆状、球形和螺旋形。细菌细胞没有经历像真核生物这样复杂的复制过程,而是通过二元裂变分裂。实例包括但不限于芽孢杆菌、大肠杆菌和沙门氏菌。
“天然”或“原生”抗原是包含表位的多肽、蛋白质或片段,其已从天然生物学来源分离,并且可以特异性结合对象中的抗原受体,特别是T细胞抗原受体(TCR)。
术语“抗原”和“抗原性”是指具有能够被抗体识别或以其他方式充当抗体-配体对的成员的能力的分子。“特异性结合”是指抗原与免疫球蛋白重链和轻链的可变区的相互作用。抗体-抗原结合可以在体内或体外发生。技术人员将理解,包括蛋白质、核酸、脂肪酸、脂质、脂多糖和多糖的大分子具有充当抗原的潜力。本领域技术人员将进一步理解,编码具有作为抗体配体的潜能的蛋白质的核酸必然编码抗原。本领域技术人员将进一步理解,抗原不仅限于全长分子,还可以包括部分分子。术语“抗原性”是形容词指具有抗原特性的分子。该术语包括具有免疫原性的物质(即免疫原)以及诱导免疫无反应性或无变应性的物质(即无能物质(anergen))。
“改变的抗原”是具有不同于相应的野生型抗原的一级序列的一级抗原。改变的抗原可以通过合成或重组方法来制备,并且包括但不限于在翻译过程中或之后例如通过磷酸化、糖基化、交联、酰化、蛋白水解切割、与抗体分子、膜分子或其他配体连接而被差异修饰的抗原肽。(Ferguson et al. (1988) Ann. Rev. Biochem. 57:285-320)。本文公开的合成或改变的抗原旨在结合作为天然表位的相同的TCR。
“免疫应答”广义上是指淋巴细胞对异物的抗原特异性应答。术语“免疫原”和“免疫原性”是指具有引发免疫反应能力的分子。所有的免疫原都是抗原,但是,并非所有的抗原都是免疫原性的。本文公开的免疫应答可以是体液的(通过抗体活性)或细胞介导的(通过T细胞激活)。该反应可以在体内或体外发生。本领域技术人员将理解,包括蛋白质、核酸、脂肪酸、脂质、脂多糖和多糖在内的多种大分子具有免疫原性的潜力。本领域技术人员将进一步理解,编码能够引发免疫应答的分子的核酸必然编码免疫原。本领域技术人员将进一步理解,免疫原不限于全长分子,而可以包括部分分子。
术语“被动免疫”是指通过抗体的转移将免疫从一个对象转移到另一个对象。被动免疫可以自然发生,就像母体抗体转移到胎儿时一样。当将抗体组合物施用于非免疫对象时,被动免疫也可以人工发生。抗体供体和受体可以是人类或非人类对象。根据实施方案,抗体可以是多克隆或单克隆的,可以在体外或体内产生,并且可以被纯化、部分纯化或未纯化。在本文所述的一些实施方案中,通过给予特异性识别或结合特定抗原的抗体或抗原结合片段,给予有需要的对象被动免疫。在一些实施方案中,通过给予编码特异性识别或结合特定抗原的抗体或抗原结合片段的分离或重组多核苷酸来给予被动免疫。
在本公开的上下文中,“配体”是多肽。在一方面,本文所用的术语“配体”是指与另一分子上的特定位点结合的任何分子。换句话说,配体在与免疫效应细胞或针对蛋白质的抗体或针对蛋白质的DNA的反应中赋予蛋白质特异性。在一方面,蛋白质内的配体位点直接与免疫效应细胞上的互补结合位点结合。
如本文所用,术语“在对象中诱导免疫应答”是本领域众所周知的术语,并且是指相对于在将抗原(或表位)引入对象之前的免疫应答(如有),在将抗原(或表位)引入对象之后,可以检测或测量到对抗原(或表位)的免疫应答增加至少约2倍、至少约5倍、至少约10倍、至少约100倍、至少约500倍或至少约1000倍或更多。对抗原(或表位)的免疫应答包括但不限于抗原特异性(或表位特异性)抗体的产生,以及在其表面表达与抗原(或表位)特异性结合的分子的免疫细胞的产生。确定是否已经诱导了针对给定抗原(或表位)的免疫应答的方法是本领域众所周知的。例如,可以使用本领域已知的多种免疫测定中的任何一种来检测抗原特异性抗体,这些免疫测定包括但不限于ELISA,其中例如样品中的抗体与固定的抗原(或表位)结合用可检测标记的二抗(例如酶标记的小鼠抗人Ig抗体)检测。
如本文所用,可互换使用的“固相支持物”或“固相载体”不限于特定类型的支持物。相反,大量的载体是可用的,并且是本领域普通技术人员已知的。固相支持物包括硅胶、树脂、衍生的塑料薄膜、玻璃珠、棉、塑料珠、氧化铝凝胶。如本文所用,“固体支持物”还包括合成的抗原呈递基质、细胞和脂质体。可以基于期望的最终用途和对各种方案的适用性来选择合适的固相支持物。例如,对于肽合成,固相支持物可以指的是树脂,例如聚苯乙烯(例如从Bachem Inc., Peninsula Laboratories等获得的PAM树脂)、POLYHIPE®树脂(从加拿大的Aminotech获得)、聚酰胺树脂(购自Peninsula Laboratories)、接枝了聚乙二醇的聚苯乙烯树脂(TentaGel®, Rapp Polymere, Tubingen, Germany)或聚二甲基丙烯酰胺树脂(获自 Milligen/Biosearch, Calif.)。
固相支持物的实例包括玻璃、聚苯乙烯、聚丙烯、聚乙烯、葡聚糖、尼龙、淀粉酶、天然和改性纤维素、聚丙烯酰胺、辉长岩和磁铁矿。载体的性质可以在某种程度上可溶或不可溶。载体材料实际上可以具有任何可能的结构构型,只要偶联的分子能够结合多核苷酸、多肽或抗体即可。因此,载体结构可以是球形的,例如珠状的,或者是圆柱形的,例如在试管的内表面或杆的外表面。替代地,表面可以是平坦的,诸如片、测试条等,或者替代地聚苯乙烯珠。本领域技术人员将知道许多用于结合抗体或抗原的其他合适的载体,或者将能够通过使用常规实验来确定它们。
术语“调节免疫应答”包括诱导(增加、引发)免疫应答;以及减少(抑制)免疫应答。免疫调节方法(或方案)是一种调节对象中的免疫应答的方法。
用于执行本发明的方式
I. 生物膜结构与疾病
维持慢性和复发性细菌感染的细菌库位于生物膜中,该生物膜是粘附在表面上的细菌群落,并且在这种状态下可以抵抗宿主免疫系统以及抗微生物剂的清除。实际上,处于生物膜状态的细菌对抗生素的抗性通常比处于自由生活或浮游状态的相同细菌高1000倍以上。Ceri et al. (1999) J Clin Microbiol. 37(6):1771-6。生物膜细菌抵抗清除的能力主要归因于半渗透性自制基质或细胞外聚合物质(EPS),它们既可作为对环境危害的物理屏障,又可为生理变化创造条件,限制新陈代谢增强这种抵抗状态。尽管EPS的成分对每种细菌都是特定的,并且包括蛋白质、多糖、脂质和核酸,但EPS的性质需要充分有益于整个细菌属产生有效的相互作用(例如作为代谢伴侣)。为此,最近的一些发现指向所有真细菌普遍存在潜在的通用EPS结构。Whitchurch及其同事(Whitchurch et al. (2002)Science. 295(5559)) 表明,细胞外DNA(eDNA)是一种普遍的EPS成分,用DNA酶处理细菌足以防止生物膜形成。尽管这一结果已被复制到多个属中,但是尽管在整个生物膜的整个生命周期中都存在eDNA的事实,但是使用DNA酶未能在生物膜播种后一到两天以上处理现存的生物膜。另外,申请人先前将DNABII蛋白鉴定为eDNA依赖性EPS的必要组成部分,DNABII蛋白是所有真细菌共有的唯一核苷相关蛋白(NAP)家族。实际上,针对DNABII蛋白质的抗体会从大量培养基中滴定DNABII蛋白质,从而使DNABII蛋白质的平衡从eDNA结合状态转变为未结合状态,这导致迄今测试的所有细菌生物膜发生灾难性崩溃,并且包括混合物种生物膜。Goodman et al. (2011) Mucosal Immunol. 4(6):625-37; Novotny et al. (2013)PLoS One. 8(6):e67629; Devaraj et al. (2015) Mol Microbiol. 96(6):1119-35;Rocco et al. (2017) Mol Oral Microbiol. 32(2):118-30; Gustave et al. (2013) JCyst Fibros. 12(4):384-9; Novotny et al. (2016) EBioMedicine. 10:33-44。重要的是,与DNA酶不同,用针对DNABII蛋白的抗体进行的处理在
细菌内的DNA是高度结构化的,并有助于调节所有形式的核酸过程,包括DNA复制、修复、转录和重组。与真核细胞不同,细菌不含组蛋白。取而代之的是,细菌DNA由一类称为核苷相关蛋白(NAP)的蛋白构成。NAP共同结合DNA以创建功能结构。Dillon et al. (2010)Nat Rev Microbiol. 8(3):185-95。在整个属中存在的多个NAP成员中,只有DNABII家族在所有真细菌中普遍存在。Dey et al. (2017) Mol Phylogenet Evol. 107:356-66。DNABII蛋白质家族起二聚体的作用(同质二聚体或异二聚体),并包括组蛋白样蛋白HU和IHF。HU弱且非特异性地结合并弯曲双链DNA(dsDNA),但对弯曲前或结构化的dsDNA3具有更高的亲和力。像HU那样的IHF结合并弯曲DNA时,强烈倾向于弯曲前/结构化的DNA。与HU不同,IHF仅由蛋白细菌表达,并且也偏爱特定的DNA共有序列。Swinger et al. (2004) Curr OpinStruct Biol. 14(1):28-35。
自从发现“转化原理”是DNA的结果以来,已知细胞外DNA(eDNA)具有生物学作用。Avery et al. (1944) J Exp Med. 79(2):137-58。实际上,eDNA对细菌生物膜的细胞外基质(细胞外聚合物,EPS)也很关键。Gunn et al. (2016) J Biol Chem. 291(24):12538-46。但是,迄今为止尚未研究生物膜eDNA的结构以及该结构对eDNA功能的重要性。
尽管生物膜通过细胞间通讯和运输系统与浮游细菌有进一步的区别,但是它们最独特的特征是它们自身的EPS,其通过充当半透屏障和通过创造新陈代谢改变/减慢的环境来保护驻留的生物膜细菌;实际上,生物膜细菌对细菌的抵抗力比其浮游对应物高1000倍以上。Ceri et al. (1999) J Clin Microbiol. 37(6):1771-6。有趣的是,每种细菌的EPS都是不同的,由多种蛋白质、脂质、多糖和核酸组成。Gunn et al. (2016) J Biol Chem.291(24):12538-46。然而,虽然生物膜可以由单一物种组成,但通常在慢性感染中以及在环境中总是由多个属组成,因此需要能够产生有效的相互作用(例如与特定的代谢伙伴共同聚集)。Stacy et al. (2016) Nat Rev Microbiol. 14(2):93-105; Wolcott et al.(2013) Clin Microbiol Infect. 19(2):107-12。这个社群概念意味着,尽管EPS组成有所不同,但每个EPS必须足够容纳以允许散布的细菌在生物膜内相互作用,并且进一步表明,生物膜EPS可能具有通用的基础结构。
多个小组已经检查了与来自人类和生态属的细菌生物膜相关的eDNA,并观察到了支架结构(图1A)。Jurcisek et al. (2017) Proc Natl Acad Sci U S A. 114(32):E6632-E41; Sena-Velez et al. (2016) PLoS One. 11(6):e0156695; Wang et al.(2015) Environ Microbiol Rep. 7(2):330-40。Whitchurch及其同事首先表明,可以通过脱氧核酸酶I(DNA酶)处理来预防铜绿假单胞菌生物膜(Whitchurch et al. (2002)Science. 295(5559)),这表明eDNA是EPS的关键结构成分。尽管DNA酶可以抑制许多属的早期生物膜形成 (Frederiksen et al. (2006) Acta Paediatr. 95(9):1070-4; Martinset al. (2012) Mycoses. 55(1):80-5; Hymes et al. (2013) J Infect Dis. 207(10):1491-7)),,尽管随着生物膜的成熟eDNA明显持续存在,但生物膜对DNA酶的抵抗力随时间而变。Goodman et al. (2011) Mucosal Immunol. 4(6):625-37; Hall-Stoodley et al.(2008) BMC Microbiol. 8:173; Izano et al. (2009) Microb Pathog. 46(4):207-13;Kaplan et al. (2012) J Antibiot (Tokyo). 65(2):73-7; Novotny et al. (2013)PLoS One. 8(6):e67629; Tetz et al. (2010) DNA Cell Biol. 29(8):399-405。尽管这种结果通常被解释为意味着eDNA对EPS的结构完整性不再重要,但申请人已证明eDNA不仅持续存在,而且成为EPS的主要基础结构。Goodman et al. (2011) Mucosal Immunol. 4(6):625-37; Novotny et al. (2013) PLoS One. 8(6):e67629; Devaraj et al.(2015) Mol Microbiol. 96(6):1119-35; Rocco et al. (2017) Mol Oral Microbiol.32(2):118-30; Brockson et al. (2014) Mol Microbiol. 93(6):1246-58。
申请人先前已经表明,普遍存在的DNABII蛋白且可能没有其他NAP(Devaraj etal. (2017) Microbiologyopen.)是eDNA的结构组成,一旦被去除,eDNA的结构就会被破坏。Goodman et al. (2011) Mucosal Immunol. 4(6):625-37; Novotny et al. (2013)PLoS One. 8(6):e67629; Devaraj et al. (2015) Mol Microbiol. 96(6):1119-35;Rocco et al. (2017) Mol Oral Microbiol. 32(2):118-30。确实,发现了DNABII蛋白与体内形成的生物膜的eDNA支架的顶点(弯曲前的DNA)特异性结合,而针对DNABII蛋白的抗体足以破坏eDNA支架的EPS的结构,并且结果导致每种物种的单物种和多物种生物膜都发生灾难性崩溃。申请人已检查(Goodman et al. (2011) Mucosal Immunol. 4(6):625-37;Novotny et al. (2013) PLoS One. 8(6):e67629; Devaraj et al. (2015) MolMicrobiol. 96(6):1119-35; Rocco et al. (2017) Mol Oral Microbiol. 32(2):118-30),无论生物膜成熟度如何。Brockson et al. (2014) Mol Microbiol. 93(6):1246-58。这种破坏将驻留细菌释放为浮游生物,从而释放出抗菌和免疫敏感状态(Goodman et al.(2011) Mucosal Immunol. 4(6):625-37; Novotny et al. (2013) PLoS One. 8(6):e67629; Brockson et al. (2014) Mol Microbiol. 93(6):1246-58),这证明了该蛋白家族在生物膜结构中的重要性和普遍性。考虑到DNABII家族与DNA的已知相互作用,出乎意料的是,申请人无法仅利用DNA和DNABII蛋白创造条件来概括细菌生物膜中3维骨架结构。
多胺通常是短有机分子,其包含多个伯胺,这些伯胺在中性pH下带正电(碱性),通常是通过氨基酸脱羧而衍生的(图1B)。Michael et al. (2016) Biochem J. 473(15):2315-29。多胺在自然界中普遍存在,在细胞内和细胞外均发现高达mM的浓度 (Tabor etal. (1985) Microbiol Rev. 49(1):81-99) ,其中亚精胺、精胺和腐胺含量最高。尽管多胺参与细菌生理学的多个过程,但由于5个原因,它们可能对eDNA支架的EPS最重要。首先,它们结合DNA并中和磷酸骨架的负电荷,结果改变了DNA结构。Bachrach et al. (2005)Curr Protein Pept Sci. 6(6):559-66; Pasini et al. (2014) Amino Acids. 46(3):595-603。第二,虽然多胺倾向于在低浓度(mM)时促进蛋白质与DNA的相互作用,但它们却倾向于在高浓度(mM)时发生干扰,其中一个例外是导致DNA形成粗纤维的DNABII-DNA相互作用。Sarkar et al. (2009) Biochemistry. 48(4):667-75; Sarkar et al. (2007)Nucleic Acids Res. 35(3):951 61。第三,已发现在生物膜形成过程中诱导了多胺的合成,其中一些作用发生在细胞外(尽管迄今为止仅在细菌膜上进行了检查)。Wilton et al.(2015) Antimicrob Agents Chemother. 60(1):544-53。第四,使多胺和DNA的混合物可视化的原子力显微镜(AFM)实验揭示了与生物膜eDNA支架高度相似的结构(图1C)。最后,多胺可以在生理浓度(100 µM)下以倾向(prone )序列诱导天然B型DNA转化为Z型DNA。Thomaset al. (1986) Nucleic Acids Res. 14(16):6721-33; Thomas et al. (1988) J MolBiol. 201(2):463-7。
B-DNA和Z-DNA是平衡存在的dsDNA的不同构象,在大多数生理条件下,B-DNA占主导。交替的嘌呤和嘧啶(尤其是dGdC)更容易以Z-DNA的形式存在于高盐(摩尔单价或二价阳离子)或负超螺旋状态下。Pohl et al. (1983) Cold Spring Harb Symp Quant Biol. 47Pt 1:113-7; Pohl et al. (1986) Proc Natl Acad Sci U S A. 83(14):4983-7。在后一种情况下,当瞬时诱导产生负超螺旋时,在转录过程中,易于形成Z-DNA的区域可以在B-DNA旁边短暂并置。Rahmouni et al. (1992) Mol Microbiol. 6(5):569-72。B-DNA碱基采用右手螺旋(10 bp/圈),而Z-DNA形成左手螺旋(12 bp/圈)。Jovin et al. (1987) Ann RevPhys Chem. 38:521-60。B-DNA有两个沟(大沟和小沟),大多数相互作用的蛋白质由于其较大的大小而识别/结合在大沟中,并区分每个核苷酸碱基的氢键供体和受体。相反,Z-DNA中不存在大沟,大多数结合接触都在凸面上发现。Jovin et al. (1987) Ann Rev PhysChem. 38:521-60。Z-DNA具有与B-DNA的小沟相对应的单个沟。有趣的是,DNABII蛋白是少数在小沟中结合的DNA结合蛋白之一(Kim et al. (2014) Acta Crystallogr D BiolCrystallogr. 70(Pt 12):3273-89),表明它们可能结合 Z-DNA。eDNA从B-DNA向Z-DNA的转变与eDNA支架EPS的4个观察结果一致。首先,向Z-DNA的转变发生在生物膜EPS中存在的条件下;在生理浓度(100 mM)的某些多胺(亚精胺和亚精胺)存在的情况下,这些倾向序列会转变为Z-DNA。第二,Z-DNA倾向于聚集并形成纤维。Chaires et al. (1988) J BiomolStruct Dyn. 5(6):1187-207。磷酸骨架的强负电荷中和作用(例如多胺)有利于Z-DNA,因为Z-DNA骨架中的磷酸酯比B-DNA中的磷酸酯更紧密,但也允许DNA聚集。第三,Z-DNA比B-DNA坚硬,持久长度几乎增加了3倍(Thomas et al. (1983) Nucleic Acids Res. 11(6):1919-30) ,与申请人在eDNA支架中观察到的直纤维一致(图1A、图2、图3)。最后,Z-DNA具有核酸酶抗性。与规范的B-DNA结合蛋白不同,只有少数已知的蛋白可识别Z-DNA(Athanasiadis et al. (2012) Semin Cell Dev Biol. 23(3):275-80) (但是已有Z-DNAvs B-DNA 区分抗体)。Bergen et al. (1987) J Immunol. 139(3):743-8。有趣的是,Z-DNA结合蛋白是同源的,相对于B-DNA以高1,000至10,000倍的亲和力结合Z-DNA (Herbertet al. (1996) J Biol Chem. 271(20):11595-8),诱导倾向序列中的Z-DNA构象,至少在ZBP1中至少是可检测微生物DNA的存在的先天免疫系统的一部分。Athanasiadis et al.(2012) Semin Cell Dev Biol. 23(3):275-80。重要的是,尽管保留了Watson Crick碱基配对的事实,但以B-DNA为底物的蛋白质(例如核酸酶)却无法识别并因此在Z构型的相同DNA序列上起作用。
申请人先前已经显示eDNA-DNABII相互作用用于维持生物膜EPS的结构完整性(Goodman et al. (2011) Mucosal Immunol. 4(6):625-37; Novotny et al. (2013)PLoS One. 8(6):e67629; Devaraj et al. (2015) Mol Microbiol. 96(6):1119-35;Rocco et al. (2017) Mol Oral Microbiol. 32(2):118-30; Devaraj et al. (2017)Microbiologyopen.; Brockson et al. (2014) Mol Microbiol. 93(6):1246-58),并且破坏这些相互作用会导致体外((Gustave et al. (2013) J Cyst Fibros. 12(4):384-9)和体内(Goodman et al. (2011) Mucosal Immunol. 4(6):625-37; Novotny et al.(2016) EBioMedicine. 10:33-44; Freire et al. (2017) Mol Oral Microbiol. 32(1):74-88)阳性结果。尽管显示DNA和DNABII蛋白质都是必需的,但它们不足以概括EPS支架结构。本文公开的是eDNA-DNABII依赖性EPS的三重eDNA依赖性支架(tripartite eDNA-dependent scaffold,TEDS),其依赖于(1)eDNA、(2)DNABII蛋白和新发现的EPS成分、(3)多胺的存在和相对位置。
在一方面,申请人表明,除了eDNA和DNABII蛋白外,多胺也是细菌生物膜TEDS结构的重要组成部分。第二,申请人表明,所有这三个组成部分共同促进了通用EPS的形成,其可促进处于保护性生物膜状态的细菌属之间的生产性相互作用。第三,申请人使用这些成分定义并概括了这种通用结构,并提供了与粗双链DNA纤维的观察结果(诱导了核酸酶抗性状态)一致的证据,并证明该状态是否需要Z-DNA作为结构终点。最后,这提供了以TEDS结构本身为目标的诊断和治疗干预措施。
由NTHI引起的急性中耳炎的毛丝鼠模型忠实地概括了人类疾病的过程和病理生理学(Bakaletz et al. (2009) Expert Rev Vaccines. 8(8):1063-82),并且依赖于中耳中顽固的生物膜。使用该模型,申请人先前显示DNABII蛋白与eDNA缔合,后者定位于eDNA链的顶点(图1A)(Goodman et al. (2011) Mucosal Immunol. 4(6):625-37) ,并且这些eDNA链看起来惊人地类似于通过AFM用DNA观察到的多胺(图1C)。Iacomino et al. (2011)Biomacromolecules. 12(4):1178-86。在体外存在mM浓度的亚精胺时,DNABII蛋白与DNA结合(图2B),这导致对DNABII蛋白与多胺之间可能产生生物膜eDNA支架结构的潜在相互作用的研究。申请人对固定和嵌入的中耳黏膜生物膜切片进行了免疫荧光共聚焦激光扫描显微镜(CLSM),以可视化生物膜EPS中与eDNA相互作用的多胺,因为申请人先前已经观察到DNABII蛋白。免疫荧光图像显示,eDNA和多胺沿着粘膜生物膜内存在的纤维共定位(图2A),并且在视觉上类似于Hud和其同事在体外观察到的与DNABII蛋白和多胺的结合(Sarkar etal. (2009) Biochemistry. 48(4):667-75; Sarkar et al. (2007) Nucleic AcidsRes. 35(3):951 61) (图2B)。
申请人研究了广泛作用的多胺生物合成抑制剂二环己胺(DCHA)是否会在体外改变NTHI生物膜的生物发生。DCHA抑制亚精胺合酶(Paulin et al. (1986) Antonie VanLeeuwenhoek. 52(6):483-90; Pegg et al. (1983) FEBS Lett. 155(2):192-6),这种酶催化腐胺转化为亚精胺。尽管DCHA不影响NTHI的生长(数据未显示),但DCHA在体外抑制生物膜的生物发生,如通过LIVE/DEAD®染色的NTHI生物膜的CLSM图像(图3A)的COMSTAT分析(Heydorn et al. (2000) Microbiology. 146 (Pt 10):2395-407)所确定的那样,降低了平均厚度和生物量 。此外,申请人发现DCHA在早期生物膜发育中抑制了eDNA支架的产生(图3B),这表明多胺生物合成是生物膜形成中必需的步骤。为了支持这些发现,申请人通过免疫荧光观察到DCHA减少了细胞外多胺掺入生物膜EPS中(图3C),并且生物膜生物发生中的缺陷可以通过添加外源亚精胺来补偿(图3A)。这些结果一起表明,聚胺结合到EPS中对于eDNA支架的发展和功能在支持强劲的生物膜生长中至关重要。
使用免疫荧光来确定DNABII蛋白是否被掺入EPS模拟结构中。将亚精胺(300 µM)和HU(1 µM)与基因组DNA(2 μg/ml)孵育。然后,用幼稚(对照)或抗DNABII IgG(一种荧光二抗)探测EPS模拟结构,用DAPI染色,然后用CLSM成像。DNABII蛋白完全掺入EPS结构中(图4A)。接下来,形成EPS模拟结构,用抗DNABII抗体处理,用DAPI染色,并用CLSM成像。从EPS模拟物中用抗DNABII抗体隔离DNABII蛋白大大降低了聚集体结构的丰度和大小(图4B),这进一步证实了DNABII的掺入及其对DNA结构稳定性的重要性。
磷酸纤维素(P11)是一种带负电荷的树脂,对带正电荷的分子(例如多胺和DNABII蛋白)具有高亲和力。为了确定这些分子的P11螯合对细菌生物膜形成的影响,申请人利用了transwell系统。NTHI在基底外侧室开始生长,同时在播种时将P11(1%w/v)添加到顶端室。在16小时,将生物膜洗涤并用LIVE/DEAD®染色,使用CLSM成像,并用COMSTAT分析。P11显著降低了平均厚度和生物量(图5)。为了确定P11阳离子的消耗是否包括多胺和/或DNABII蛋白,外源添加1 mM亚精胺和1 µM HU来启动基底外侧室中NTHI的生长,而将P11以1%w/v的浓度添加到顶端室中。亚精胺和HU都是生物膜基质所需的结构成分,并且仅一起防止了P11对生物膜的破坏(图5)。单独的HU和亚精胺是不足够的(数据未显示)。
申请人评估了Pulmozyme®(一种重组人DNA酶)的抗生物膜作用,其与标准疗法联合用于治疗囊性纤维化(CF)患者以改善肺功能。Yang et al. (2017) Paediatr RespirRev. 21:65-7。在播种时(生物膜预防)或在预先形成的生物膜中(生物膜破坏)添加Pulmozyme®(图6)。所得生物膜用LIVE/DEAD®染色,并使用CLSM和COMSTAT分析进行评估。与未处理的生物膜相比,播种时添加DNA酶导致NTHI和UPEC生物膜的平均厚度和生物量显著降低(图6)。然而,DNA酶不能破坏成熟的生物膜(图6),这表明eDNA随着生物膜的发展而丧失了对核酸酶消化的敏感性。申请人提出,随着生物膜的成熟,eDNA在空间上不受核酸酶的保护,和/或eDNA具有新颖的结构,使其对核酸酶消化具有顽固性。
用抗DNABII和抗亚精胺抗体探测的NTHI生物膜的免疫荧光表明,多胺在体外(图7A)和体内(图7B)与DNABII蛋白共定位。申请人假设DNABII和多胺协同地与eDNA相互作用以赋予成熟的生物膜以DNA酶抗性。为了在体外测试协同作用,申请人在存在或不存在HU(50或100 nM)和DNA酶(0.5个单位)的情况下,将NTHI基因组DNA(gDNA)与亚精胺(10、20、50或100 µM)一起孵育。通过琼脂糖凝胶电泳和UV照射评估降解。虽然亚精胺(> 100 µM)和HU(1 µM)分别保护gDNA,但较低含量的亚精胺(<50 µM)和HU(<100 nM)的混合协同地抑制gDNA消化(图7C),表明核酸酶抗性是由于生物膜基质中DNABII蛋白和多胺的组合。接下来,申请人想测试EPS模拟结构是否对DNA酶具有抗性。将HU(1 µM)和亚精胺(300 mM)与gDNA(2μg/ml)孵育40小时以创建EPS支架模拟物,然后用DNA酶(5个单位)处理。如CLSM所示,类似于成熟生物膜,这些DNABII-多胺依赖性DNA结构对DNA酶具有抗性(图7D)。
聚胺导致结合后Z-DNA倾向序列将B-Z平衡转变成Z-DNA构型(Thomas et al.(1986) Nucleic Acids Res. 14(16):6721-33; Thomas et al. (1988) J Mol Biol.201(2):463-7),而DNABII蛋白则弯曲并浓缩DNA。由于申请人的体外DNA酶降解试验中HU和亚精胺结合的抑制作用协同作用(图7),以及Z-DNA抵抗DNA酶的能力,申请人假设EPS模仿结构将包含Z-DNA。如图4和图7中形成的EPS结构用gDNA处理16小时。用抗Z-DNA抗体进行免疫荧光CLSM的探测,其确认了Z-DNA的存在(注意白色;图8A)。为了确定DNABII蛋白是否影响B-Z平衡,申请人使用了圆二色性(CD)。 HU和聚(dGdC)DNA底物的混合物表现出B-DNA单独的椭圆峰(250和280 nm)的倒置(Jang et al. (2015) Sci Rep. 5:9943),类似于特征性Z-DNA光谱(图8B)。
为了进一步表征Z-DNA和多胺的缔合,申请人在40小时生物膜上进行了免疫荧光。用抗DNABII和抗亚精胺或抗Z-DNA抗体探测后,用CLSM对所示的细菌病原体的生物膜进行成像。尽管在每种细菌病原体的生物膜EPS中均检测到Z-DNA(图9),但物种之间存在明显的等级关系(UPEC =表皮葡萄球菌<肺炎克雷伯菌
由于多胺在体外(图7A和图9)和体内(图7B)在NTHI生物膜EPS内与HU共定位,因此申请人评估了HU在NTHI生物膜EPS中的多胺和Z-DNA掺入中的作用。 通过免疫荧光比较HU缺陷NTHI(
本文提供了用于治疗对象中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的试剂。在一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在不存在DNA酶的情况下提供试剂。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,用于治疗对象中的生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂。在一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在不存在DNA酶的情况下提供试剂。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于治疗对象中的生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的两种或更多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于治疗对象中的生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的三种或更多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于治疗对象中的生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的四种或更多种、或五种或更多种、或六种或更多种、或七种或更多种、或八种或更多种、或九种或更多种、或十种或更多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。 本公开内容还涉及用于防止在易于发展生物膜的对象中形成生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的试剂。在一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在不存在DNA酶的情况下提供试剂。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,用于防止在易于发展生物膜的对象中形成生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂。在一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在不存在DNA酶的情况下提供试剂。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于防止在易于发展生物膜的对象中形成生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的两种或更多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于防止在易于发展生物膜的对象中形成生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的三种或更多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于防止在易于发展生物膜的对象中形成生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的四种或更多种、或五种或更多种、或六种或更多种、或七种或更多种、或八种或更多种、或九种或更多种、或十种或更多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。 本公开进一步涉及用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的试剂和抑制生物体复制的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂。在另一方面,用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的两种或多种试剂和抑制生物体复制的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的三种或多种试剂和抑制生物体复制的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰生物膜中多胺与DNA结合的四种或更多种、或五种或更多种、或六种或更多种、或七种或更多种、或八种或更多种、或九种或更多种、或十种或更多种试剂和抑制生物体复制的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。 对于上述任何方法,多胺可以选自:腐胺、精胺、尸胺、1,3-二氨基丙烷或亚精胺。在一个实施方案中,对于上述方法,干扰多胺与生物膜中DNA结合的试剂是tRNA。在另一个实施方案中,该试剂是多胺合成的抑制剂或抑制多胺与DNA结合的试剂。在第二实施方案中,该试剂包括多胺类似物二氟甲基鸟氨酸、反式-4-甲基环己胺、沙多齐特、甲基乙二醛双[脒基腙](MGBG)、1-氨基氧基-3-氨基丙烷、奥沙利铂、顺铂、二环己胺、其任何衍生物、或其盐,或基本上由其组成,或由其组成。在一方面,这些化合物的衍生物保持相同的质荷比。在第三实施方案中,该试剂包括从生物膜中消耗阳离子的试剂,任选地阳离子交换树脂、氨基多羧酸、冠醚、氮杂冠、或穴醚,或基本上由其组成,或由其组成。在第四实施方案中,从生物膜中消耗阳离子的试剂选自:磺酸盐,磺丙基,磷酸纤维素,P11磷酸纤维素,硫酸肝素,或其衍生物或类似物。在一方面,从生物膜消耗阳离子的试剂的衍生物或类似物是具有净负电荷的树脂。在第五实施方案中,该试剂干扰生物膜或其局部环境中B-DNA向Z-DNA转化。在第六实施方案中,所述试剂包含抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。在第七实施方案中,所述试剂包含核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[c]菲啶生物碱、奎那克林、9-氨基吖啶、或其衍生物,或基本上由其组成,或由其组成。在第八实施方案中,所述试剂包含氯喹或其衍生物,或基本上由其组成,或由其组成。在一方面,化合物的衍生物保持插入DNA碱基之间的能力。在一方面,所述试剂不是HGMB1蛋白或其片段。在一方面,从生物膜中消耗阳离子的试剂具有净负电荷。在另一方面,从生物膜中消耗阳离子的试剂具有净中性电荷。 本文还提供了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)的患者中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。本文公开了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)和/或TB的患者中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的在生物膜或其局部环境中干扰B-DNA向Z-DNA转化的一种或多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,本文提供了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)和/或TB的患者中的生物膜的方法,所述方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的在生物膜或其局部环境中干扰B-DNA向Z-DNA转化的两种或多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,本文公开了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)和/或TB的患者中的生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的在生物膜或其局部环境中干扰B-DNA向Z-DNA转化的三种或多种试剂。在另一方面,用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)的患者中的生物膜的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的在生物膜或其局部环境中干扰B-DNA向Z-DNA转化的四种或更多种、或五种或更多种、或六种或更多种、或七种或更多种、或八种或更多种、或九种或更多种、或十种或更多种试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一个实施方案中,所述试剂干扰生物膜或其局部环境中B-DNA向Z-DNA转化。在第二实施方案中,所述试剂包括抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。在第三实施方案中,所述试剂包括核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[ 本文还提供了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)和/或TB的患者中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。HMGB1的生物活性片段可以包含以下中一种或多种:A box、B box、和/或AB box、C末端片段或N末端片段,或基本上由其组成,或由其组成。在一个具体的实施方案中,HMGB1的生物活性片段可以包含能够结合DNA的B Box结构域,或基本上由其组成,或由其组成。在一方面,本文提供了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)和/或TB的患者中的生物膜的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的氯喹和抗B-DNA抗体或其片段或衍生物。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。本公开还涉及用于治疗在正在接受或已经接受化学疗法的患者中的因施用基于铂的化学疗法而发生的产生生物膜的感染的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,所述试剂包含氯喹或其衍生物,或基本上由其组成,或由其组成。在另一方面,所述试剂包含抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。在另一方面,所述试剂包含氯喹或其衍生物,或基本上由其组成,或由其组成。在一个实施方案中,所述试剂包括核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[ 本文还涉及用于治疗在正在接受或已经接受化学疗法的患者中的因施用基于铂的化学疗法而发生的产生生物膜的感染的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。HMGB1的生物活性片段可以包含以下中一种或多种:A box、B box、和/或AB box、C末端片段或N末端片段,或基本上由其组成,或由其组成。在一个具体的实施方案中,HMGB1的生物活性片段可以包含能够结合DNA的B Box结构域,或基本上由其组成,或由其组成。在一方面,本文提供了用于治疗在正在接受或已经接受化学疗法的患者中的因施用基于铂的化学疗法而发生的产生生物膜的感染的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:施用有效量的氯喹和抗B-DNA抗体或其片段或衍生物。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。 上述方法可以进一步包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰eDNA与DNA结合蛋白结合的试剂和/或抗菌剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在一方面,所述方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:向对象施用有效量的干扰eDNA与DNA结合蛋白结合的试剂和/或抗菌剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,所施用的DNA酶是Pulmozyme。在另一方面,所述干扰eDNA与DNA结合蛋白结合的试剂包括抗DNABII抗体、抗IHF抗体和/或抗HU抗体、或其每一个的片段中的一种或多种,或基本上由其组成,或由其组成。在一个实施方案中,所述干扰eDNA与DNA结合蛋白结合的试剂具有净负电荷。在第二个实施方案中,所述干扰eDNA与DNA结合蛋白结合的试剂具有净中性电荷。在第三个实施方案中,所述干扰eDNA与DNA结合蛋白结合的试剂具有净正电荷。 本文描述了用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的试剂接触。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在一方面,用于抑制生物膜的稳定性的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的一种或多种试剂接触。在一方面,在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在另一方面,用于抑制生物膜的稳定性的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的两种或更多种试剂接触。在一方面,在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在另一方面,用于抑制生物膜的稳定性的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的三种或更多种试剂接触。在一方面,在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在另一方面,用于抑制生物膜的稳定性的方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰生物膜中多胺与DNA结合的四种或更多种、或五种或更多种、或六种或更多种、或七种或更多种、或八种或更多种、或九种或更多种、或十种或更多种试剂接触。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。所述接触可以是体外或体内的。 本公开还涉及用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与干扰生物膜中多胺与DNA结合的试剂在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的消耗阳离子的试剂包被表面。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在一方面,用于抑制生物膜的稳定性的方法可以包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与干扰生物膜中多胺与DNA结合的试剂在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的消耗阳离子的一种或多种试剂包被表面。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。 在另一方面,用于抑制生物膜的稳定性的方法可以包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与干扰生物膜中多胺与DNA结合的试剂在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的消耗阳离子的两种或更多种试剂包被表面。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。 在另一方面,用于抑制生物膜的稳定性的方法可以包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与干扰生物膜中多胺与DNA结合的试剂在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的消耗阳离子的三种或更多种试剂包被表面。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在另一方面,用于抑制生物膜的稳定性的方法可以包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与干扰生物膜中多胺与DNA结合的试剂在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的消耗阳离子的四种或更多种、或五种或更多种、或六种或更多种、或七种或更多种、或八种或更多种、或九种或更多种、或十种或更多种试剂包被表面。在一个实施方案中,在一方面,在不存在DNA酶的情况下接触干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在第二实施方案中,所述试剂包括抗B-DNA抗体或其片段或衍生物,或基本上由其组成,或由其组成。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。在第三实施方案中,所述试剂包括核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[ 本文进一步描述了用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的HMGB1蛋白或其生物活性片段和抗B-DNA抗体或其片段或衍生物包被表面。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。 在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。HMGB1的生物活性片段可以包含以下中一种或多种:Abox、B box、和/或AB box、C末端片段或N末端片段,或基本上由其组成,或由其组成。在一个具体的实施方案中,HMGB1的生物活性片段可以包含能够结合DNA的B Box结构域,或基本上由其组成,或由其组成。本文还涉及用于抑制生物膜的稳定性的方法,该方法包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的氯喹和抗B-DNA抗体或其片段或衍生物在体外接触,其中所述接触包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:用有效量的氯喹和抗B-DNA抗体或其片段或衍生物包被表面。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,在接触试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在一方面,多克隆或单克隆抗B-DNA抗体或其片段或衍生物在亲和力/亲和力上比Z形式DNA识别B形式DNA强至少10倍。 上述方法可以进一步包括以下步骤,或基本上由以下步骤组成,或由以下步骤组成:使生物膜与有效量的干扰eDNA与DNA结合蛋白结合的试剂和/或抗菌剂接触。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在一方面,干扰eDNA与DNA结合蛋白结合的试剂包含抗DNABII抗体、抗IHF抗体和/或抗HU抗体、或其每一个的片段中的一种或多种,或基本上由其组成,或由其组成。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在一个实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净负电荷。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在第二实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净中性电荷。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。在第三实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净正电荷。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。 本文提供了用于抑制生物膜的稳定性的方法,其包括使生物膜与有效量的干扰生物膜中多胺与DNA结合的试剂接触。在一方面,在不存在DNA酶的情况下接触试剂。在另一方面,根据所述方法接触DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在接触所述试剂之后接触DNA酶。在一个特定方面,DNA酶是Pulmozyme。接触可以是体外或体内的。 本文还提供了用于治疗对象中的生物膜的方法,其包括向感染了生物膜的对象施用有效量的干扰生物膜中多胺与DNA结合的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,施用DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。 本文还提供了用于防止在易于发展生物膜的对象中形成生物膜的方法,其包括向对象施用有效量的干扰生物膜中多胺与DNA结合的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,施用DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。 本文进一步提供了用于在有需要的对象中治疗由产生生物膜的细菌引起的感染的方法,其包括向对象施用有效量的干扰生物膜中多胺与DNA结合的试剂和抑制生物体复制的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,施用DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。 在一方面,上述试剂是多胺合成的抑制剂或抑制多胺与DNA结合的试剂。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。多胺的非限制性实例包括:腐胺、精胺、尸胺、1,3-二氨基丙烷或亚精胺。在另一方面,该试剂包括多胺类似物、二氟甲基鸟氨酸、反式-4-甲基环己胺、沙多齐特(Boc Sciences)、甲基乙二醛双[脒基腙](甲基-GBG)、1-氨基氧基-3-氨基丙烷(AKos Consulting & Solutions)、奥沙利铂、顺铂、二环己胺、其任何衍生物、或其盐(除非另有说明,否则该段落的所有试剂均可从Millipore Sigma商购获得)。在一方面,这些化合物的衍生物保持相同的质荷比。 在另一方面,上述试剂包括从生物膜中消耗阳离子的试剂,任选地阳离子交换树脂、氨基多羧酸、冠醚、氮杂冠、或穴醚(每类试剂的各种代表性化合物可从MilliporeSigma获得)。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。阳离子交换树脂的非限制性实例包括:磺酸盐,磺丙基,磷酸纤维素,P11磷酸纤维素,硫酸肝素,或其衍生物或类似物。在一方面,从生物膜消耗阳离子的试剂具有净负电荷。在一方面,从生物膜消耗阳离子的试剂具有净中性电荷。 在一个实施方案中,本文提供了用于抑制生物膜的稳定性的方法,其包括使生物膜与干扰多胺与生物膜中DNA结合的试剂在体外接触,并且所述接触包括用消耗阳离子的试剂包被表面。在一个方面,在不存在DNA酶的情况下施用试剂。在另一方面,施用DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。 在以上方法的一个方面,在不存在DNA酶的情况下施用干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在另一方面,施用DNA酶。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。这样的例子包括抗B-DNA抗体或其片段或衍生物。在另一方面,所述试剂包括选自以下的试剂:核黄素、溴化乙锭、双(甲锭)精胺(Dervan et al. (1978) 100(6):1968-1970)、柔红霉素、TMPyP4、季苯并[c]菲啶生物碱(Rajecky et al. (2015); Le et al. (2004) 69(8):2768-2772) 、奎那克林、9-氨基吖啶、或其衍生物(除非另外指出,否则该另一方面的所有试剂可从Millipore Sigma 商购获得 )。所述试剂不是HGMB1蛋白或其片段。 本文还提供了用于治疗患有系统性红斑狼疮(SLE)和/或囊性纤维化(CF)的患者中的生物膜的方法,该方法包括施用有效量的干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,施用DNA酶。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。这样的例子包括抗B-DNA抗体或其片段或衍生物。在另一方面,所述试剂包括选自以下的试剂:核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[c]菲啶生物碱、奎那克林、9-氨基吖啶、或其衍生物。在一方面,该方法在不存在DNA酶的情况下进行。在一方面,CF的治疗在不存在DNA酶的情况下进行。所述试剂不是HGMB1蛋白或其片段。 在另一方面,本发明还提供了用于治疗在正在接受或已经接受化学疗法的患者中的因施用基于铂的化学疗法而发生的产生生物膜的感染的方法,该方法包括施用有效量的干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,施用DNA酶。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。这样的例子包括抗B-DNA抗体或其片段或衍生物。在另一方面,所述试剂包括选自以下的试剂:核黄素、溴化乙锭、双(甲锭)精胺、柔红霉素、TMPyP4、季苯并[c]菲啶生物碱、奎那克林、9-氨基吖啶、或其衍生物。所述试剂不是HGMB1蛋白或其片段。 上述方法可以进一步包括使生物膜与有效量的干扰eDNA与DNA结合蛋白结合的试剂和/或抗菌剂(在体外)接触或向对象施用该试剂。在一方面,在不存在DNA酶的情况下施用试剂。在另一方面,施用DNA酶。在另一方面,该试剂不是HMGB1蛋白、其片段或其每一个的等效物。在另一方面,在施用所述试剂之后施用DNA酶。在一个特定方面,DNA酶是Pulmozyme。在一个实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净正电荷。在一个实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净负电荷。在一个实施方案中,干扰eDNA与DNA结合蛋白结合的试剂具有净中性电荷。 当在体外实践时,该方法可用于筛选或确认与本文公开的多肽、多核苷酸、抗体、宿主细胞、小分子和组合物具有相同、相似或相反能力的试剂。或者,它们可用于确定哪种药物最适合治疗微生物感染或治疗是否有效。例如,通过具有两个样品,例如其包含DNABII多肽和微生物DNA以及待测试剂,可以筛选新的试剂或联合疗法。第二个样品包含DNABII多肽和微生物DNA以及已知具有活性的试剂,例如抗IHF抗体或小分子以用作阳性对照。在另一方面,提供了几个样品并且将试剂以增加的稀释度添加到系统中,以确定在临床环境中可能有效治疗对象的最佳剂量。如对本领域技术人员显而易见的,可以提供包含DNABII多肽和微生物DNA的阴性对照。在另一方面,DNABII多肽和微生物DNA被可检测地标记,例如用当彼此紧密接触时将会发射信号的发光分子。在相似的条件下将样品包含有效时间,以使试剂抑制、竞争或滴定DNABII多肽与微生物DNA之间的相互作用,然后分析样品中发光分子的信号发射。如果样品发出信号,则该试剂不能有效抑制结合。 在另一方面,在微型腔室载玻片系统中实践了体外方法,其中可以从人/动物中分离引起感染的微生物(例如细菌)分离株,然后对其进行体外培养以使其生长成生物膜。将试剂(例如抗DNABII或IHF抗体)或测试试剂或潜在试剂单独添加或与另一种试剂组合添加到培养物中,同时增加或不增加潜在试剂或例如抗DNABII或IHF(或其他抗体、小分子、药剂等)的试剂的稀释度以找到在递送到存在感染的对象中时可能会有效地治疗该患者的最佳剂量。对本领域技术人员显而易见的是,可以同时进行阳性和阴性对照。 在另一方面,该方法在流动池中使用试剂(例如抗DNABII或IHF抗体)和/或潜在试剂(单独或与另一种试剂组合)在高通量平台中实践。将试剂(例如抗DNABII或IHF抗体)或潜在试剂生物膜单独添加或与另一种试剂组合添加到培养物中,同时增加或不增加潜在试剂或例如抗DNABII或IHF(或其他抗体、小分子、药剂等)的试剂的稀释度以找到在递送到存在感染的对象中时可能会有效地治疗该患者的最佳剂量。对生物膜分离物进行超声处理,以将生物膜细菌从与微生物DNA结合的DNABII多肽(例如IHF)中分离出来。通过平台上的抗DNABII或IHF抗体分离DNABII多肽-DNA复合物。然后用例如盐洗液释放微生物DNA,并用于鉴定添加的生物膜细菌。然后例如通过PCR测序鉴定释放的DNA。如果未释放DNA,则该试剂将成功执行或结合微生物DNA。如果在样品中发现了DNA,则该试剂不会干扰DNABII多肽与微生物DNA的结合。对本领域技术人员显而易见的是,可以同时进行阳性和/或阴性对照。 上述方法也可以用作诊断测试,因为给定的细菌物种对一种试剂逆转其生物膜的反应可能比另一种更好,这种快速的高通量测定系统可以使本领域的技术人员检测一组可能的抗DNABII或IHF样试剂以鉴定群组中最有效的。 这些方法的优点在于,医院中的大多数临床微生物实验室已经配备装备来使用在液体培养物中(或浮游性地)生长的细菌来执行这些类型的测定(即MIC、MBC值的测定)。对本领域技术人员显而易见的是,细菌在引起疾病时通常不会生长出浮游细菌。相反,它们以稳定的生物膜的形式生长,并且这些生物膜对抗生素、抗体或其他治疗剂的治疗具有明显的抵抗力。这种抗性是大多数MIC/MBC值无法准确预测体内功效的原因。因此,通过确定试剂的“剂量”可以在体外逆转细菌生物膜(如上所述),即使作为个性化医学的应用,申请人的临床前测定也将是临床疗效的更可靠预测指标。 除临床环境外,该方法还可用于鉴定引起感染的微生物和/或在工业环境中确认有效药剂。因此,该试剂可以用于在工业环境中治疗、抑制或滴定生物膜。 在上述方法的另一方面,按顺序地或同时地添加已知抑制潜在感染的生长的抗生素或抗微生物剂,以确定感染是否可以被抑制。在添加缺失的复合物之前,也可以将试剂添加到微生物DNA或DNABII多肽中,以检测生物膜抑制。 当在非人类动物(例如毛丝鼠)中体内实践时,该方法提供了临床前筛选,以鉴定可单独使用或与其他试剂结合使用以分解生物膜的试剂。 在另一方面,本文提供了通过向对象施用有效量的试剂从而抑制、预防或破坏微生物生物膜来抑制、预防或破坏对象生物膜的方法。这样的对象的非限制性实例包括哺乳动物,例如宠物和人类患者。 本文公开的试剂和组合物可以与其他抗微生物剂和/或表面抗原同时或相继施用。在一个特定方面,例如通过直接注射或通过吸入在感染部位局部施用。施用的其他非限制性实例包括通过一种或多种方法,其包括经皮、尿道、舌下、直肠、阴道、眼、皮下、肌内、腹膜内、鼻内、通过吸入或口服。 可以通过本文公开的方法治疗的微生物感染和疾病包括无乳链球菌( 感染也可能发生在口腔中(龋齿、牙周炎),并且是由变形链球菌( 无乳链球菌( 因此,适用于本文公开的方法的施用途径包括鼻内、肌内、尿道、气管内、皮下、真皮内、透皮、局部施用、静脉内、直肠、鼻、口服、吸入和其他肠内和肠胃外施用途径 。如果需要,可以组合给药途径,或者根据药剂和/或期望的效果进行调整。活性剂可以单剂量或多剂量施用。这些方法和适合于递送的途径的实施方案包括全身性或局部性途径。通常,适用于本文公开的方法的给药途径包括但不限于直接注射、肠内、肠胃外或吸入途径。 除吸入给药以外的肠胃外给药途径包括但不限于局部、透皮、皮下、肌内、眶内、包膜内、脊柱内、胸骨内和静脉内途径,即除通过消化道以外的任何给药途径。可以进行肠胃外给药以实现抑制剂的全身或局部递送。在需要全身递送的情况下,给药通常涉及药物制剂的侵入性或全身吸收的局部或粘膜给药。 本文公开的药剂也可以通过肠内给药递送给对象。肠内给药途径包括但不限于口服和直肠给药(例如使用栓剂)。 通过皮肤或粘膜施用活性物质的方法包括但不限于合适药物制剂的局部施用、经皮传输、透皮传输、注射和表皮施用。对于透皮传输,吸收促进剂或离子电渗疗法是合适的方法。离子电渗疗法可以使用可商购的“贴剂”来实现,该贴剂通过电脉冲连续不断地将其产品连续几天或几天以上地递送。 在本文公开的方法的各种实施方案中,通过吸入、注射或口服连续地以天为基础每天至少一次(QD)(在各种实施方案中每天两次(BID)、三次(TID)、甚至四次 )施用试剂。通常,治疗有效日剂量可以为至少约1 mg、或至少约10 mg、或至少约100 mg、或约200至约500 mg,有时取决于化合物,高达约1 g至约2.5 g。 可以根据本文公开的方法,使用胶囊、片剂、口服混悬剂、用于肌内注射的混悬剂、用于静脉内输注的混悬剂、用于局部施用的软膏或乳膏、或用于关节内注射的混悬剂来完成给药。 本文所述的组合物的剂量、毒性和治疗功效可以通过细胞培养或实验动物中的标准药学方法来确定,例如,以确定LD50(致死人群的50%的剂量)和ED50( 在50%的人群中治疗有效的剂量)。毒性和治疗效果之间的剂量比是治疗指数,可以表示为LD50/ED50之比。在某些实施方案中,组合物表现出高治疗指数。尽管可以使用表现出毒副作用的化合物,但应注意设计一种将这类化合物靶向受影响组织部位的递送系统,以最大程度地减少对未感染细胞的潜在损害,从而减少副作用。 从细胞培养测定法和动物研究中获得的数据可用于配制用于人类的剂量范围。在某些实施方案中,此类化合物的剂量在循环浓度范围内,其包括几乎没有毒性或无毒性的ED50。取决于所用剂型和所用给药途径,剂量可以在此范围内变化。对于在该方法中使用的任何化合物,可以从细胞培养测定中初步估算出治疗有效剂量。可以在动物模型中制定剂量,以达到包括在细胞培养物中确定的IC50(即,达到症状最大抑制一半的试验化合物的浓度)的循环血浆浓度范围。这些信息可用于更准确地确定对人体有用的剂量。血浆水平可通过例如高效液相色谱法进行测量。 在一些实施方案中,足以实现治疗或预防作用的组合物的有效量为每次施用每千克体重约0.000001 mg至每次施用每千克体重约10,000 mg。合适地,剂量范围是从每次施用每公斤体重约0.0001 mg到每次施用每公斤体重约100 mg。可以以初始剂量提供给药,然后以一个或多个“加强”剂量提供。初始剂量后一天、两天、三天、一周、两周、三周、一、二、三、六或十二个月可以提供加强剂量。在一些实施方案中,在评估对象对先前施用的反应之后施用加强剂量。 本领域技术人员将理解,某些因素可影响有效治疗对象所需的剂量和时机,包括但不限于疾病或病症的严重程度、先前的治疗、患者的总体健康和/或年龄、以及其他疾病。此外,用治疗有效量的本文所述的治疗组合物治疗对象可以包括单一治疗或一系列治疗。 抗体及其衍生物 本公开还提供了结合和/或特异性识别和结合B DNA的抗体,用于本文公开的方法。抗体可以是本文所述的各种抗体中的任何一种,这样的非限制性实例包括多克隆抗体、单克隆抗体、嵌合抗体、人抗体、贴面化抗体( veneered antibody)、双抗体、人源化抗体、抗体衍生物、重组人源化抗体或其各自的衍生物或片段。在一方面,该片段包含抗体的CDR,或基本上由其组成,或由其组成。在一方面,抗体被可检测地标记或进一步包含与其偶联的可检测标记。还提供了产生本文公开的单克隆抗体的杂交瘤细胞系。本文进一步提供了组合物,其包含一种或多种上述实施方案,或基本上由其组成,或由其组成。还提供了编码抗体和片段的氨基酸序列的多核苷酸,以及产生重组或化学合成抗体多肽及其片段的方法。抗体多肽可以在真核或原核细胞中产生,或通过本领域已知的和本文描述的其他方法产生。 该方法的变化包括佐剂的修饰、施用的途径和部位、每个部位的注射量以及每只动物的部位的数量,以优化动物的生产和人道处理。例如,佐剂通常用于改善或增强对抗原的免疫应答。大多数佐剂提供注射位点抗原贮库,从而允许抗原积聚释放到引流淋巴结中。其他佐剂包括在大表面积上促进蛋白质抗原分子浓缩的表面活性剂和免疫刺激分子。用于产生多克隆抗体的佐剂的非限制性实例包括弗氏佐剂、Ribi佐剂系统和Titermax、可以使用本领域已知的方法产生多克隆抗体,其中一些描述于美国专利号7,279,559;7,119,179;7,060,800;6,709,659;6,656,746;6,322,788;5,686,073;和5,670,153。 单克隆抗体可以使用本领域已知的和文献中充分描述的常规杂交瘤技术来产生。例如,通过以下来产生杂交瘤:融合合适的永生细胞系(例如,骨髓瘤细胞系,例如但不限于Sp2/0、Sp2/0-AG14、NSO、NS1、NS2、AE-1, L.5、P3X63Ag8、653、Sp2、SA3、Sp2 MAI、Sp2 SS1、Sp2 SA5、U397、MIA 144、ACT IV、MOLT4、DA-1、JURKAT、WEHI, K-562, COS, RAJI、NIH 313、HL-60、MLA 144、NAMAIWA, NEURO 2A、CHO, PerC.6, YB2/O)等、或杂种骨髓瘤、其融合产物、或衍生自其的任何细胞或融合细胞、或本领域已知的任何其他合适的细胞系(参见以下网址的技术,例如atcc.org, lifetech.com,最新访问时间为2007年11月26日),其具有产生抗体的细胞,例如但不限于:分离的或克隆的脾脏、外周血、淋巴、扁桃体、或其他免疫或包含B细胞的细胞、或表达重链或轻链恒定或可变或构架或CDR序列(内源或异源核酸)(重组或内源)的任何其他细胞、病毒、细菌、藻类、原核、两栖动物、昆虫、爬行动物、鱼、哺乳动物、啮齿动物、马、绵羊、山羊、绵羊、灵长类动物、真核生物、基因组DNA、cDNA、rDNA、线粒体DNA或RNA、叶绿体DNA或RNA、hnRNA、mRNA、tRNA、单链、双链或三链的、杂交的等或其任何组合。产生抗体的细胞也可以从已经用目的抗原免疫然后筛选目的活性的人或其他合适动物的外周血或脾或淋巴结中获得。任何其他合适的宿主细胞也可以用于表达编码本发明的抗体、其指定片段或变体的异源或内源核酸。融合细胞(杂交瘤)或重组细胞可以使用选择性培养条件或其他合适的已知方法分离,并通过有限稀释或细胞分选或其他已知方法克隆。 可以使用产生或分离具有必要特异性的抗体的其他合适方法,包括但不限于使用本领域已知的方法从肽或蛋白质文库中选择重组抗体的方法(例如但不限于噬菌体、核糖体、寡核苷酸、cDNA等展示文库;例如可从各种商业供应商获得的,例如MorphoSys(Martinsreid/Planegg, Del.)、BioInvent (Lund, Sweden)、Affitech (Oslo,Norway))。专利文献中描述了本领域已知的方法,其中一些包括美国专利号4,704,692、5,723,323、5,763,192、5,814,476、5,817,483、5,824,514和5,976,862。替代方法依赖于如本领域已知的和 /或如本文所述能够产生人类抗体库的转基因动物(例如SCID小鼠,Nguyenet al. (1977) Microbiol. Immunol. 41:901-907 (1997);Sandhu et al. (1996)Crit, Rev. Biotechnol. 16:95-118;Eren et al. (1998) Mumma 93:154-161)的免疫。此类技术包括但不限于:核糖体展示 Wanes et al. (1997) Proc. Natl. Acad. Sci.USA 94:4937-4942; Hanes et al. (1998) Proc. Natl. Acad. Sci. USA 95:14130-14135);单细胞抗体产生技术(例如,选择的淋巴细胞抗体方法(“ SLAM”)(美国专利号5,627,052;Wen et al. (1987) J. Immunol 17:887-892; Babcook et al. (1996) Proc.Natl. Acad. Sci. USA 93:7843-7848);凝胶微滴和流式细胞术(Powell et al. (1990)Biotechnol. 8:333-337;One Cell Systems, (Cambridge, Mass.);Gray et al. (1995)J. Imm. Meth. 182:155-163;以及Kenny et al. (1995) Bio. Technol. 13:787-790);B细胞选择(Steenbakkers et al. (1994) Molec. Biol. Reports 19:125-134)。 本公开的抗体衍生物也可以通过以下方法来制备:将编码本文公开的抗体的多核苷酸递送至合适的宿主以提供转基因动物或哺乳动物(例如山羊、牛,马、绵羊等在),其在奶中产生这种抗体 。这些方法是本领域已知的,并且例如描述于美国专利号5,827,690、5,849,992、4,873,316、5,849,992、5,994,616、5,565,362和5,304,489。 术语“抗体衍生物”包括对抗体或片段的线性多肽序列的翻译后修饰。例如,美国专利号6,602,684 B1描述了一种用于产生修饰的乙二醇形式的抗体的方法,包括完整的抗体分子、抗体片段或融合蛋白,其包含与免疫球蛋白的Fc区等效的区域,具有增强的铁介导的细胞毒性,并因此产生糖蛋白。 本文公开的抗体还包括这样的衍生物,其通过任何类型的分子共价附接至抗体而修饰,使得共价附接不会阻止抗体产生抗独特型应答。抗体衍生物包括但不限于已经通过糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、通过已知的保护/封闭基团衍生化、蛋白水解切割、与细胞配体或其他蛋白质的连接等修饰的抗体。此外,衍生物可包含一种或多种非经典氨基酸。 抗体衍生物也可以通过以下方法来制备:递送本文公开的多核苷酸以提供在植物部分中或在从中培养的细胞中产生此类抗体、特定部分或变体的转基因植物和培养的植物细胞(例如但不限于烟草、玉米和浮萍)。例如Cramer et al. (1999) Curr. Top.Microbol. Immunol. 240:95-118和其中引用的参考文献描述了表达大量重组蛋白的转基因烟草叶的生产,例如使用诱导型启动子。转基因玉米已用于以商业生产水平表达哺乳动物蛋白,其生物学活性等同于其他重组系统中产生的或从天然来源纯化的生物活性。参见例如Hood et al. (1999) Adv. Exp. Med. Biol. 464:127-147及其中引用的参考文献。还已经从包括抗体片段的转基因植物种子中大量生产抗体衍生物,例如单链抗体(scFv),包括烟草种子和马铃薯块茎。参见,例如Conrad et al. (1998) Plant Mol. Biol. 38:101-109 及其中引用的参考文献。因此,根据已知方法,也可以使用转基因植物产生抗体。 抗体衍生物也可以通过以下方法来产生:例如添加外源序列以修饰免疫原性或减少、增强或修饰结合、亲和力、开启速率、关闭速率、活动性、特异性、半衰期或任何其他合适的特性。通常,部分或全部非人或人CDR序列得以保留,而可变区和恒定区的非人序列被人或其他氨基酸或来自其他同种型的可变区或恒定区代替。 通常,CDR残基直接且最实质地参与影响抗原结合。可以使用任何已知方法来进行抗体的人源化或工程改造,例如但不限于美国专利号5,723,323、5,976,862、5,824,514、5,817,483、5,814,476、5,763,192、5,723,323、5,766,886、5,714,352、6,204,023、6,180,370、5,693,762、5,530,101、5,585,089、5,225,539和4,816,567中所述的方法。 可基于使用标准分子生物学技术制备的参考单克隆抗体的序列来制备本公开的嵌合、人源化或灵长类化的抗体。可以从感兴趣的杂交瘤中获得编码重链和轻链免疫球蛋白的DNA,并使用标准分子生物学技术对其进行改造,使其包含非参考(例如人)免疫球蛋白序列。例如,为了产生嵌合抗体,可以使用本领域已知的方法(美国专利号4,816,567)将鼠类可变区连接至人恒定区。为了产生人源化抗体,可以使用本领域已知的方法(美国专利号5,225,539和美国专利号5,530,101、5,585,089、5,693,762和6,180,370)将鼠CDR区插入人框架。类似地,为了产生灵长类化的抗体,可以使用本领域已知的方法(WO 93/02108和WO99/55369)将鼠的CDR区插入灵长类框架中。 制备部分至完全人抗体的技术是本领域已知的,并且可以使用任何此类技术。根据一个实施方案,在转基因小鼠中制备完全人抗体序列,该转基因小鼠已被工程化以表达人重链和轻链抗体基因。已经制备了可以产生不同类别的抗体的这种转基因小鼠的多种菌株。可以融合产生所需抗体的转基因小鼠的B细胞,以制备杂交瘤细胞系,以连续产生所需抗体。(参见例如,Russel et al. (2000) Infection and Immunity April 2000:1820-1826;Gallo et al. (2000) European J. of Immun. 30:534-540;Green (1999) J. ofImmun. Methods 231:11-23;Yang et al. (1999A) J. of Leukocyte Biology 66:401-410;Yang (1999B) Cancer Research 59(6):1236-1243;Jakobovits (1998) AdvancedDrug Reviews 31:33-42;Green and Jakobovits (1998) J. Exp. Med. 188(3):483-495;Jakobovits (1998) Exp. Opin. Invest. Drugs 7(4):607-614;Tsuda et al.(1997) Genomics 42:413-421;Sherman-Gold (1997) Genetic Engineering News 17(14); Mendez et al. (1997) Nature Genetics 15:146-156;Jakobovits (1996) Weir's Handbook of Experimental Immunology, The Integrated Immune System Vol. IV,194.1-194.7;Jakobovits (1995) Current Opinion in Biotechnology 6:561-566;Mendez et al. (1995) Genomics 26:294-307;Jakobovits (1994) Current Biology 4(8):761-763;Arbones et al. (1994) Immunity 1(4):247-260;Jakobovits (1993)Nature 362(6417):255-258;Jakobovits et al. (1993) Proc. Natl. Acad. Sci. USA90(6):2551-2555;以及美国专利号6,075,181)。 本文公开的抗体也可以被修饰以产生嵌合抗体。嵌合抗体是指抗体重链和轻链的各个域都被一种以上的DNA编码的抗体。参见例如美国专利号4,816,567。 替代地,本文公开的抗体也可以被修饰以产生贴面化抗体。贴面化抗体是这样的抗体,其中一个物种的抗体的外部氨基酸残基被明智地替换或“贴面”为第二物种的抗体,以使第一个物种的抗体在第二个物种中不具有免疫原性,从而降低免疫原性抗体。由于蛋白质的抗原性主要取决于其表面的性质,因此可以通过替换暴露的残基来降低抗体的免疫原性,所述残基不同于另一种哺乳动物物种抗体中通常发现的残基。明智地替换外部残留物应该对内部域或域间接触几乎没有或没有影响。因此,由于限于可变区框架残基的改变,配体结合性质应不受影响。该过程被称为“贴面”,因为仅抗体的外表面或皮肤被改变,支持残基保持不受干扰。 “贴面”的过程利用了人类抗体可变域的可用序列数据,其由Kabat et al.(1987) Sequences of Proteins of Immunological interest, 4th ed., Bethesda,Md., National Institutes of Health编译(此数据库的更新 ),以及其他可访问的美国和外国数据库(核酸和蛋白质)。 用于产生贴面化抗体的方法的非限制性实例包括EP519596、美国专利号6,797,492,并且描述于Padlan et al. (1991) Mol. Immunol. 28(4-5):489-498。 术语“抗体衍生物”还包括“双抗体”,其是具有两个抗原结合位点的小抗体片段,其中片段在同一多肽链中包含连接至轻链可变域(VL)的重链可变域(VH) 。 (参见例如EP404,097、WO 93/11161、以及Hollinger et al. (1993) Proc. Natl. Acad. Sci. USA90:6444-6448)。通过使用太短以至于不允许同一条链上两个结构域之间配对的接头,这些结构域被迫与另一条链的互补结构域配对并产生两个抗原结合位点。(还参见Chen等人的美国专利号6,632,926,其公开了这样的抗体变体,其具有一个或多个氨基酸插入亲本抗体的高变区中,并且对靶抗原的结合亲和力比亲本抗体对抗原的结合亲和力强至少约两倍)。 术语“抗体衍生物”还包括工程化的抗体分子、片段和单个结构域,例如scFv、dAb、纳米抗体、微型抗体、单一体和Affibodies & Hudson (2005) Nature Biotech 23(9):1126-36、美国专利 申请公开号2006/0211088、PCT国际申请公开号WO 2007/059782、美国专利 5,831,012。 术语“抗体衍生物”还包括“线性抗体”。 制备线性抗体的方法在本领域中是已知的,并在Zapata et al. (1995) Protein Eng. 8(10):1057-1062中描述。简而言之,这些抗体包含一对串联的Ed区段(V 可通过已知方法从重组细胞培养物中回收和纯化本文公开的抗体,所述方法包括但不限于蛋白A纯化、硫酸铵或乙醇沉淀、酸提取、阴离子或阳离子交换色谱、磷酸纤维素色谱、疏水相互作用色谱、亲和色谱、羟基磷灰石色谱和凝集素色谱。高效液相色谱法(“HPLC”)也可用于纯化。 本公开内容的抗体包括天然纯化的产物、化学合成程序的产物以及通过重组技术从真核宿主(包括例如酵母、高等植物、昆虫和哺乳动物细胞)或可替代地从如上所述的原核生物宿主产生的产物。许多抗体生产系统描述于Birch & Radner (2006) Adv. DrugDelivery Rev. 58: 671-685。 如果被测试的抗体与蛋白质或多肽结合,则被测试的抗体和本公开内容提供的抗体是等效的。还可以通过确定被测试的抗体是否阻止本文公开的抗体结合与该抗体正常反应的蛋白质或多肽,而无需过多实验就可以确定抗体是否具有与本文公开的抗体相同的特异性。如果所测试的抗体与本文公开的抗体竞争,如本文公开的单克隆抗体的结合减少所显示,则这两种抗体很可能与相同或密切相关的表位结合。或者,可以将本文公开的抗体与通常与之反应的蛋白质预孵育,并确定所测试的抗体结合抗原的能力是否受到抑制。如果所测试的抗体被抑制,则极有可能具有与本文公开的抗体相同或密切相关的表位特异性。 术语“抗体”还意图包括所有免疫球蛋白同种型和亚类的抗体。单克隆抗体的特定同种型可以通过从初始融合中选择直接制备,或者通过使用sib选择技术来使用Steplewski et al. (1985) Proc. Natl. Acad. Sci. USA 82:8653 or Spira et al.(1984) J. Immunol. Methods 74:307中描述的方法分离类别转换变体,以从分泌不同同种型单克隆抗体的亲本杂交瘤中进行间接制备。或者,可以使用重组DNA技术。 具有本文所述单克隆抗体特异性的其他单克隆抗体的分离也可以由本领域普通技术人员通过产生抗独特型抗体来完成。Herlyn et al. (1986) Science 232:100。抗独特型抗体是识别存在于感兴趣的单克隆抗体上的独特决定簇的抗体。 在本文公开的一些方面,对抗体进行可检测或治疗性标记是有用的。合适的标签如上所述。偶联针对这些试剂的抗体的方法是本领域已知的。仅出于举例说明的目的,可以用可检测的部分(例如放射性原子、发色团、荧光团)等标记抗体。这样的标记抗体可以用于体内或分离的测试样品中的诊断技术。 抗体与低分子量半抗原的偶联可以增加测定中抗体的敏感性。然后可以通过第二反应特异性地检测半抗原。例如,通常使用半抗原,例如生物素(其可与抗生物素蛋白反应)或二硝基苯酚、吡哆醛和荧光素(其可与特定的抗半抗原抗体反应)。参见上文的Harlow和Lane(1988)。 本发明的抗体的可变区可以通过突变VH和/或VL CDR 1、CDR 2和/或CDR 3区域内的氨基酸残基来修饰以改善抗体的一种或多种结合特性(例如亲和力)。可以通过定点诱变或PCR介导的诱变引入突变,并且可以在适当的体外或体内测定中评估对抗体结合或其他目的功能特性的影响。在某些实施方案中,引入保守修饰并且通常改变CDR区内不超过1、2、3、4或5个残基。突变可以是氨基酸取代、添加或缺失。 可以对抗体进行框架修饰以降低免疫原性,例如,通过将一个或多个框架残基“突变回”相应的种系序列。 另外,本文公开的抗体可以被工程化以包括Fc区内的修饰,以改变抗体的一种或多种功能特性,例如血清半定量、补体固定、Fc受体结合和/或抗原依赖性细胞毒性。此类修饰包括但不限于铰链区中半胱氨酸残基的数目的改变,以促进轻链和重链的组装或增加或降低抗体的稳定性(美国专利号5,677,425),或者Fc铰链区中的氨基酸突变以降低抗体的生物学半衰期(美国专利号6,165,745)。 另外,本文公开的抗体可以被化学修饰。可以例如通过修饰抗体序列内一个或多个糖基化位点以增加抗体对抗原的亲和力来改变抗体的糖基化(美国专利号5,714,350和6,350,861)。或者,为了增加抗体依赖性细胞介导的细胞毒性,可以通过在具有改变的糖基化机制的宿主细胞中表达抗体来获得具有减少的岩藻糖基残基量的次岩藻糖基化抗体或具有增加的二等分GlcNac结构的抗体。(Shields, R. L. et al. (2002) J. Biol. Chem.277:26733-26740; Umana et al. (1999) Nat. Biotech. 17:176-180)。 本文公开的抗体可以被聚乙二醇化以增加生物学半衰期,其通过在一个或多个PEG基团变得连接至抗体或抗体片段的条件下使抗体或其片段与聚乙二醇(PEG)或PEG的反应性酯或醛衍生物反应。可以通过与反应性PEG分子(或类似的反应性水溶性聚合物)的酰化反应或烷基化反应来进行抗体聚乙二醇化。如本文所用,术语“聚乙二醇”旨在涵盖已用于衍生化其他蛋白质的任何形式的PEG,例如单(C1-C10)烷氧基-或芳氧基-聚乙二醇或聚乙二醇-马来酰亚胺。待聚乙二醇化的抗体可以是无糖基化的抗体。聚乙二醇化蛋白质的方法是本领域已知的,并且可以应用于本文公开的抗体(EP 0154316和EP 0401384)。 另外,可以通过将抗体的抗原结合区与血清蛋白(例如人血清白蛋白)缀合或融合来化学修饰抗体,以增加所得分子的半衰期。例如在EP 0322094和EP 0486525中描述了这种方法。 本公开的抗体或其片段可以与诊断剂缀合并且可以诊断地用于例如监测疾病的发展或进程并确定给定治疗方案的功效。诊断剂的实例包括酶、辅基、荧光材料、发光材料、生物发光材料、放射性材料、使用各种正电子发射断层摄影术的正电子发射金属、以及非放射性顺磁性金属离子。可以使用本领域已知的技术,将可检测物质直接偶联或偶联至抗体或其片段,或通过接头间接偶联或偶联。合适的酶的例子包括辣根过氧化物酶、碱性磷酸酶、β-半乳糖苷酶、或乙酰胆碱酯酶。合适的修复基团复合物的例子包括链霉亲和素/生物素和抗生物素蛋白/生物素。合适的荧光材料的例子包括伞形酮、荧光素、异硫氰酸荧光素、若丹明、二氯三嗪胺荧光素、丹磺酰氯或藻红蛋白。发光材料的实例包括鲁米诺。生物发光材料的实例包括萤光素酶、萤光素和水母发光蛋白。合适的放射性物质的例子包括 此外,本公开的抗体或其片段可以与治疗剂缀合。合适的治疗剂包括:紫杉醇,细胞松弛素B,短杆菌肽D,溴化乙锭,依米丁,丝裂霉素,依托泊苷,替诺泊苷,长春新碱,长春碱,秋水仙碱,阿霉素,柔红霉素,二羟基蒽醌二酮,米托蒽醌,光神霉素(mithramycin),放线菌素D,1-去氢睾酮,糖皮质激素,普鲁卡因,丁卡因,利多卡因,普萘洛尔和嘌呤霉素,抗代谢药(例如甲氨蝶呤、6-巯基嘌呤、6-硫鸟嘌呤、阿糖胞苷、氟达拉宾、5-氟尿嘧啶、达卡巴嗪、羟基脲、天冬酰胺酶、吉西他滨、克拉屈滨),烷化剂(例如氮芥、thioepa、苯丁酸氮芥、美法仑、卡莫司汀(BSNU)、洛莫司汀(CCNU)、环磷酰胺、白消安、二溴甘露醇、链脲佐菌素、达卡巴嗪(DTIC)、丙卡巴肼、丝裂霉素C、顺铂和其他铂衍生物(例如卡铂)),抗生素(例如放线菌素、博来霉素、柔红霉素(旧称道诺霉素)、阿霉素、伊达比星、光神霉素、丝裂霉素、米托蒽醌、普卡霉素、氨茴霉素(AMC)),白喉毒素和相关分子(例如白喉A链及其活性片段和杂种分子),蓖麻毒蛋白毒素(例如蓖麻毒蛋白A或去糖基化的蓖麻毒蛋白A链毒素),霍乱毒素,志贺样毒素(SLT-I、SLT-II、SLT-IIV),LT毒素,C3毒素,志贺毒素,百日咳毒素,破伤风毒素,大豆Bowman-Birk蛋白酶抑制剂,假单胞菌外毒素,芦荟素,皂苷,蒴莲根毒素(modeccin),gelanin,相思豆毒素A链,蒴莲根毒素A链,α-八叠球菌素(sarcin),Aleurites fordii蛋白,香石竹毒素(dianthin )蛋白,美洲商陆( 其他合适的共轭分子包括核糖核酸酶(RNA酶)、DNA酶I、反义核酸、抑制性RNA分子(例如siRNA分子)、免疫刺激性核酸、适体、核酶、三链体形成分子和外部指导序列。适体是长度为15-50个碱基的小核酸,其折叠成限定的二级和三级结构,例如茎环或G四联体,并且可以结合小分子(例如ATP(美国专利号5,631,146)和茶碱(美国专利号5,580,737))以及大分子(例如逆转录酶(美国专利号5,786,462)和凝血酶(美国专利号5,543,293))。核酶是能够在分子内或分子间催化化学反应的核酸分子。核酶通常通过靶底物的识别和结合以及随后的切割来切割核酸底物。具有三链体形成功能的核酸分子可以通过形成三链体与双链或单链核酸相互作用,其中三链DNA形成依赖于Watson-Crick和Hoogsteen碱基配对的复合物。三链体分子可以高亲和力和特异性结合靶区域。 功能性核酸分子可以充当靶分子具有的特定活性的效应子、抑制剂、调节剂和刺激剂,或者功能性核酸分子可以具有独立于任何其他分子的de novo 活性。 治疗剂可以使用多种可用方法中的任何一种直接或间接地与抗体连接。例如,通过二硫键形成、使用交联剂 (例如N-琥珀酰3-(2-吡啶基二硫代)丙酸酸酯(SPDP))、或通过抗体的Fc区中的糖部分,将试剂附着在还原抗体成分的铰链区。(Yu et al. 1994 Int. J.Cancer 56: 244; Upeslacis et al., “Modification of Antibodies by ChemicalMethods,” in Monoclonal antibodies: principles and applications, Birch et al.(eds.), pages 187-230 (Wiley-Liss, Inc. 1995); Price, “Production andCharacterization of Synthetic Peptide-Derived Antibodies,” in Monoclonalantibodies: Production, engineering and clinical application, Ritter et al.(eds.), pages 60-84 (Cambridge University Press 1995))。 将治疗剂与抗体缀合的技术是众所周知的(Amon et al. “MonoclonalAntibodies For Immunotargeting Of Drugs In Cancer Therapy,” in MonoclonalAntibodies And Cancer Therapy; Reisfeld et al. (eds.), pp. 243-56 (Alan R.Liss, Inc. 1985); Hellstrom et al. “Antibodies For Drug Delivery,” inControlled Drug Delivery (2nd Ed.); Robinson et al. (eds.), pp. 623-53(Marcel Dekker, Inc. 1987); Thorpe “Antibody Carriers Of Cytotoxic Agents InCancer Therapy: A Review,” in Monoclonal Antibodies '84: Biological AndClinical Applications, Pinchera et al. (eds.), pp. 475-506 (1985); “Analysis,Results, And Future Prospective Of The Therapeutic Use Of RadiolabeledAntibody in Cancer Therapy,” in Monoclonal Antibodies For Cancer DetectionAnd Therapy, Baldwin et al. (eds.), pp. 303-16 (Academic Press 1985), 以及Thorpe et al. “The Preparation And Cytotoxic Properties Of Antibody-ToxinConjugates,” (1982) Immunol. Rev. 62:119-58)。 本文公开的抗体或其抗原结合区可与另一种功能分子(例如另一种抗体或受体的配体)连接,以产生与至少两种或更多种不同结合位点或目标分子结合的双特异性或多特异性分子。抗体与一个或多个其他结合分子(例如另一种抗体、抗体片段、肽或结合模拟物)的连接可以例如通过化学偶联、遗传融合或非共价结合来完成。除了第一和第二靶表位之外,多特异性分子还可包括第三结合特异性。 可以使用本领域已知的方法制备双特异性和多特异性分子。例如,高特异性分子的每个结合单元可以分开产生,然后彼此缀合。当结合分子是蛋白质或肽时,多种偶联剂或交联剂可用于共价结合。交联剂的实例包括蛋白A、碳二亚胺、N-琥珀酰亚胺基-S-乙酰基-硫代乙酸酯(SATA)、5,5'-二硫代双(2-硝基小苯甲酸)(DTNB)、邻苯二甲酰亚胺(oPDM)、N-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯(SPDP)和磺基琥珀酰亚胺基4-(N-马来酰亚胺基甲基)环已烷-1-羧酸酯(磺基-SMCC)(Karpovsky et al. (1984) J. Exp. Med. 160:1686; Liu et al. (1985) Proc. Natl. Acad. Sci. USA 82:8648)。当结合分子是抗体时,它们可以通过两条重链的C末端铰链区的巯基键结合。 本公开的抗体或其片段可以与对结合有抗体的细胞有毒的部分连接以形成“耗竭”抗体。 本文公开的抗体也可以附着于固体支持物,其对于免疫测定或靶抗原的纯化特别有用。这样的固体支持物包括但不限于玻璃、纤维素、聚丙烯酰胺、尼龙、聚苯乙烯、聚氯乙烯或聚丙烯。 抗体也可以与许多不同的载体结合。因此,本公开内容还提供了包含抗体和另一种活性或惰性物质的组合物。众所周知的载体的例子包括玻璃、聚苯乙烯、聚丙烯、聚乙烯、葡聚糖、尼龙、淀粉酶、天然和改性纤维素、聚丙烯酰胺、琼脂糖和磁铁。为了本文公开的目的,载体的性质可以是可溶的或不溶的。本领域技术人员将知道用于结合单克隆抗体的其他合适的载体,或者将能够使用常规实验确定这种载体。 在本文提供的抗体的一些方面,该抗体是全长抗体。 在本文提供的抗体的一些方面,该抗体是单克隆抗体。 在本文提供的抗体的一些方面,该抗体是嵌合的或人源化的。 在本文提供的抗体的一些方面,该抗体选自Fab、F(ab)'2、Fab’、scF 在本文提供的抗体的一些方面,该抗体包含Fc结构域。在本文提供的抗体的一些方面,该抗体是非人类动物(例如大鼠、绵羊、牛、犬、猫或兔)抗体。在本文提供的抗体的一些方面,该抗体是人或人源化抗体或在人中是非免疫原性的。 在本文提供的抗体的一些方面,该抗体包含人抗体框架区。 在其他方面,本文提供的抗体的CDR中的一个或多个氨基酸残基被另一氨基酸取代。就相同氨基酸家族内的取代而言,该取代可以是“保守的”。天然存在的氨基酸可分为以下四个家族,并且在这些家族中将进行保守取代。 1)具有碱性侧链的氨基酸:赖氨酸,精氨酸,组氨酸。 2)具有酸性侧链的氨基酸:天冬氨酸,谷氨酸。 3)具有不带电荷的极性侧链的氨基酸:天冬酰胺,谷氨酰胺,丝氨酸,苏氨酸,酪氨酸。 4)具有非极性侧链的氨基酸:甘氨酸,丙氨酸,缬氨酸,亮氨酸,异亮氨酸,脯氨酸,苯丙氨酸,蛋氨酸,色氨酸,半胱氨酸。 在另一方面,一个或多个氨基酸残基被添加至抗体的一个或多个CDR或从抗体的一个或多个CDR中缺失。这样的添加或缺失发生在CDR的N或C末端或CDR内的位置。 通过通过氨基酸的添加、缺失或取代来改变抗体的CDR的氨基酸序列,可以获得各种效果,例如对靶抗原的增加的结合亲和力。 应当理解,包含这种变化的CDR序列的本公开的抗体仍然以与所公开的抗体相似的特异性和敏感性概况结合DNABII蛋白。这可以通过结合测定法进行测试。 另一方面,如使用适当的测定和筛选所确定的,抗体的特征在于具有免疫优势和免疫保护性。 抗体的功能分析 本文公开的抗体可以用于纯化本文公开的多肽和鉴定生物学等效的多肽和/或多核苷酸。它们也可以用于鉴定修饰本文公开的多肽的功能的试剂。这些抗体包括多克隆抗血清、单克隆抗体和衍生自这些制剂的各种试剂,其是本领域技术人员熟悉并在上文中描述过的。 中和由鉴定的基因编码的蛋白质的活性的抗体也可以在体内和体外使用,以通过将这种中和抗体加入体内和体外测试系统中来证明其功能。它们还可用作调节本文公开的多肽活性的药物。 各种抗体制剂也可用于分析方法,例如ELISA测定法或Western印迹法,以证明测试细胞在体外或体内由鉴定的基因编码的蛋白质表达。也可以通过使用适当的多克隆抗血清和衍生自实验样品的样品来鉴定代谢过程中蛋白酶降解产生的此类蛋白质的片段。 本文公开的抗体可单独或与基于肽或蛋白质的疫苗或基于树突细胞的疫苗组合用于疫苗接种或加强疫苗接种。 组合物 本公开进一步提供了组合物,其包含以下中的一种、两种或三种或更多种,或基本上由其组成,或由其组成:干扰生物膜中多胺与DNA结合的试剂,从生物膜中消耗阳离子的试剂,干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂,干扰eDNA与DNA结合蛋白结合的试剂和/或抗菌剂。在一方面,所述组合物不包括HMGB1蛋白、其片段或其每一个的等效物,或基本上不由其组成,或不由其组成。在另一方面,其包括DNA酶,或基本上由其组成,或由其组成。在一方面,其不包括DNA酶,或基本上不由其组成,或不由其组成。在一个实施方案中,所述组合物包括干扰生物膜中多胺与DNA结合的试剂以及干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂,或基本上由其组成,或由其组成。在第二实施方案中,所述组合物包括从生物膜中消耗阳离子的试剂、干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂以及干扰eDNA与DNA结合蛋白结合的试剂,或基本上由其组成,或由其组成。在第三实施方案中,所述组合物包括从干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂以及干扰eDNA与DNA结合蛋白结合的试剂,或基本上由其组成,或由其组成。在第四实施方案中,所述组合物包括干扰生物膜中多胺与DNA结合的试剂以及干扰eDNA与DNA结合蛋白结合的试剂,或基本上由其组成,或由其组成。在第五实施方案中,所述组合物包括干扰生物膜中多胺与DNA结合的试剂以及干扰eDNA与DNA结合蛋白结合的试剂和/或抗菌剂,或基本上由其组成,或由其组成。在第六实施方案中,所述组合物包括干扰生物膜中多胺与DNA结合的试剂、从生物膜中消耗阳离子的试剂以及干扰eDNA与DNA结合蛋白结合的试剂,或基本上由其组成,或由其组成。在第七实施方案中,所述组合物包括干扰生物膜中多胺与DNA结合的试剂、从生物膜中消耗阳离子的试剂以及干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂,或基本上由其组成,或由其组成。 本公开的组合物可以进一步包括药学上可接受的载体,或基本上由其组成,或由其组成。 在一方面,所述干扰生物膜中多胺与DNA结合的试剂包括以下中的一种或多种:多胺类似物二氟甲基鸟氨酸、反式-4-甲基环己胺、沙多齐特、甲基乙二醛双[脒基腙](MGBG)、1-氨基氧基-3-氨基丙烷、奥沙利铂、顺铂和/或二环己胺、其任何衍生物、或其盐。在另一方面,所述从生物膜中消耗阳离子的试剂包括以下中的一种或多种:阳离子交换树脂、氨基多羧酸、冠醚、氮杂冠、或穴醚、磺酸盐、磺丙基、磷酸纤维素、P11磷酸纤维素和/或硫酸肝素、或其衍生物或类似物。在另一方面,所述干扰生物膜或其局部环境中B-DNA向Z-DNA转化的试剂包括以下中的一种或多种:HMGB1蛋白、其片段或其每一个的等效物,抗B-DNA抗体或其片段或衍生物,和/或氯喹、或其任何衍生物。在一个特定的方面,所述干扰eDNA与DNA结合蛋白结合的试剂包括以下中的一种或多种:抗DNABII抗体、抗IHF抗体和/或抗HU抗体、或其每一个的片段。 进一步提供了组合物。所述组合物包含载体和本文公开的分离的多肽、本文公开的分离的多核苷酸、本文公开的载体、本文公开的分离的宿主细胞,本文公开的小分子或抗体中的一种或多种。载体可以是固体支持物或药学上可接受的载体中的一种或多种。组合物可以进一步包含佐剂或其他适合作为疫苗施用的组分。在一方面,组合物与一种或多种药学上可接受的赋形剂、稀释剂、载体和/或佐剂一起配制。另外,本公开的组合物的实施方案包括一种或多种本文公开的分离的多肽、本文公开的分离的多核苷酸、本文公开的载体、小分子、本文公开的分离的宿主细胞或本公开的抗体,其与一种或多种药学上可接受的物质配制而成。 对于口服制剂,如本文所述的分离或重组的多肽、如本文所述的分离或重组的多核苷酸、如本文所述的载体、如本文所述的分离的宿主细胞、如本文所述的小分子或抗体中的任何一种或多种,可以单独使用或以本文公开的药物制剂使用,所述药物制剂包含化合物或基本上由其组成,所述化合物与合适的添加剂组合以制备片剂、散剂、颗粒剂或胶囊剂,例如与常规添加剂组合,例如如乳糖、甘露醇、玉米淀粉或马铃薯淀粉;与粘合剂组合,例如结晶纤维素、纤维素衍生物、阿拉伯胶、玉米淀粉或明胶;与崩解剂组合,例如玉米淀粉、马铃薯淀粉或羧甲基纤维素钠;与润滑剂组合,例如滑石粉或硬脂酸镁;如果需要,还可以与稀释剂、缓冲剂、润湿剂、防腐剂和调味剂组合。药学上相容的粘合剂和/或佐剂材料可以作为组合物的一部分包括在内。片剂、丸剂、胶囊剂、锭剂等可包含以下任何成分或类似性质的化合物:粘合剂,例如微晶纤维素、黄蓍胶或明胶;赋形剂,例如淀粉或乳糖;崩解剂,例如藻酸,Primogel或玉米淀粉;润滑剂,例如硬脂酸镁或Sterotes;助流剂,例如胶体二氧化硅;甜味剂,例如蔗糖或糖精;或调味剂,例如薄荷、水杨酸甲酯或橙色调味剂。 适用于口服给药的药物制剂和单位剂型特别适用于治疗患者自我给药的慢性病、感染和疗法。在一方面,该制剂对儿科给药是特异性的。 本公开提供了药物制剂,其中可以将本文公开的分离的多肽、本文公开的分离的多核苷酸、本文公开的载体、本文公开的分离的宿主细胞或本文公开的抗体中的一种或多种配制为注射用制剂。根据本发明,通过将它们溶解、悬浮或乳化在水性或非水性溶剂中,例如植物油或其他类似的油、合成的脂肪酸甘油酯、高级脂肪酸的酯或丙二醇;如果需要,还可与常规添加剂一起使用,例如增溶剂、等渗剂、悬浮剂、乳化剂、稳定剂和防腐剂或其他抗菌剂。这样的非限制性实例是抗微生物剂,例如其他疫苗组分,例如表面抗原,例如OMPP5、OMP 26、OMP P2或IV型皮林蛋白(Jurcisek and Bakaletz (2007) J. ofBacteriology 189(10):3868-3875和Murphy, T F, Bakaletz, L O and Smeesters, P R(2009) The Pediatric Infectious Disease Journal, 28:S121-S126)和抗菌剂。对于静脉内给药,合适的载体包括生理性抑菌水、Cremophor EL™ (BASF, Parsippany, N.J.)或磷酸盐缓冲盐水(PBS)。在所有情况下,用于肠胃外给药的组合物必须是无菌的,并且应该以易于注射的程度流动。 本公开内容提供的气雾剂制剂可以通过吸入施用,并且可以基于推进剂或基于非推进剂。例如,本文公开的药物制剂的实施方案包括配制进加压的可接受的推进剂(例如二氯二氟甲烷、丙烷、氮气等)的本文公开的化合物。为了通过吸入给药,化合物可以以气雾喷雾的形式从加压容器或分配器中递送,其包含合适的推进剂,例如气体,例如二氧化碳、或喷雾器。非推进剂的非限制性示例是借助于机械力从密闭容器中喷出的泵喷雾剂(即,用手指推动活塞或通过压缩容器,例如通过施加到容器壁上的压力或壁本身施加的弹力,例如通过弹性气囊)。 本文公开的栓剂可以通过将本文公开的化合物与多种碱(例如乳化碱或水溶性碱)中的任一种混合来制备。本文公开的化合物的这种药物制剂的实施方案可以通过栓剂经直肠给药。栓剂可包括诸如可可脂、碳蜡和聚乙二醇的载体,它们在体温下熔化,但在室温下固化。 可提供用于口服或直肠施用的单位剂型,例如糖浆剂、酏剂和混悬剂,其中每个剂量单位(例如按茶匙的、按汤匙的、片剂或栓剂)均包含预定量的包含一种或多种本文公开的化合物的组合物。类似地,用于注射或静脉内给药的单位剂型可以在组合物中包含本文公开的化合物,其为在无菌水、生理盐水或另一种药学上可接受的载体中的溶液。 本文公开的药物制剂的实施方案包括其中一种或多种本文公开的分离的多肽、本文公开的分离的多核苷酸、本文公开的载体、用于本公开的小分子、本文公开的分离的宿主细胞、或本文公开的抗体被配制成可注射的组合物的实施方案。本文公开的可注射药物制剂被制备为液体溶液或混悬液;或为固体形式,适合在注射前溶解或悬浮在液体载体中。根据本文公开的药物制剂的其他实施方案,所述制剂也可以被乳化或将活性成分封装在脂质体载体中。 在一个实施方案中,配制本文公开的分离的多肽、本文公开的分离的多核苷酸、本文公开的载体、本文公开的分离的宿主细胞、或本文公开的抗体中的一种或多种,以通过连续递送系统进行递送。术语“连续输送系统”在本文中可与“受控输送系统”互换使用,并且包括本领域已知多种的与导管、注射装置等组合的连续(例如受控)输送装置(例如泵)。 机械或机电输液泵也可以适用于本公开。这样的设备的示例包括例如在美国专利号4,692,147、4,360,019、4,487,603、4,360,019、4,725,852、5,820,589、5,643,207、6,198,966等中描述的。通常,本文公开的化合物的递送可以使用多种可再填充的泵系统中的任一种来完成。泵随提供着时间的推移一致的受控释放。在一些实施方案中,本文公开的化合物在药物不可渗透的储库中呈液体制剂形式,并以连续方式递送至个体。 在一个实施方案中,药物输送系统是至少部分可植入的装置。可使用本领域公知的方法和装置将可植入装置植入任何合适的植入位置。植入部位是对象体内引入和定位药物输送装置的部位。植入部位包括但不限于对象体内的皮下、皮下、肌内或其他合适部位。在一些实施例中使用皮下植入部位是因为方便了药物输送装置的植入和移除。 适用于本公开的药物释放装置可以基于多种操作模式中的任何一种。例如,药物释放装置可以基于扩散系统、对流系统或易蚀系统(例如基于侵蚀的系统)。例如,药物释放装置可以是电化学泵、渗透泵、电渗透泵、蒸气压泵或渗透爆破基质,例如,其中将药物掺入聚合物中并且聚合物提供随着药物浸渍的聚合物材料(例如可生物降解的、药物浸渍的聚合物材料)的降解而释放的药物制剂。在其他实施方案中,药物释放装置基于电扩散系统、电解泵、泡腾泵、压电泵、水解系统等。 基于机械或机电输液泵的药物释放装置也可以适用于本公开。这样的设备的示例包括例如在美国专利号4,692,147、4,360,019、4,487,603、4,360,019、4,725,852等中描述的。通常,可以使用多种可再填充、不可交换的泵系统中的任何一种来完成本发明的治疗方法。可以使用泵和其他对流系统,因为它们通常会随着时间的推移更加一致、受控地释放。由于渗透泵具有更一致的控制释放和相对较小的尺寸的组合优点,所以在一些实施例中使用了渗透泵(参见,例如,PCT国际申请公开号WO 97/27840和美国专利号5,985,305和5,728,396)。适用于本公开的示例性渗透驱动装置包括但不必限于美国专利号3,760,984、3,845,770、3,916,899、3,923,426、3,987,790、3,995,631、3,916,899、4,016,880、4,036,228、4,111,202、4,111,203、4,203,440、4,203,442、4,210,139、4,327,725、4,627,850、4,865,845、5,057,318、5,059,423、5,112,614、5,137,727、5,234,692、5,234,693、5,728,396等中描述的。可以适用于本公开的另一示例性装置是Synchromed输液泵(Medtronic)。 在一些实施例中,药物输送装置是可植入装置。可以使用本领域众所周知的方法和装置将药物输送装置植入任何合适的植入部位。如本文所述,植入部位是对象体内引入和定位药物输送装置的部位。植入部位包括但不限于对象体内的皮下、皮下、肌内或其他合适部位。 用于本文公开的化合物的合适的赋形剂载体是例如水、盐水、右旋糖、甘油、乙醇等、以及它们的组合。另外,如果需要,媒介物可以包含少量的辅助物质,例如润湿剂或乳化剂或pH缓冲剂。制备这些剂型的方法对于本领域技术人员而言是已知的,或者在考虑本公开后将是显而易见的。参见,例如,Remington's Pharmaceutical Sciences, MackPublishing Company, Easton, Pa., 17th edition, 1985。无论如何,待施用的组合物或制剂将包含足以在正在接受治疗的对象中达到所需状态的一定量的化合物。 本公开内容的组合物包括包含持续释放或控制释放基质的组合物。另外,本公开的实施方案可以与使用持续释放制剂的其他治疗结合使用。如本文所用,持续释放基质是由通常是聚合物的材料制成的基质,其可通过酶或基于酸的水解或通过溶解而降解。一旦插入体内,基质就会被酶和体液作用。缓释基质理想地选自生物相容性材料,例如脂质体、聚丙交酯(聚乳酸)、聚乙交酯(乙醇酸的聚合物)、聚丙交酯乙交酯(乳酸和乙醇酸的共聚物)、聚酸酐、聚原酸酯、多肽、透明质酸、胶原蛋白、硫酸软骨素、羧酸、脂肪酸、磷脂、多糖、核酸、聚氨基酸、氨基酸(例如苯丙氨酸、酪氨酸、异亮氨酸)、多核苷酸、聚乙烯丙烯、聚乙烯吡咯烷酮和硅酮。示例性的可生物降解的基质包括聚丙交酯基质、聚乙交酯基质和聚丙交酯乙交酯(乳酸和乙醇酸的共聚物)基质。 在另一个实施方案中,试剂(以及组合组合物)以控释系统递送。例如,本文公开的化合物可以使用静脉内输注、可植入的渗透泵、透皮贴剂、脂质体或其他给药方式来给药。在一个实施方案中,可以使用泵(Sefton (1987) CRC Crit. Ref. Biomed. Eng. 14:201;Buchwald et al. (1980) Surgery 88:507; Saudek et al. (1989) N. Engl. J. Med.321:574)。 在另一个实施方案中,使用聚合物材料。在另一个实施方案中,将控释系统放置在治疗目标即肝脏附近,因此仅需要全身剂量的一小部分。在又一个实施方案中,将控释系统放置在治疗靶标附近,因此仅需要全身的一部分。Langer (1990) Science 249:1527-1533在综述中讨论了其他控释系统。 在另一个实施方案中,本公开的组合物(以及分开或一起的组合组合物)包括通过将本文所述的抑制剂浸渍到吸收性材料(例如缝线、绷带和纱布)中或涂覆到固相材料(例如手术钉书钉、拉链和导管)的表面上以递送组合物而形成的。鉴于本公开,这种类型的其他输送系统对于本领域技术人员将是显而易见的。 本公开提供了用于将一种或多种试剂施用给宿主(例如人)以治疗微生物感染的方法和组合物。在各种实施方案中,本文公开的这些方法几乎涵盖任何适用于药物递送的可用方法和途径,包括体内和离体方法,以及全身和局部施用途径。 筛选试验 本公开提供了筛选等效试剂的方法,例如本文所述的针对多克隆抗体的等效单克隆抗体和调节本文所述的活性试剂和药物组合物的活性或本文公开的多核苷酸编码的多肽或肽产物的功能的各种试剂。为了本公开的目的,“试剂”旨在包括但不限于生物或化学化合物,例如简单或复杂的有机或无机分子、肽、蛋白质(例如抗体)、多核苷酸(反义)或核酶。可以合成各种各样的化合物,例如聚合物,例如多肽和多核苷酸,以及基于各种核心结构的合成有机化合物,这些也包括在术语“试剂”中。另外,各种天然来源可以提供用于筛选的化合物,例如植物或动物提取物等。应当理解,尽管并非总是明确指出,但是该试剂单独使用或与另一种试剂结合使用,该另一种试剂具有与本发明筛选所鉴定的试剂相同或不同的生物学活性。 如对于本领域技术人员显而易见的,可以通过注意基因型变化、表型变化或微生物滴度的降低,在微量滴定板中培养合适的细胞,并且可以同时测定几种试剂。 当试剂是除DNA或RNA以外的组合物时(例如如上所述的小分子),可以将试剂直接添加到细胞培养物中或添加到培养基中进行添加。对本领域技术人员显而易见的是,必须添加 “有效”量,其可以凭经验确定。 当试剂是抗体或抗原结合片段时,可以在进行竞争性ELISA的条件下使试剂与本文所述的靶抗原和多克隆抗体接触或孵育。 这样的方法是技术人员已知的。 当试剂是抗体或抗原结合片段时,可以在进行竞争性ELISA的条件下使试剂与本文所述的靶抗原和多克隆抗体接触或孵育。这样的方法是技术人员已知的。 该测定法也可以在对象中进行。当对象是诸如大鼠、毛丝鼠、小鼠或猿猴之类的动物时,该方法提供了方便的动物模型系统,该系统可在对人类患者进行药剂的临床测试之前使用。在该系统中,如果与具有相同感染的未经治疗的动物相比,每种疾病的症状或微生物感染的症状得到缓解或消除,则候选药物就是潜在的药物。拥有健康且未经治疗的细胞或动物的单独阴性对照组也很有用,这为进行比较提供了基础。 所述试剂和组合物可通过按照常规方法(例如药物组合物中的活性成分)给药而用于药物的制造以及用于治疗人和其他动物。 联合疗法 本公开的组合物和相关方法可以与其他疗法的给药组合使用。这些包括但不限于DNase酶、抗生素、抗微生物剂或其他抗体的施用。在一方面,在不存在DNase酶的情况下施用药剂。 在其他实施方案中,所述方法和组合物可以与抗生素和/或抗微生物剂组合。抗菌剂是杀死或抑制微生物(例如细菌、真菌或原生动物)生长的物质。尽管生物膜通常对抗生素的作用具有抗性,但是本文所述的组合物和方法可用于使涉及生物膜的感染对用于治疗感染的传统治疗方法敏感。在其他实施方案中,将抗生素或抗微生物剂与本文所述的方法和组合物结合使用可减少抗微生物和/或生物膜还原剂的有效量。与本公开内容的方法组合使用的抗微生物剂和抗生素的一些非限制性实例包括阿莫西林、阿莫西林-克拉维酸盐、头孢地尼、阿奇霉素和磺胺甲恶唑-甲氧苄啶。可以通过传统方法容易地确定抗微生物剂和/或抗生素与生物膜还原剂的治疗有效剂量。在一些实施方案中,抗微生物剂与生物膜减少剂组合的剂量是平均有效剂量,该平均有效剂量已被证明对其他细菌感染是有效的,例如其中感染的病因学不包括生物膜的细菌感染。在其他实施方案中,剂量是平均有效剂量的0.1、0.15、0.2、0.25、0.30、0.35、0.40、0.45、0.50、0.55、0.60、0.65、0.70、0.75、0.8、0.85、0.9、0.95、1.1、1.2 、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.5、3.0或5倍。可以在添加抗DNABII抗体之前、同时或之后添加抗生素或抗微生物剂。 在其他实施方案中,所述方法和组合物可以与治疗细菌感染的抗体组合。与本文所述的方法和组合物结合使用的抗体的一个实例是针对不相关的外膜蛋白(即OMP P5)的抗体。单独使用该抗体进行治疗不会在体外使生物膜减薄。与该抗体和生物膜还原剂的联合治疗所产生的效果要大于单独使用相同浓度的任何一种试剂所能达到的效果。当与生物膜还原剂或减少生物膜的方法结合使用时可能产生协同作用的其他抗体包括抗rsPilA、抗OMP26、抗OMP P2和抗全OMP制剂。 本文所述的组合物和方法可用于使涉及生物膜的细菌感染对有效治疗无生物膜的细菌感染有效的普通治疗方式敏感,否则在治疗涉及生物膜的细菌感染方面无效。在其他实施方案中,本文描述的组合物和方法可以与有效治疗涉及生物膜的细菌感染的治疗方式组合使用,但是这种另外的疗法和生物膜减少剂或方法的组合产生协同作用,使得可以减少生物膜还原剂或其他治疗剂的有效剂量。在其他情况下,这种另外的疗法和生物膜减少剂或方法的组合产生协同作用,从而增强了治疗。可以通过治疗感染所需的更短时间来证明治疗的增强。 可以在用于减少生物膜的方法或组合物之前、同时或之后添加额外的治疗,并且可以包含在相同的结构中或作为单独的制剂。 试剂盒 本文提供的试剂盒包含本文公开的组合物和使用说明,或基本上由其组成,或由其组成。在一方面,使用说明提供了进行本文公开的任何方法的指导。在一方面,用于所公开的方法的一种或多种、两种或更多种或三种或更多的试剂独立地或一起包装在试剂盒中。 还要求保护试剂盒,该试剂盒包含进行本文所述的体外和体内方法所必需的试剂和说明。因此,本公开提供了用于执行这些方法的试剂盒,其可以包括如本文公开的以及用于进行本文公开的方法的说明书,例如收集组织和/或进行筛选、和/或分析结果和/或给予有效量的本文定义的试剂。这些可以单独使用或与其他合适的抗菌剂结合使用。 以下实施例旨在说明而非限制本文公开的实施方案。
多胺是几乎所有活生物体产生和利用的普遍存在的小的脂肪族聚阳离子。Michael et al. (2016) J Biol Chem. 291(29):14896-903; D'Agostino et al.(2005) FEBS J. 272(15):3777-87。它们源自氨基酸,在多种细胞功能中起着重要的作用,这些功能对生长和增殖至关重要,包括转录、翻译、转录调节、自噬和抗逆性。Miller-Fleming et al. (2015) J Mol Biol 427(21):3389-406。多胺合成有多种途径,它们在代谢库中的存在因物种而异。Michael et al. (2016) Biochem J. 473(15):2315-29。由于它们的静电介导的相互作用具有非特异性,多胺的合成受到转录、翻译和蛋白质降解机制的严格控制。Miller-Fleming et al. (2015) J Mol Biol 427(21):3389-406。 生命有机体产生五种主要的多胺分子:精胺、亚精胺、腐胺、尸胺和1,3-二氨基丙烷。Miller-Fleming et al. (2015) J Mol Biol 427(21):3389-406。其他类型的多胺以更具物种特异性的方式生产。由于决定阳离子特性和电荷分布的胺基的长度和数量不同,每种多胺的属性略有不同。Michael et al. (2016) J Biol Chem. 291(29):14896-903。这种差异允许多胺活性具有某种程度的特异性,并指导多胺聚集体的组装。D'Agostino etal. (2005) FEBS J. 272(15):3777-87; D'Agostino et al. (2006) IUBMB Life 58(2):75-82。 多胺功能是浓度依赖性的,这由与多胺浓度失调相关的多种疾病状态所证明。Miller-Fleming et al. (2015) J Mol Biol 427(21):3389-406。由于多胺水平的改变,一般的代谢过程可能会中断,而特定过程可能会因多胺水平的特定或整体变化而改变。例如,特定的多胺浓度可以介导微生物生物膜生产的不同结果。在多种物种中,已经证明细胞内多胺水平调节生物膜的生物发生,并且这种调节可能是由于特定多胺的细胞内感测。Karatan et al. (2013) Biotechnol Lett. 35(11):1715-7; McGinnis et al. (2009)FEMS Microbiol Lett. 299(2):166-74; Wortham et al. (2010) Environ Microbiol.12(7):2034-47。哪些多胺介导这些表型可以是物种特异性的。在缺乏产生特定多胺能力的突变菌株中,外源添加无法合成的多胺可恢复生物膜的形成,而添加其他多胺则不能。另外,文献中充斥着细菌生物膜形成的例子,这些细菌生物膜形成被内源性或高浓度的外源特定多胺及其衍生物抑制,而其他多胺则没有作用。Karatan et al. (2013) BiotechnolLett. 35(11):1715-7; Goytia et al. (2013) FEMS Microbiol Lett. 343(1):64-9;Cardile et al. (2017) Adv Exp Med Biol. 973:53-70; Wang et al. (2016) JBacteriol. 198(19):2682-91; Qu et al. (2016) Microbiologyopen. 5(3):402-12;Konai et al. (2015) Bioconjug Chem. 26(12):2442-53; Si et al. (2015) ApplMicrobiol Biotechnol. 99(24):10861-70; Dewangan et al. (2014) AntimicrobAgents Chemother. 58(9):5435-47; Ding et al. (2014) Appl Environ Microbiol.80(4):1498-506; Planet et al. (2013) MBio. 4(6):e00889-13。但是,抑制一个物种的相同多胺可能不会抑制生物膜的生物发生,甚至可能需要其他细菌物种中的生物膜生产。Karatan et al. (2013) Biotechnol Lett. 35(11):1715-7; McGinnis et al. (2009)FEMS Microbiol Lett. 299(2):166-74; Wortham et al. (2010) Environ Microbiol.12(7):2034-47; Wang et al. (2016) J Bacteriol. 198(19):2682-91; Hobley et al.(2017) J Biol Chem. 292(29):12041-53; Ou et al. (2017) Mol Med Rep. 15(1):21-20; Nesse et al. (2015) Appl Environ Microbiol. 81(6):2226-32; Ramon-Perez etal. (2015) Microb Pathog. 79:8-16; Hobley et al. (2014) Cell. 156(4):844-54;Sakamoto et al. (2012) Int J Biochem Cell Biol. 44(11):1877-86; Burrell etal. (2010) J Biol Chem. 285(50):39224-38; Lee et al. (2009) J Biol Chem. 284(15):9899-907; Patel et al. (2006) J Bacteriol. 188(7):2355-63。此外,虽然多胺在真菌生物膜发育中的作用尚不明确,但多胺合成中的白色念珠菌突变体对于生物膜生产是有缺陷的,用多胺合成抑制剂处理白色念珠菌会对生物膜的生长产生负面影响。Chen etal. (2014) Mol Biosyst. 10(1):74-85; Liao et al. (2015) Int J AntimicrobAgents. 46(1):45-52。 eDNA稳定化的一种潜在来源是生物膜基质中多胺的存在。已观察到多胺可调节DNA结构(Pasini et al. (2014) Amino Acids. 46(3):595-603),并保护DNA免受外部修饰剂或危险条件的侵害。D'Agostino et al. (2005) FEBS J. 272(15):3777-87; Baezaet al. (1991) Orig Life Evol Biosph. 21(4):225-42; Nayvelt et al. (2010)Biomacromolecules. 11(1):97-105。多胺在细胞内染色质稳定中的作用已被充分证明。Pasini et al. (2014) Amino Acids. 46(3):595-603。在此,申请人假设细胞外多胺稳定了细菌生物膜的eDNA结构,并表明改变多胺含量和多胺在细胞外结合生物膜基质中eDNA的能力是破坏细菌生物膜群落的一种手段。申请人观察到多胺的合成或拮抗作用的抑制破坏了已建立的细菌生物膜,并且补充了多胺耗竭的细菌恢复了eDNA的结构。
为了确定生物膜基质中是否存在多胺,作为模型人类病原体,申请人在体外培养了不可分型的流感嗜血菌(NTHi)生物膜,并用针对腐胺、精胺或亚精胺的抗体进行了免疫荧光。使用共聚焦激光扫描显微镜(CLSM;图12)可视化生物膜细胞外基质内的多胺定位。通过免疫荧光显微镜在整个生物膜中检测到所有三种多胺。
二环己基胺通过竞争性抑制机制,即通过结合到蛋白质上与腐胺底物相同的位点来抑制亚精胺合酶。因此,申请人假设二环己胺将会抑制NTHi生物膜的发育。首先,申请人证实了直到10 mM,二环己胺都不影响NTHi的生长(图13)。但是,通过COMSTAT分析评估,添加50 μM二环己胺可显著抑制NTHi生物膜形成,将生物膜厚度和生物量减少约40%,同时增加生物膜粗糙度(图14)。随后的免疫荧光显微镜检查在用二环己胺处理的残留NTHi生物膜中亚精胺的存在表明,生物膜基质中存在的亚精胺统计学上显著相应降低(图15)。二环己胺还减少了早期生物膜形成过程中产生的eDNA支架结构的数量和复杂性(图16)。向二环己基胺处理的NTHi生物膜中外源添加1 mM亚精胺可恢复正常的生物膜厚度、生物量和粗糙度(图14),表明二环己胺在生物膜细胞外基质中充当亚精胺合酶和/或外源亚精胺的竞争性抑制剂。这些数据表明,通过抑制多胺合成或与多胺结合位点的拮抗结合,针对细胞外多胺与生物膜基质中的eDNA结合的化合物具有预防生物膜生物发生和破坏已建立的生物膜的潜力。
据疾病控制和预防中心估计,所有细菌感染性疾病中>80%的发病机理包括在疾病过程的发病机理中必要的生物膜状态。生物膜由附着于非生物和生物表面的细菌细胞组成,这些细菌细胞已经发展成为结构化种群,该种群嵌入包含核酸、蛋白质、脂质、生物聚合物(Davies (2003) Nat Rev Drug Discov. 2(2):114-22)和二价阳离子(Cavaliere etal., (2014) Microbiology Open. 3(4):557-567)的细胞外聚合物(EPS)中。 EPS起到了抵抗恶劣环境和抗菌剂(例如抗生素和宿主免疫效应物)的保护性屏障作用(Devaraj etal., (2013)同上)。生物膜基质的关键结构和体系结构成分是细胞外DNA(eDNA)和DNA结合蛋白(IHF和HU)的DNABII家族。DNABII蛋白与eDNA具有高亲和力,可稳定生物膜。靶向DNABII的抗体可诱导生物膜瓦解,并在体内和体外释放出细菌(Novotny et al. (2016) 在许多行业中,微生物形成生物膜的能力是高度成问题的并且无处不在。例如,医院获得的机械性心脏瓣膜、导尿管和静脉导管的设备相关感染是生物膜形式的细菌污染的结果(Donlan, (2001) Emerging Infectious Diseases. 7(2):277-281)。在农业和食品加工设施中控制生物膜的形成对于预防疾病和广泛的食物损失也很重要。Chmielewskiand Frank (2006) Compr Rev Food Sci F 2:22–32。废水处理设施还发展了生物膜介导的问题,例如生物污垢、阻止适当的膜过滤的EPS和微生物的积累(这会导致水污染)(Woodet al., (2016) PNAS. E2802-E2811)。 用阳离子交换树脂(磺酸盐、磺丙基、磷酸纤维素或肝素琼脂糖凝胶)包被表面和/或处理生物膜利用带负电的树脂化学性质来靶向EPS的带正电的成分(即多胺、二价金属阳离子和DNABII蛋白)。这导致生物膜的破坏和生物和非生物表面的预防。 在这里,申请人证明了阳离子交换树脂P11磷酸纤维素和肝素琼脂糖凝胶阻止了生物膜的形成,并且能够破坏预先形成的生物膜。
申请人质疑不能穿透生物膜的带负电荷的树脂是否可以用来滴定细菌生物膜形成普遍需要的带正电荷的分子(即多胺、二价金属阳离子和DNABII蛋白)。选择了两种树脂,例如磷酸纤维素(P11)和肝素琼脂糖,它们都是用于离子交换色谱的阳离子交换剂,而且还用于DNABII蛋白的亲和纯化(Nash et al. (1987) 为了确定阳离子交换树脂对预先形成的生物膜的抗生物膜活性(即破坏现存生物膜的能力),启动NTHI生长并维持24小时,然后用0(sBHI对照)、0.1、1或5%(w/v)的P11磷酸纤维素处理16小时(图18A)。为了确定阳离子交换树脂防止生物膜形成的抗生物膜活性,在0(sBHI对照)、0.1、1和5%(w/v)的P11磷酸纤维素(图18B)或5%(w/v)肝素琼脂糖凝胶(图19)的存在下开始并维持NTHI生长。生物膜用盐水洗涤并用LIVE/DEAD®染色,用共聚焦扫描显微镜(CSLM)可视化,并通过COMSTAT分析以确定平均厚度和生物量。如图18所示,P11磷酸纤维素对预先形成的生物膜(能够破坏)和生物膜形成(能够防止)均具有负面影响,这通过平均厚度和生物量的减少来揭示。肝素琼脂糖凝胶也对生物膜形成具有负面影响,其中观察到平均厚度和生物量的降低(图18)。这些数据表明阳离子交换树脂可以在体外破坏和阻止生物膜的形成。
为了确定DNABII蛋白的去除是否部分负责观察到的生物膜破坏和预防,在0(sBHI对照)或1%P11磷酸纤维素(w/v)的存在下,开始并维持NTHI生长24小时,以1或5 ug/mL外源加入HU蛋白16小时。用盐水洗涤生物膜,并用LIVE /DEAD®染色,用CSLM显像,并通过COMSTAT分析以确定平均厚度和生物量。如图19所示,HU可以部分补偿体外磷酸纤维素的负面作用。这些数据表明HU被阳离子交换树脂作为目标。
为了确定Mg
为了确定亚精胺是否可恢复阳离子耗尽的生物膜,在0(sBHI对照)或1%P11磷酸纤维素(w/v)的存在下开始并维持NTHI生长24小时,外源添加亚精胺(0-5 mM)16小时。用盐水洗涤生物膜,并用LIVE/DEAD®染色,用CSLM显像,并通过COMSTAT分析以确定平均厚度和生物量。如图21所示,亚精胺可以部分补偿体外磷酸纤维素的负面作用。这些数据表明,P11磷酸纤维素会消耗生物膜基质中的多胺。
为了确定生物膜形成的减少是否取决于直接的细胞接触,在Transwell系统的基底腔室中开始NTHI生长,该基底板在分隔顶部和基底外侧腔室的膜内包含0.4 μm的孔径。这允许小分子(例如蛋白质(DNABII)、多胺和二价金属阳离子)在两个室之间扩散,但细菌细胞则不会。在接种时,顶室包含0(sBHI对照)、0.5、1、或1.5%(w/v)的P11磷酸纤维素,并保持16小时。用盐水洗涤生物膜,并用LIVE/DEAD®染色,用CSLM显像,并通过COMSTAT分析以确定平均厚度和生物量。如图22所示,生物膜平均厚度和生物量的剂量依赖性降低不依赖于与阳离子交换树脂的直接接触。
为了确定多胺(亚精胺)是否在Transwell系统中不直接接触而被P11磷酸纤维素耗尽,在含有0、100、500或1000 μM亚精胺的基底外侧腔中开始NTHi生长,而将0(sBHI对照)或1.5 %(w/v)P11磷酸纤维素加入到顶端室中并保持16小时。用盐水洗涤生物膜,并用LIVE/DEAD®染色,用CSLM显像,并通过COMSTAT分析以确定平均厚度和生物量。如图23所示,单独的亚精胺可以剂量依赖的方式恢复阳离子耗竭的生物膜。
可以通过剂量依赖性方式通过外源添加HU(图20)和亚精胺(图22和图24)来恢复P11磷酸纤维素阳离子耗竭的生物膜。如图17A所示,免疫荧光CSLM表明亚精胺和DNABII(HU)也共定位在由多种致病细菌物种形成的生物膜中。因此,申请人希望确定HU和亚精胺是否在体外协同作用。NTHi在含有100μM亚精胺或500nM HU或两种成分的组合的Transwell系统的基底外侧腔室中开始生长。同时,将0或1.5%的P11磷酸纤维素添加至顶端室并保持16小时。用盐水洗涤生物膜,并用LIVE/DEAD®染色,用CSLM显像,并通过COMSTAT分析以确定平均厚度和生物量。如图25所示,与sBHI对照生物膜相比,亚最佳浓度的亚精胺和HU的组合以协同方式恢复了阳离子耗竭的生物膜。
用0、0.1、1或5%的P11磷酸纤维素和5%的肝素琼脂糖(w/v)的溶液涂覆玻璃室载玻片。NTHI生长在包被的载玻片上开始并保持40小时。用盐水洗涤生物膜,并用LIVE/DEAD®染色,用CSLM显像,并通过COMSTAT分析以确定平均厚度和生物量。图26表明,P11磷酸纤维素和肝素琼脂糖均以剂量依赖性方式防止生物膜形成。这些数据表明,用阳离子交换树脂涂覆表面可以防止细菌附着和随后的体外生物膜形成。
生物膜由仅细菌、仅酵母或两者的微生物群落组成。对于细菌性病原体,抗生素是治疗的第一线。驻留在生物膜中的细菌对所有细菌感染中约80%的发病机理有重要贡献,并且已知对传染病的慢性和复发性也有贡献。同样,生物膜中的细菌对细菌的抵抗力比其自由生活的细菌高出1000倍。这种抗药性主要归因于一种细胞外基质,它保护了驻留的微生物免受敌对环境的侵害。生物膜介导的细菌感染的长期性和复发性要求对抗生素的过度使用,进而导致全球范围内多种耐药性细菌的清醒出现。这种不断增长的抗生素抗性表型导致抗生素治疗失败。而且,抗生素的主要副作用是它们会对共生菌群产生负面影响,这可能使宿主遭受多种副作用以及对继发感染的易感性。以前,申请人将DNA和DNABII蛋白质家族鉴定为迄今为止研究的所有细菌生物膜基质中的通用靶标。申请人显示DNABII蛋白可以用治疗性抗体去除,从而导致生物膜的结构崩溃和驻留细菌的释放。相反,酵母生物膜不表达DNABII蛋白,但仍包含细胞外DNA(eDNA),这也是其基质的一部分。这种新方法是对当前治疗技术的一种改进,因为它不依赖具有杀菌活性的化合物(其对细菌施加压力以形成耐药机制),而是靶向生物膜细胞外基质结构本身,从而导致生物膜的破坏并将常驻细菌释放到易受伤害的状态。所有生物膜,无论构成的细菌或酵母菌如何,均会产生DNA依赖性结构,而这些结构不是以B-DNA(在细胞内占主导地位的规范形式)存在,生物膜基质内存在的最重要的结构性eDNA以Z-DNA的形式存在。重要的是,Z-DNA具有更高的刚性,使其成为更好的结构材料,Z-DNA完全抵抗核酸酶(降解B-DNA的酶)。因此,任何潜在疗法的靶标都是生物膜基质先前无法识别的结构元素,当其改变(即转变回B-DNA)或破坏时,将增强当前疗法的功效。重要的是,结合Z-DNA的蛋白质也可以稳定Z-DNA。由于Z-DNA是生物膜的天然成分,因此与Z-DNA结合的抗体将促进生物膜的生长。相反,结合B DNA的分子可稳定B DNA,并阻止B-DNA转化为Z-DNA,从而防止生物膜形成。重要的是,自然诱导针对Z-DNA的抗体形成的疾病状态将促进生物膜的形成,例如系统性红斑狼疮(SLE)和/或囊性纤维化(CF)。因此,在SLE患者中发生的慢性感染部分是SLE衍生的Z-DNA抗体的结果。此外,某些破坏DNA的化学治疗剂例如铂基化学疗法也将DNA转变为Z型。因此,这些试剂有助于稳定或促进生物膜的生长。指导破坏Z-DNA的疗法是针对SLE和/或囊性纤维化(CF)和慢性感染的化学疗法加重的新疗法。 生物膜是聚集或粘附于表面的微生物的集合,其显示出社区的架构和行为(社区内的信号、运输、分工等)。这种社区架构的部分特色是自制的细胞外基质,可保护驻留的微生物免受有害条件的侵害,该条件在宿主体内包括免疫系统和抗菌剂。在动物和植物中,以及在工业上,例如在工业设备的结垢中,生物膜都占疾病的很大一部分,并因此是研究的重点,这是由于其在医疗、农业和工业环境中的重要性。Visick et al. (2016) JBacteriol. 198(19):2553-63; Hoiby et al. (2017) APMIS. 125(4):272-5。由于多种因素,包括保护性细胞外基质的产生,根除或治疗病原性生物膜特别困难。Hoiby et al.(2017) APMIS. 125(4):272-5。生物膜基质可变地包含多糖、蛋白质和细胞外DNA(eDNA)。细菌生物膜的eDNA是普遍的,对于细胞外基质的稳定性和保护功能至关重要。Okshevskyet al. (2015) Crit Rev Microbiol. 41(3):341-52; Wilton et al. (2015)Antimicrob Agents Chemother. 60(1):544-53。通过DNA降解或去除稳定该结构的DNA结合蛋白来破坏生物膜eDNA的结构,会导致生物膜的灾难性崩溃,并使驻留细菌释放到更易受伤害的状态。Brandstetter et al. (2013) Nasopore. Laryngoscope. 123(11):2626-32; Brockson et al. (2014) Mol Microbiol. 93(6):1246-58; Devaraj et al.(2015) Mol Microbiol. 96(6):1119-35; Freire et al. (2017) Mol Oral Microbiol.32(1):74; Gustave et al. (2013) J Cyst Fibros. 12(4):384-9; Novotny et al.(2017) Clin Vaccine Immunol. 24(6); Novotny et al. (2016) EBioMedicine. 10:33-44; Rocco et al. (2017) Mol Oral Microbiol. 32(2):118-30; Novotny et al.(2013) PLoS One. 8(6):e67629; Baelo et al. (2015) J Control Release. 209:150-8; Brown et al. (2015) Front Microbiol. 6:699; Frederiksen et al. (2006) ActaPaediatr. 95(9):1070-4; Martins et al. (2012) Mycoses. 55(1):80-5; Goodman etal. (2011) Mucosal Immunol. 4(6):625-37。 基于eDNA在生物膜完整性中的核心作用,核酸酶治疗具有明显的治疗潜力。实际上,许多细菌利用内源性分泌的核酸酶来调节生物膜的结构并介导生物膜的扩散。 Cho etal. (2015) Infect Immun. 83(3):950-7; Kiedrowski et al. (2011) PLoS One. 6(11):e26714; Liu et al. (2017) Front Cell Infect Microbiol. 7:97; Steichen etal. (2011) Infect Immun. 79(4):1504-11。然而,当在生物膜形成开始时施用时,外源核酸酶通常仅显示有效的生物膜预防活性,其中已建立的生物膜需要高浓度的核酸酶以观察任何适度的生物膜破坏。Hall-Stoodley et al. (2008) BMC Microbiol. 8:173; Izanoet al. (2009) Microb Pathog. 46(4):207-13; Kaplan et al. (2012) J Antibiot(Tokyo). 65(2):73-7; Tetz et al. (2010) DNA Cell Biol. 29(8):399-405。在慢性肺部感染是发病率和死亡率的主要来源的囊性纤维化患者中,治疗性核酸酶主要用作针对宿主衍生eDNA而不是细菌产生的eDNA造成的阻塞以形成生物膜群落的粘液溶解剂,并且仅在生命早期开始建立慢性感染之前才观察到这些核酸酶(即Pulmozyme)会改变微生物群落。Frederiksen et al. (2006) Acta Paediatr. 95(9):1070-4。为什么细菌生物膜eDNA对核酸酶降解具有抗性仍是一个悬而未决的问题。 对于为什么细菌生物膜eDNA不能被外源核酸酶充分降解存在多种假设。在这些假设中,细菌可能会主动将eDNA的结构改变为对核酸酶不敏感的形式。耐核酸酶的一种这样的DNA结构元件是Z-DNA,Z-DNA是替代的左手螺旋形式。Z-DNA仅在体外特定条件下和体内有限的细胞内条件下进行了描述,尽管有证据表明Z-DNA在正常细胞生理中的作用,尤其是鉴定可调节各种细胞内过程的Z-DNA结合蛋白。Wang et al. (2007) Front Biosci. 12:4424-38; Barraud et al. (2012) Curr Top Microbiol Immunol. 353:35-60。为了将平衡从规范的B-DNA转变为Z-DNA形式,还需要其他因素(Choi et al. (2011) Chem SocRev. 40(12):5893-909; Yang, et al. (2012) Curr Med Chem. 2012;19(4):557-68),包括双螺旋的负超螺旋、高阳离子浓度以抵消带负电的磷酸-脱氧核糖主链的排斥、以及Z-DNA结合蛋白相互作用以稳定结构。金属阳离子和聚阳离子多胺都能够诱导Z-DNA的形成(D'Agostino et al. (2006) IUBMB Life. 58(2):75-82; Balasundaram et al. (1991)Mol Cell Biochem. 100(2):129-40),尤其是在聚(dG-dC)区域中。Thomas et al. (1991)J Biol Chem. 266(10):6137-41。另一方面,Z-DNA向B-DNA的过渡由嵌入剂 (Kim et al.(1993) Biopolymers. 33(11):1677-86; Mirau et al. (1983) Nucleic Acids Res. 11(6):1931-41) 以及潜在的真核染色质蛋白HMGB1催化。Waga et al. (1988) BiochemBiophys Res Commun. 153(1):334-9。 生物膜基质由DNABII家族的细菌染色质蛋白稳定。这些蛋白质在eDNA支架中占据了弯曲的DNA结构,它们的去除导致生物膜的破坏和驻留细菌的释放。 Brandstetter etal. (2013) Nasopore. Laryngoscope. 123(11):2626-32; Brockson et al. (2014)Mol Microbiol. 93(6):1246-58; Devaraj et al. (2015) Mol Microbiol. 96(6):1119-35; Freire et al. (2017) Mol Oral Microbiol. 32(1):74-88; Gustave et al.(2013) J Cyst Fibros. 12(4):384-9; Novotny et al. (2017) Clin VaccineImmunol. 24(6); Novotny et al. (2016) EBioMedicine. 10:33-44; Rocco et al.(2017) Mol Oral Microbiol. 32(2):118-30; Novotny et al. (2013) PLoS One. 8(6):e67629; Goodman et al. (2011) Mucosal Immunol. 4(6):625-37。另外,已经观察到多胺调节DNA结构(Pasini et al. (2014) Amino Acids. 46(3):595-603)并保护其免受外部修饰剂的侵害。Baeza et al. (1991) Orig Life Evol Biosph. 21(4):225-42;D'Agostino et al. (2005) FEBS J. 272(15):3777-87; Nayvelt et al. (2010)Biomacromolecules. 11(1):97-105。在此,申请人表明,胞外多胺与DNABII蛋白协同作用可诱导细菌生物膜内eDNA中的Z-DNA结构,并确定Z-DNA向B-DNA的还原是否会破坏细菌生物膜结构以使其对常规疗法敏感,或者本身破坏生物膜。
不可分型的流感嗜血杆菌(NTHi)是急性和慢性呼吸道感染的常见原因,并且由该生物体引起的疾病依赖于生物膜的形成来保持其慢性。Duell et al. (2016) FEBS Lett.590(21):3840-53。 同样,尿路致病性大肠杆菌(UPEC)是泌尿道感染的主要病原体,并且由于其引起慢性膀胱炎的能力也依赖于生物膜的产生。Blango et al. (2010) AntimicrobAgents Chemother. 54(5):1855-63。当存在核酸酶时,在不存在或不存在核酸酶的条件下体外生长的NTHi和UPEC生物膜显示出对生物膜生物发生的显著损害(图27)。相反,用这些核酸酶处理预先建立的生物膜并没有减少生物膜的厚度或生物量,这表明随着生物膜的成熟,生物膜基质中的eDNA对核酸酶的抗性得以建立。 申请人使用针对Z-DNA的抗体随着其发育在体外探测NTHi和UPEC生物膜。抗体染色和免疫荧光显微镜检查显示,随着生物膜的成熟,Z-DNA累积在生物膜基质的底部(图28),与成熟生物膜中核酸酶的抗性增加相对应。还发现主要的真菌病原体白色念珠菌在其生物膜细胞外基质内含有Z-DNA(图29)。此外,Z-DNA的积累对于生物膜构建过程至关重要,因为用特异性结合(并因此稳定)Z-DNA刺激的NTHi生物膜生物发生的抗体处理新生的生物膜,而针对B-DNA的抗体却没有(图30)。 Z-DNA可能赋予对核酸酶降解生物膜细胞外基质的抗性,这可以通过检查核酸酶处理过的生物膜中存在的eDNA含量和DNA形式来证明。核酸酶处理大大降低了NTHi生物膜细胞外基质中的B-DNA可视化,而Z-DNA的存在变得更加明显(图31)。
已知由聚(dG-dC)组成的寡核苷酸底物易于形成Z-DNA(即由于高NaCl浓度或多胺的存在),因此被保护免受核酸酶降解(图32 )。因此,通过分光光度法测量λ 由于NTHi DNABII蛋白在生物膜基质完整性中起核心作用(Brockson et al.(2014) Mol Microbiol. 93(6):1246-58; Novotny et al. (2017) Clin VaccineImmunol. 24(6); Novotny et al. (2016) EBioMedicine. 10:33-44; Goodman et al.(2011) Mucosal Immunol. 4(6):625-37),申请人推测它们可能有助于DNA核酸酶抗性。实际上,NTHi HU能够在体外赋予对聚(dG-dC)底物的核酸酶抗性,并且能够将聚(dGdC)底物转变为Z-DNA形式(图34)。Jang et al. (2015) Sci Rep. 5:9943。由于观察到NTHi HU和多胺在生物膜基质中的共定位,申请人测试了它们在抑制核酸酶降解的能力上是否表现出协同作用。在不能单独保护聚(dG-dC)底物的聚胺和HU的浓度下,两者的组合表现出赋予核酸酶抗性的协同能力(图35)。在由于NTHI引起的实验性中耳炎期间,在中耳体内形成的生物膜中也观察到了多胺和DNABII蛋白的共定位,以及因此产生的核酸酶抗性表型(图35)。此外,掺入生物膜细胞外基质的DNABII和多胺的水平随物种而不同,并且与通过免疫荧光检测的Z-DNA水平相关(图36)。这些结果一起表明,细菌DNABII蛋白和内源性多胺相互作用在一起,以维护细菌生物膜基质,可能是通过转化为生物膜eDNA支架内的Z-DNA结构元素并使其稳定。
为了利用申请人的新发现,即细菌细菌膜中Z-DNA随时间积累,并且这可以解释为什么成熟的生物膜对核酸酶破坏具有抗性,申请人寻求测试能够将Z-DNA平衡转变为B-DNA的分子是否可以调节生物膜。据报道,真核染色质蛋白HMGB1可将Z-DNA转化为B-DNA。Wagaet al. (1988) Biochem Biophys Res Commun. 153(1):334-9。因此,用HMGB1处理的NTHi生物膜的免疫荧光显微镜检查显示Z-DNA含量降低(图37)。 DNABII蛋白通过HMGB1处理从生物膜中释放出来(图38),通过在没有DNABII介导的Z-DNA稳定的情况下使Z-DNA还原为B-DNA来破坏eDNA结构的稳定性,并导致生物膜的显著破坏。因此,用HMGB1或非炎性HMGB1变体治疗在毛丝鼠中耳中由NTHI诱导的急性中耳炎导致粘附生物膜的有效分离(图39)。HMGB1还有效破坏由伯克霍尔德氏菌(一种与囊性纤维化肺生物膜感染相关的致命机会病原体)在体外形成的生物膜,而HMGB1的预防性给药可防止小鼠肺中的鲍氏芽孢杆菌聚集生物膜形成(图40)。在多胺的保护性浓度的存在下,将HMGB1添加到聚(dG-dC)底物中恢复了核酸酶敏感性(图41)。 这些组合数据表明,多胺和DNABII蛋白将一部分DNA转化为Z-构象,不仅为生物膜细胞外基质中的结构材料奠定了基础,而且解释了其对常规核酸酶破坏成熟生物膜的抗性。Cho et al. (2015) Infect Immun. 83(3):950-7。这些数据一起表明,可以将Z-DNA转换为B-DNA或将Z-DNA构型的生物膜eDNA转换为B-DNA的方法和物质(例如通过去除或抑制DNABII蛋白或多胺)可以恢复核酸酶的敏感性,潜在地允许开发能够增强核酸酶针对体内成熟且顽强的生物膜的活性的药物。这也表明,微生物生物膜都可以通过其Z-DNA含量进行鉴定,从而使Z-DNA定量成为诊断生物膜感染的潜在有用的生物标志物。
该实验提供了用于临床前测试用于治疗囊性纤维化的药物、药剂和方法的猪模型。参见Stoltz et al. (2010) Science Translational Medicine 2(29):29-31。囊性纤维化是一种常染色体隐性遗传疾病,归因于编码CF跨膜电导调节剂(称为CFTR)阴离子通道的基因突变。在该模型中,经过专门繁殖后携带“CFTR”基因缺陷的猪被称为CF猪,其自然发展出CF肺病的标志性特征,包括多种细菌感染下呼吸道。用诸如多肽或其他免疫原性试剂之类的试剂免疫猪,从而诱导抗体的形成,该抗体将消灭肺中的细菌生物膜。该实验类似于如实验1所示主动免疫后递送IHF抗体以消除位于毛丝鼠中耳内的生物膜。可以通过雾化将抗IHF(或其他试剂)抗体递送到这些猪的肺部,以评估疾病征象和相关病理的改善。
使用单突变体的
使用Keio Collection,将 多胺在TEDS和生物膜发育中的作用,通过证明多胺与eDNA和DNABII蛋白协同作用来创建TEDS来确定。图42A-42D显示,要挽救阳离子交换剂(P11)介导的生物膜预防,DNABII蛋白和多胺都是必需的。而图44显示DNABII蛋白、多胺和eDNA(B-和Z-DNA)随着时间稳定地积累在NTHI生物膜的EPS内。此外,如图46所示, DNABII、多胺、Z-DNA和B-DNA成分都存在于多种细菌生物膜的EPS中。 申请人还在图47A-B中发现,多胺、Z-DNA和B-DNA组分全部存在于被NTHI生物膜感染的毛丝鼠中耳的部分内。另外,申请人已经表明,添加RNase A以剂量依赖性方式刺激了生物膜的形成,这可能是由于多胺的释放引起的,多胺是B-DNA向Z-DNA细胞外转化的关键催化剂(图52A),并且 tRNA(而不是GMP)的添加以剂量依赖的方式破坏了早期NTHI生物膜,这可能是由于从生物膜EPS中螯合了多胺(图52B)。
从细菌生物膜中收集条件培养基,以确定多胺(腐胺、尸胺、亚精胺、精胺)的浓度。定量测定了8种难降解生物膜形成病原体(以下称为8种标准菌株)的多胺释放量:ES*KAPE病原体、NTHI和UPEC的临床分离株(*表示使用表皮葡萄球菌作为代表性致病性葡萄球菌(Sabate et al. (2017) Front Microbiol. 8:1401),因为金黄色葡萄球菌产生可以混淆免疫荧光测定的抗体结合蛋白A;在易于处理的情况下,该分析适用于金黄色葡萄球菌)和突变变体,其显示出生物膜EPS结构发生了变化,从而使生物膜表型与细胞外多胺浓度相关。生物膜在于不同时间(0、4、8、16和24小时)收集的富培养基和CDM以及条件培养基中生长。将无细胞的条件培养基(过滤)和培养基样品在液体N
在确定体外生物膜整个发育过程中存在哪些多胺之后,进行测试以区分这些多胺是否影响eDNA结构。申请人已经鉴定出一系列核酸酶,这些核酸酶在播种时添加时可防止生物膜形成(类似于或优于Pulmozyme®),但对亚精胺具有不同的敏感性(表1)。利用随着时间推移的eDNA支架和生物膜形成试验,申请人利用这些酶作为多胺浓度的定性探针,并区分多胺介导的对核酸酶活性的抑制或eDNA结构介导的对核酸酶活性的抑制(即转化为核酸酶抗性形式,例如Z-DNA)。在每种情况下,在用核酸酶处理之前,生物膜应生长指定的时间(0、4、8、16和24小时),并孵育至40小时的最终时间。作为生物膜环境中核酸酶活性的对照,确定了针对在条件培养基中从生物膜中温育的质粒底物的核酸酶活性(0、4、8、16和24h),为此申请人将确定多胺成分及其各自的浓度。如前所述评估和分析8种标准菌株的生物膜破坏。在图35A-D中,申请人显示DNABII蛋白与多胺协同作用以诱导DNA酶抗性。 表1. 阻止生物膜形成的核酸酶及其亚精胺敏感性
尽管申请人已经表明eDNA、DNABII蛋白和多胺对于维持生物膜的TEDS都是必不可少的,但是可以确定相互作用以有效地最大化生物膜形成的每种成分的比例。当细菌形成生物膜时,浮游生物/生物膜细菌达到平衡。实际上,申请人之前已经表明,DNABII蛋白限制了UPEC生物膜(Devaraj et al. (2015) Mol Microbiol. 96(6):1119-35),并且浓度不断提高的DNABII蛋白以剂量依赖性方式将浮游细菌驱动进入生物膜状态。 Devaraj et al.(2015) Mol Microbiol. 96(6):1119-35。缺乏HU的UPEC会产生较小的生物膜,但在使浮游细菌进入生物膜状态中对外源DNABII的反应更强。Devaraj et al. (2015) MolMicrobiol. 96(6):1119-35。同时,在外源DNABII加入后,耗尽eDNA(通过DNA酶)的UPEC生物膜不再增加生物膜的生物量。相反,任何添加外源DNABII蛋白质或DNA的尝试都会对NTHI生物膜产生负面影响(数据未显示)。申请人假设,当生物膜EPS缺乏DNA、DNABII蛋白和多胺,使得这3种成分彼此次优存在时,细菌会优先分配为浮游状态。同样,类似于UPEC,将一种或多种限制成分补充至最佳比例应可改善细菌向生物膜状态的分配。相反,当所有三种成分均处于适当的比例时,正如申请人认为的NTHI一样,这些成分中任何一种的添加都会破坏适当的比例,并对生物膜的形成产生负面影响。在这里,将外源DNABII蛋白滴定至天然UPEC生物膜,并测量生物膜和浮游细菌的比例。一旦生物膜被外源DNABII蛋白质饱和,则多胺和染色体DNA都将发生变化,以进一步将细菌划分为生物膜状态。 当生物膜在CDM中生长时,通过改变与细胞外环境中所测得的浓度相比小至多10倍和更高的浓度,对鉴定出的每种主要多胺(或其组合)进行测试。申请人假设,如果以适当的比例添加DNA、多胺和DNABII蛋白,它将驱使浮游细菌进入生物膜。因此,还测试了WTUPEC和NTHI及其同基因的HU缺陷菌株(尽管仍存在IHF,但HU缺陷菌株仍具有生物膜缺陷表型),以检测HU、DNA和多胺的最佳比例。通过稀释平板测定浮游生物相与生物膜相中细菌的相对比例,评估通过体外生物膜形成试验添加组合生物膜成分的效果。Devaraj et al.(2015) Mol Microbiol. 96(6):1119-35。申请人在图42A-D中示出,P11的添加以剂量依赖的方式阻止生物膜的形成。外源性亚精胺和HU
如上所示,在物理上分离的腔室中的生物膜可被阳离子交换剂P11破坏。重要的是,多胺和DNABII蛋白都必须加回到生物膜腔中以抵消这种破坏,而每种成分本身都是无效的。由于精胺和亚精胺比腐胺或尸胺更有效地调节DNA结构(Kabir et al. (2013) PLoSOne. 8(7):e70510),因此按上述方法在多种浓度下对多胺进行单独和组合测试,以确定是否需要特定的多胺来促进TEDS生产。在整个实验过程中,以规定的浓度(如上所述以及较低和较高的5倍)添加HU。该实验用如上所述的8种标准菌株进行,使用上述体外生物膜测定法定量了各种多胺对生物膜形成的影响。
可以应用遗传方法来通过检查UPEC多胺合成和输出基因的缺陷对细胞外多胺稳态水平的贡献来研究TEDS的形成。鉴定出产生缺乏生物膜表型的突变的单个和组合,并与外源多胺互补。选择UPEC作为模型系统是基于其临床重要性(Subashchandrabose et al.(2015) Microbiol Spectr. 3(4))、已发表的eDNA-DNABII依赖性EPS的特征(Devaraj etal. (2015) Mol Microbiol. 96(6):1119-35)和对大肠杆菌多胺途径的广泛公开理解(Tabor et al. (1985) Microbiol Rev. 49(1):81-99),尽管未检查生物膜表型(其他细菌生物膜表型的综合遗传分析较少,主要集中在多胺的细胞内作用)。Di Martino et al.(2013) Int J Med Microbiol. 303(8):484-91; Karatan et al. (2013) BiotechnolLett. 35(11):1715-7。确定了哪些多胺在生物膜形成过程中存在以及以何种组合存在以及临床上重要的生物膜形成病原体。揭示了
随着生物膜的成熟,生物膜EPS中的eDNA变得耐DNA酶。已知Z-DNA具有DNA酶抗性,并随着生物膜的成熟而积累在TEDS中。 检查特定的混合生物膜中和人类样品中的其他病原体,以确定Z-DNA在细菌生物膜中普遍存在的程度。
检查了11种生物膜形成病原体(8种标准的和另外3种(
检查双物种生物膜以确定在微生物环境下,Z-DNA含量、分布以及与多胺和DNABII蛋白的相互作用如何随着生物膜的年龄而变化。已知在疾病中发生并且申请人具有体外经验的多菌种组合包括NTHI +卡他莫拉菌(代表多菌种OM生物膜)、牙龈卟啉单胞菌+戈登菌(代表多菌种牙周生物膜)29和NTHI+铜绿假单胞菌(代表细菌性CF和慢性化脓性OM生物膜)。按照临床观察指导接种微生物生物膜培养物。例如NTHI生物膜是在铜绿假单胞菌接种之前建立的,因为铜绿假单胞菌通常是NTHI感染部位的继发性入侵者。如前所述,通过免疫荧光分析所得的微生物生物膜的Z-DNA含量以及生物膜EPS中多胺和DNABII蛋白的存在。
HU缺陷的NTHI菌株形成无多胺和eDNA的垫状生物膜。该菌株与常见的共同感染病原体铜绿假单胞菌和粘膜炎莫拉氏菌以及WT NTHI(携带表达gfp的基因以将其与HU突变体区分开)配对,确定每种WT细菌的eDNA、HU和多胺是否可以弥补NTHI突变体的不足,包括Z-DNA的形成。如上所述通过CLSM定量生物膜形成,并通过免疫荧光CLSM分析EPS组分和结构。在图46A中,显示了用幼稚或抗-HU(灰色)和抗亚精胺(Spd,浅灰色)抗体探测的所示生物膜的免疫荧光CLSM图像,共定位用白色表示。
如申请人先前对DNABII蛋白和多胺所做的那样(Gustave et al. (2013) J CystFibros. 12(4):384-9; Idicula et al. (2016) Laryngoscope. 126(8):1946-51),检查了来自人类生物膜相关疾病的临床样本,包括从患有OM的儿童回收的积液(Idicula etal. (2016) Laryngoscope. 126(8):1946-51)、CF痰液(Gustave et al. (2013) J CystFibros. 12(4):384-9)、以及来自CRS成人的抽吸物,并通过免疫荧光显微镜检查是否存在Z-DNA。申请人可以访问上述每种方法的混合性别临床样品,申请人具有相应的微生物学数据。用不同浓度的Z-DNA特异性抗体探查每个切片(测试相等数量的男性和女性临床样品),同时将免疫荧光与纯抗体对照进行比较。还研究了Z-DNA检测如何通过先前描述的免疫荧光在空间上与多胺和DNABII蛋白相关联。在图46A中,申请人显示了用幼稚或抗B-DNA(深灰色)和抗Z-DNA抗体(白色)探测的所示细菌中生长40小时的生物膜的免疫荧光CLSM图像。一种特征明确的Z-DNA特异性单克隆抗体(克隆Z2258、59、78)用于检测Z-DNA。DNABII、多胺、Z-DNA和B-DNA成分都存在于多种细菌生物膜的EPS中。Z-DNA是处于不同稳态水平的多个细菌生物膜EPS的组成部分。
在此,申请人确定Z-DNA在TEDS中起何种程度的整体结构作用。
根据文献和申请人的数据,当Z-DNA积累在老化的生物膜中时,可能会残留大量B型DNA(数据未显示),这意味着整个生物膜中都存在B-Z界面。Rich和他的同事通过将人Z-DNA结合蛋白ADAR1的N端ZɑZ-DNA结合域与II型限制性核酸内切酶FokI的C端催化F
可以通过添加嵌入剂(例如溴化乙锭、氯喹、DAPI (Kwakye-Berko et al. (1990)Mol Biochem Parasitol. 39(2):275-8; Shafer et al. (1984) Nucleic Acids Res.12(11):4679-90; Kim et al. (1993) Biopolymers. 33(11):1677-86)或DNA结合分子(例如纺锤菌素、支链多胺),将B-DNA和Z-DNA之间的平衡进一步驱入B形式。Muramatsu etal. (2016) J Chem Phys. 145(23):235103; Zimmer et al. (1983) FEBS Lett. 154(1):156-60。如果Z-DNA是为了验证TEDS结构完整性的基础,则这些试剂应破坏生物膜。为验证这一假设,申请人将会使用体外生物膜测定法来评估添加Z-DNA至B-DNA催化剂是否会阻碍生物膜形成,通过CLSM评估生物膜抑制,通过稀释板改变浮游生物/生物膜的分配,通过免疫荧光显微镜观察Z-DNA含量的变化。可以通过微型板稀释来确定每种化合物的最小抑制浓度(MIC),然后可以确定亚膜浓度下生物膜破坏和Z-DNA还原的剂量依赖性。针对8种标准菌株对这些分子进行了测试,并与申请人观察到Z-DNA检测相对流行的任何其他物种一起,进一步研究了具有最佳抗生物膜活性的3种分子。
通过增加聚阳离子(例如多胺)的水平(Jovin et al. (1987) Ann Rev PhysChem. 38:521-60),通过添加将Z-DNA易感序列驱动为Z-DNA的衍生自Zɑ结构域蛋白家族的成员(Athanasiadis et al. (2012) Semin Cell Dev Biol. 23(3):275-80),或通过在交替的嘌呤嘧啶区(例如HhaI、M.SssI和M.CviPI甲基转移酶)中胞嘧啶的C5甲基化90,可以使B-DNA和Z-DNA之间的平衡可以朝Z-DNA驱动。虽然生物膜形成的程度可能不会改变,但生物膜形成的动力学和整体结构可能会发生变化。如上所述,仅在没有FokI核酸酶融合的情况下,产生了来自hADAR1的重组Zaa结构域,并且通过CD确认了重组蛋白的Z-DNA结合活性。在生物膜接种和接种到8种标准菌株后4、8、16、24、48和96小时的生物膜形成过程中添加Zaa域(0、0.5、5或50 µM)或甲基转移酶(例如0、0.1、1或10 U/mL的HhaI和S-腺苷甲硫氨酸),其他同基因的HU缺陷型NTHI(阴性对照,其生物膜中不包含明显的eDNA)通过使用CLSM的体外生物膜测定进行评估,通过稀释板改变浮游生物/生物膜分配,并通过免疫荧光观察Z-DNA积累。
尽管申请人已经表明HU可以将DNA驱动成Z-DNA,但是还进行了其他分析,以确定是否DNABII蛋白、多胺和DNA协同作用以形成Z-DNA。
Hud和其同事的广泛研究(Sarkar et al. (2009) Biochemistry. 48(4):667-75; Sarkar et al. (2007) Nucleic Acids Res. 35(3):951-61)已经表明,在亚精胺存在下,IHF和HU都会结合DNA并将其结构改变为粗纤维。确定了在存在或不存在多胺(0、0.1、0.5、1和5 mM的以下物质:腐胺、亚精胺、精胺、尸胺或以上确定的组合)的情况下确定的DNA底物(Holliday接合处DNA,这是包含IHF共有序列(WATCAANNNNTTR,其中W是A或T,N是任何核苷酸,R是嘌呤)的35 bp双链体,相同序列的加扰版本35 bp(dGdC)易于Z-DNA转换而35bp(dAdT)不会)是否会形成Z-DNA。首先通过预期通过250和280 nm进行特征反转的椭圆度测量(通过CD光谱法)进行Z-DNA转换。然后研究通过southwestern分析(EMSA,然后用抗Z-DNA和抗DNABII抗体进行免疫印迹分析)判断的DNABII蛋白与每种底物的结合。Goodman etal. (1989) Nature. 341(6239):251-4。具有CD确认的Z-DNA转化的底物(0.2 nM)与25-500 nM DNABII蛋白在50 mM HEPES缓冲液pH 7.0中于室温孵育30分钟。反应混合物通过在6%非变性聚丙烯酰胺凝胶上的电泳进行拆分。使用ImageQuant软件来量化谱带强度。测量了平衡解离常数(K
确定IHF和HU是否直接结合Z-DNA。进行了具有DNABII蛋白和35 bp溴化(dGdC)DNA底物的EMSA,该底物是稳定的Z-DNA构象异构体。Herbert et al. (1993) Nucleic AcidsRes. 21(11):2669-72。由于溴化DNA底物不需要多胺来稳定Z-DNA状态,因此该实验扩展了早期发现。标记底物DNA,并通过与如所述的ɑ
尽管已知eDNA和DNABII是生物膜破坏的靶标,但是本申请人在存在或不存在常规抗微生物剂的情况下集中于多胺和Z-DNA。申请人假设针对TEDS组分和结构的试剂将与常规抗微生物剂协同作用。
靶定了作为生物膜破坏方法的TEDS的所有三个组成部分或结构(单独地或组合地)。申请人假设通过治疗TEDS中的多个靶标,申请人可以证明具有协同作用并因此比单独靶向一个靶标具有更高的潜力。
使用已知的细菌多胺合成抑制剂(二环己胺(DCHA)(Mattila et al. (1984)Biochem J. 223(3):823-30)、二氟甲基鸟氨酸(DFMO)(Muth et al. (2014) J Med Chem.57(2):348-63)、甲基乙二醛-双(环戊基脒腙)(MGBCP)(Takaji et al. (1997) Lett ApplMicrobiol. 25(3):177-80)),测试了每种化合物在不同的成熟年龄(0、8、24和48 h)破坏8种标准菌株形成的生物膜的能力。确定每种病原体的DCHA、DFMO和MGBCP的MIC,并测试低于MIC的100倍稀释系列的破坏活性。每种抑制剂均单独测试,并与DNA酶(Pulmozyme®;0、0.1、1 U/ml)或针对HU的富含IgG的抗血清(0、150、300 μg/ ml)结合使用。通过LIVE/DEAD®染色和CLSM分析生物膜,并事先定量生物膜参数。与单独的治疗相比,评估每种治疗组合的有效性,并评估无治疗的对照。此外,检查了留在生物膜和浮游生物中的CFU的分区,以确定是否存在任何杀死常驻细菌的情况或只是破坏了生物膜。然后测试来自10位患者的CF(以人类多微生物生物膜样群落为例(Gustave et al. (2013) J Cyst Fibros. 12(4):384-9))样本上的所有8种病原体的5种最有效组合破坏顽固性痰液生物量的能力。Gustaveet al. (2013) J Cyst Fibros. 12(4):384-9。简要地说,将痰液样本处理24小时并成像。如前所述,与未处理的对照相比,通过浊度增加判断的痰液样本破裂被用于量化治疗效果。Gustave et al. (2013) J Cyst Fibros. 12(4):384-9。
使用所确定的最佳Z-DNA抑制剂/破坏剂(或其最佳组合),测试了处于亚MIC浓度下的每一种单独地或与抗DNABII抗体或DNA酶组合以破坏由不同成熟年龄的8种标准菌株形成的生物膜的能力。如果以上利用的Z-DNA特异性核酸酶抑制生物膜形成,则这些核酸酶与抗DNABII抗体或DNA酶共同施用。分析了生物膜破坏活性和CF痰液破坏活性的最有效组合。
使用多胺合成抑制剂和Z-DNA抑制剂,测试了每一种组合来破坏由不同成熟年龄(0、8、24和48 h)的8种标准菌株中的每一个形成的生物膜的能力。还测试了多胺抑制剂和Z-DNA抑制剂与抗DNABII抗体或DNA酶的最佳双重处理。评估每种治疗组合的生物膜破坏情况。前三个组合针对CF痰液进行了测试。
检查了当与抗微生物剂组合时,靶向TEDS的所有3种组分或结构以破坏生物膜的有效性。
使用最有效的生物膜破坏组合,确定是否可以通过共同施用抗微生物剂来杀死剩余的常驻生物膜细菌和它们各自的浮游细菌。使用上面确定的各种浓度和组合的抗DNABII抗体、DNA酶、多胺抑制剂和Z-DNA转化剂,测试了添加适当的抗生素来破坏生物膜并杀死8种标准菌株。以MIC上下10倍共同施用临床上针对每种病原体的抗生素(粪肠球菌-0.1、1、10 μg/ ml氨苄青霉素(Weinstein et al. (2001) J Clin Microbiol. 39(7):2729-31);金黄色葡萄球菌-0.025、0.25、2.5 μg/ml克林霉素(LaPlante et al. (2008) AntimicrobAgents Chemother. 52(6):2156-62);表皮葡萄球菌-0.2、2、20 mg/ml万古霉素(Pinheiroet al. (2016) Microb Drug Resist. 22(4):283-93);肺炎克雷伯菌-0.4、4、40 μg/ml头孢他啶/阿维巴坦(Sader, et al. (2017) Antimicrob Agents Chemother. 61(9));鲍曼不动杆菌-0.3、3、30 μg/ml美罗培南(Liang et al. (2011) J Microbiol ImmunolInfect. 44(5):358-63);铜绿假单胞菌-0.4、4、40 μg/ml大肠菌素(Hindler et al.(2013) J Clin Microbiol. 51(6):1678-84);肠杆菌-0.4、4、40 μg/ml头孢吡肟(Riveraet al. (2016) Antimicrob Agents Chemother. 60(6):3854-5);NTHI- 0.1/0.05、1/0.5、10 µg/ml / 5 mg/ml阿莫西林/克拉维酸锂31)。评估每种治疗组合的生物膜破坏和剩余的可行分配,并针对CF痰液测试组合。 随着这个对通用TEDS的更好理解,申请人设想了将生物膜作为独立方法处理或将该方法与靶向物种特异性生物膜成分的策略(例如铜绿假单胞菌多糖藻酸盐Pel和Psl(Gunn et al. (2016) J Biol Chem. 291(24):12538-46))组合来优化生物膜破坏对特定病原体的手术效果。 申请人还提供了结核病(TB)的临床前模型。参见Ordway et al. (2010) Anti.Agents and Chemotherapy 54:1820。微生物结核分枝杆菌是导致全球流行病加剧的原因。当前数据表明,每年约有800万新的TB病例,约有270万人死于TB。除了这种微生物作为艾滋病毒个体的共同感染(约有4500万人感染了艾滋病毒,据估计还有约1/3的人同时感染了结核分枝杆菌)之外,其特别麻烦的是,分离株已变得对多种药物具有高度耐药性,并且在25年以上的时间里还没有引入任何新的结核病药物。在该动物模型中,将SPF豚鼠维持在屏障菌落中,并通过雾化喷雾进行感染,以将约20 cfu的结核分枝杆菌菌株Erdman K01细菌递送到其肺中。在攻击后第25、50、75、100、125和150天,处死动物并测定细菌负荷并恢复组织以进行组织病理学评估。与不产生经典TB症状的小鼠不同,以这种方式攻击的豚鼠会形成组织性肉芽肿,并伴有中央坏死,这是人类疾病的标志。此外,豚鼠与人类一样,会发展为引流性淋巴结的严重脓性肉芽肿和坏死性淋巴结炎,这是原发性病变复合物的一部分。使用该模型可进行临床前筛查,以确认和鉴定减少和/或消除所产生的结核分枝杆菌生物膜的治疗及预防策略,这些生物膜已观察到在攻击后会在这些动物的肺部形成,并且被认为有助于疾病的发病机理和慢性病。
导管/留置装置生物膜感染的多种动物模型是已知的。参见Otto (2009) NatureReviews Microbiology 7:555。尽管通常被认为是正常的皮肤菌群,但微生物表皮葡萄球菌已成为许多人认为是关键的机会病原体,在医院感染的病原体中排名第一。首先,这种细菌负责在留置医疗器械上发生的大多数感染,这些医疗器械在器械插入过程中被这种常见的皮肤定植器污染。尽管通常不会危及生命,但与这些生物膜感染的治疗相关的困难及其发生频率使它们成为严重的公共卫生负担。在美国,仅与由表皮葡萄球菌引起的与血管导管相关的血流感染的治疗相关的当前成本每年为20亿美元。除表皮葡萄球菌外,粪肠球菌和金黄色葡萄球菌也是在留置医疗设备中发现的污染物。有几种与导管有关的表皮葡萄球菌感染的动物模型,包括兔、小鼠、豚鼠和大鼠,所有这些模型均用于研究发病机理的分子机制,并有助于进行预防和/或治疗的研究。大鼠颈静脉导管已用于评估干扰粪肠球菌、金黄色葡萄球菌和表皮葡萄球菌生物膜形成的疗法。生物膜的减少通常通过以下三种方法进行测量:(i)对导管进行超声处理并计算CFU,(ii)切下导管的切片或简单地放在板上并刻划,或者(iii)生物膜可以用结晶紫或另一种染料染色,洗脱以及测量为CFU的替代指标的OD。
本文所述的方法可用于赋予非免疫对象被动免疫。可以通过转移特异性识别或结合特定抗原的抗体或抗原结合片段来赋予针对给定抗原的被动免疫力。抗体供体和受体可以是人类或非人类对象。另外地或可替代地,抗体组合物可以包含分离的或重组的多核苷酸,其编码特异性识别或结合特定抗原的抗体或抗原结合片段。 在免疫原性组合物的施用对接受对象构成风险、接受对象免疫受损、或接受对象需要立即免疫的情况下,可以赋予被动免疫。免疫原性组合物可以按照与所选给药方式一致的方式制备。组合物可包含完整抗体、抗原结合片段、多克隆抗体、单克隆抗体、体内生成的抗体、体外生成的抗体、纯化或部分纯化的抗体、或完整血清。施用可以包括单剂量的抗体组合物,或初始施用之后的一个或多个加强剂量。可以在初始剂量后的一天、两天、三天、一周、两周、三周、一、二、三、六或十二个月、或任何其他时间点提供加强剂量。可以在评估对象的抗体效价后给予加强剂量。 等效物 应当理解,尽管已经结合以上实施例描述了本公开,但是前述描述和实施例旨在示出而不是限制本公开的范围。在本公开范围内的其他方面、优点和修改对于本公开所属领域的技术人员而言将是显而易见的。 除非另有定义,否则本文所用的所有的技术和科学的术语都具有如本发明的所属领域中的普通技术人员中的一个通常所理解的相同的含义。本文提供的所有核苷酸序列均以5'至3'方向呈现。 可以在缺少在本文没有具体公开的任何元素或限制的情况下,适当地实施本文说明性地描述的本技术。因此,例如,术语“包括”、“包含”、“含有”等应该被广泛且没有限制地理解。此外,本文采用的术语和表达已经被用作描述而非限制,在这些术语和表达的使用中,没有意图排除所示和所述特征或其部分的任何等效物,但是应该认识到,在本发明技术的范围之内各种变化都是可能的。 因此,应该理解,尽管已经通过特定实施例和可选特征来具体公开了本公开,但是本领域技术人员可以对本文所公开的实施例进行修改、改进和变化,这些修改、改进和变化被认为在本公开的范围内。这里提供的材料、方法和示例代表特定实施例,是示例性的,并且不意图限制本公开的范围。 本文广泛地和一般地描述了本技术。落入一般性公开之内的较窄的种类和亚属分组中的每一个也都形成本公开的一部分。这包括了具有从该属中移除任何主题的附带条件或负面限制的本技术的一般性描述,无论被删除的材料是否在本文中被具体描述。 此外,在以马库什组描述本技术的特征或方面的情况中,本领域技术人员将认识到,还借此以该马库什组中的任何单独成员或成员的亚组描述了本公开的实施方案。 在本文中提及的所有公开文献、专利申请、专利和其他参考文献,其整体都以引用的方式明确地合并入本文中,至如同每个都单独地以引用的方式合并的相同程度。如发生冲突,以本说明书(包括定义)为准。 参考文献 1.Dillon SC, Dorman CJ. Bacterial nucleoid-associated proteins,nucleoid structure and gene expression.Nat Rev Microbiol. 2010;8(3):185-95.doi: 10.1038/nrmicro2261. PubMed PMID: 20140026. 2.Dey D, Nagaraja V, Ramakumar S. Structural and evolutionaryanalyses reveal determinants of DNA binding specificities of nucleoid-associated proteins HU and IHF. Mol Phylogenet Evol. 2017;107:356-66. doi:10.1016/j.ympev.2016.11.014. PubMed PMID: 27894997. 3.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA.Curr Opin Struct Biol. 2004;14(1):28-35. doi: 10.1016/j.sbi.2003.12.003.PubMed PMID: 15102446. 4.Avery OT, Macleod CM, McCarty M. Studies on the Chemical Nature ofthe Substance Inducing Transformation of Pneumococcal Types : Induction ofTransformation by a Desoxyribonucleic Acid Fraction Isolated fromPneumococcus Type Iii. J Exp Med. 1944;79(2):137-58. PubMed PMID: 19871359;PMCID: PMC2135445. 5.Gunn JS, Bakaletz LO, Wozniak DJ. What's on the Outside Matters:The Role of the Extracellular Polymeric Substance of Gram-negative Biofilmsin Evading Host Immunity and as a Target for Therapeutic Intervention. J BiolChem. 2016;291(24):12538-46. doi: 10.1074/jbc.R115.707547. PubMed PMID:27129225; PMCID: PMC4933457. 6.Dongari-Bagtzoglou A. Pathogenesis of mucosal biofilm infections:challenges and progress. Expert Rev Anti Infect Ther. 2008;6(2):201-8. doi:10.1586/14787210.6.2.201. PubMed PMID: 18380602; PMCID: PMC2712878. 7.Steele L, Fawal H. 4th Decennial International Conference onnosocomial and healthcare-associated infections: a challenge for change. Am JInfect Control. 2000;28(3):207-10. doi: 10.1067/mic.2000.107275. PubMed PMID:10840339. 8.Apicella MA. Bacterial otitis media, the chinchilla middle ear, andbiofilms. J Infect Dis. 2009;199(6):774-5. PubMed PMID: 19434910. 9.Foxman B, Barlow R, D'Arcy H, Gillespie B, Sobel JD. Urinary tractinfection: self-reported incidence and associated costs. Ann Epidemiol. 2000;10(8):509-15. PubMed PMID: 11118930. 10.Mancl KA, Kirsner RS, Ajdic D. Wound biofilms: lessons learnedfrom oral biofilms. Wound Repair Regen. 2013;21(3):352-62. doi: 10.1111/wrr.12034. PubMed PMID: 23551419; PMCID: PMC3648594. 11.Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of theESKAPE pathogens. Expert Rev Anti Infect Ther. 2013;11(3):297-308. doi:10.1586/eri.13.12. PubMed PMID: 23458769. 12.Ceri H, Olson ME, Stremick C, Read RR, Morck D, Buret A. TheCalgary Biofilm Device: new technology for rapid determination of antibioticsusceptibilities of bacterial biofilms. J Clin Microbiol. 1999;37(6):1771-6.PubMed PMID: 10325322; PMCID: PMC84946. 13.Stacy A, McNally L, Darch SE, Brown SP, Whiteley M. Thebiogeography of polymicrobial infection. Nat Rev Microbiol. 2016;14(2):93-105. doi: 10.1038/nrmicro.2015.8. PubMed PMID: 26714431; PMCID: PMC5116812. 14.Wolcott R, Costerton JW, Raoult D, Cutler SJ. The polymicrobialnature of biofilm infection. Clin Microbiol Infect. 2013;19(2):107-12. doi:10.1111/j.1469-0691.2012.04001.x. PubMed PMID: 22925473. 15.Jurcisek JA, Brockman KL, Novotny LA, Goodman SD, Bakaletz LO.Nontypeable Haemophilus influenzae releases DNA and DNABII proteins via aT4SS-like complex and ComE of the type IV pilus machinery. Proc Natl Acad SciU S A. 2017;114(32):E6632-E41. doi: 10.1073/pnas.1705508114. PubMed PMID:28696280; PMCID: PMC5559034. 16.Sena-Velez M, Redondo C, Graham JH, Cubero J. Presence ofExtracellular DNA during Biofilm Formation by Xanthomonas citri subsp. citriStrains with Different Host Range. PLoS One. 2016;11(6):e0156695. doi:10.1371/journal.pone.0156695. PubMed PMID: 27248687; PMCID: PMC4889101. 17.Wang S, Liu X, Liu H, Zhang L, Guo Y, Yu S, Wozniak DJ, Ma LZ. Theexopolysaccharide Psl-eDNA interaction enables the formation of a biofilmskeleton in Pseudomonas aeruginosa. Environ Microbiol Rep. 2015;7(2):330-40.doi: 10.1111/1758-2229.12252. PubMed PMID: 25472701; PMCID: PMC4656019. 18.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS.Extracellular DNA required for bacterial biofilm formation. Science. 2002;295(5559):1487. doi: 10.1126/science.295.5559.1487. PubMed PMID: 11859186. 19.Frederiksen B, Pressler T, Hansen A, Koch C, Hoiby N. Effect ofaerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients withcystic fibrosis. Acta Paediatr. 2006;95(9):1070-4. doi: 10.1080/08035250600752466. PubMed PMID: 16938752. 20.Martins M, Henriques M, Lopez-Ribot JL, Oliveira R. Addition ofDNase improves the in vitro activity of antifungal drugs against Candidaalbicans biofilms. Mycoses. 2012;55(1):80-5. doi: 10.1111/j.1439-0507.2011.02047.x. PubMed PMID: 21668524; PMCID: PMC3175262. 21.Hymes SR, Randis TM, Sun TY, Ratner AJ. DNase inhibits Gardnerellavaginalis biofilms in vitro and in vivo. J Infect Dis. 2013;207(10):1491-7.doi: 10.1093/infdis/jit047. PubMed PMID: 23431033; PMCID: PMC3627197. 22.Goodman SD, Obergfell KP, Jurcisek JA, Novotny LA, Downey JS,Ayala EA, Tjokro N, Li B, Justice SS, Bakaletz LO. Biofilms can be dispersedby focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol. 2011;4(6):625-37. doi: 10.1038/mi.2011.27. PubMed PMID: 21716265. 23.Hall-Stoodley L, Nistico L, Sambanthamoorthy K, Dice B, Nguyen D,Mershon WJ, Johnson C, Hu FZ, Stoodley P, Ehrlich GD, Post JC.Characterization of biofilm matrix, degradation by DNase treatment andevidence of capsule downregulation in Streptococcus pneumoniae clinicalisolates. BMC Microbiol. 2008;8:173. doi: 10.1186/1471-2180-8-173. PubMedPMID: 18842140; PMCID: PMC2600794. 24.Izano EA, Shah SM, Kaplan JB. Intercellular adhesion and biocideresistance in nontypeable Haemophilus influenzae biofilms. Microb Pathog.2009;46(4):207-13. doi: 10.1016/j.micpath.2009.01.004. PubMed PMID: 19490830;PMCID: PMC2691864. 25.Kaplan JB, LoVetri K, Cardona ST, Madhyastha S, Sadovskaya I,Jabbouri S, Izano EA. Recombinant human DNase I decreases biofilm andincreases antimicrobial susceptibility in staphylococci. J Antibiot (Tokyo).2012;65(2):73-7. doi: 10.1038/ja.2011.113. PubMed PMID: 22167157; PMCID:PMC3288126. 26.Novotny LA, Amer AO, Brockson ME, Goodman SD, Bakaletz LO.Structural stability of Burkholderia cenocepacia biofilms is reliant on eDNAstructure and presence of a bacteriuml nucleic acid binding protein. PLoSOne. 2013;8(6):e67629. doi: 10.1371/journal.pone.0067629. PubMed PMID:23799151; PMCID: PMC3682984. 27.Tetz VV, Tetz GV. Effect of extracellular DNA destruction by DNaseI on characteristics of forming biofilms. DNA Cell Biol. 2010;29(8):399-405.doi: 10.1089/dna.2009.1011. PubMed PMID: 20491577. 28.Devaraj A, Justice SS, Bakaletz LO, Goodman SD. DNABII proteinsplay a central role in UPEC biofilm structure. Mol Microbiol. 2015;96(6):1119-35. doi: 10.1111/mmi.12994. PubMed PMID: 25757804; PMCID: PMC4464964. 29.Rocco CJ, Davey ME, Bakaletz LO, Goodman SD. Natural antigenicdifferences in the functionally equivalent extracellular DNABII proteins ofbacterial biofilms provide a means for targeted biofilm therapeutics. MolOral Microbiol. 2017;32(2):118-30. doi: 10.1111/omi.12157. PubMed PMID:26988714; PMCID: PMC5023461. 30.Devaraj A, Buzzo J, Rocco CJ, Bakaletz LO, Goodman SD. The DNABIIfamily of proteins is comprised of the only nucleoid associated proteinsrequired for nontypeable Haemophilus influenzae biofilm structure.Microbiologyopen. 2017. doi: 10.1002/mbo3.563. PubMed PMID: 29230970. 31.Brockson ME, Novotny LA, Mokrzan EM, Malhotra S, Jurcisek JA,Akbar R, Devaraj A, Goodman SD, Bakaletz LO. Evaluation of the kinetics andmechanism of action of anti integration host factor-mediated disruption ofbacterial biofilms. Mol Microbiol. 2014;93(6):1246-58. doi: 10.1111/mmi.12735. PubMed PMID: 25069521; PMCID: PMC4160410. 32.Michael AJ. Biosynthesis of polyamines and polyamine-containingmolecules. Biochem J. 2016;473(15):2315-29. doi: 10.1042/BCJ20160185. PubMedPMID: 27470594. 33.Tabor CW, Tabor H. Polyamines in microorganisms. Microbiol Rev.1985;49(1):81-99. PubMed PMID: 3157043; PMCID: PMC373019. 34.Bachrach U. Naturally occurring polyamines: interaction withmacromolecules. Curr Protein Pept Sci. 2005;6(6):559-66. PubMed PMID:16381604. 35.Pasini A, Caldarera CM, Giordano E. Chromatin remodeling bypolyamines and polyamine analogs. Amino Acids. 2014;46(3):595-603. doi:10.1007/s00726-013-1550-9. PubMed PMID: 23836422. 36.Sarkar T, Petrov AS, Vitko JR, Santai CT, Harvey SC, Mukerji I,Hud NV. Integration host factor (IHF) dictates the structure of polyamine-DNAcondensates: implications for the role of IHF in the compaction of bacterialchromatin. Biochemistry. 2009;48(4):667-75. doi: 10.1021/bi8019965. PubMedPMID: 19132923. 37.Sarkar T, Vitoc I, Mukerji I, Hud NV. Bacterial protein HUdictates the morphology of DNA condensates produced by crowding agents andpolyamines. Nucleic Acids Res. 2007;35(3):951 61. doi: 10.1093/nar/gkl1093.PubMed PMID: 17259223; PMCID: PMC1807954. 38.Wilton M, Charron-Mazenod L, Moore R, Lewenza S. Extracellular DNAAcidifies Biofilms and Induces Aminoglycoside Resistance in Pseudomonasaeruginosa. Antimicrob Agents Chemother. 2015;60(1):544-53. doi: 10.1128/AAC.01650-15. PubMed PMID: 26552982; PMCID: PMC4704225. 39.Thomas TJ, Messner RP. A left-handed (Z) conformation of poly(dA-dC).poly(dG-dT) induced by polyamines. Nucleic Acids Res. 1986;14(16):6721-33. PubMed PMID: 3748821; PMCID: PMC311676. 40.Thomas TJ, Messner RP. Structural specificity of polyamines inleft-handed Z-DNA formation. Immunological and spectroscopic studies. J MolBiol. 1988;201(2):463-7. PubMed PMID: 3418706. 41.Pohl FM. Salt-induced transition between two double-helical formsof oligo (dC-dG). Cold Spring Harb Symp Quant Biol. 1983;47 Pt 1:113-7.PubMed PMID: 6574834. 42.Pohl FM. Dynamics of the B-to-Z transition in supercoiled DNA.Proc Natl Acad Sci U S A. 1986;83(14):4983-7. PubMed PMID: 3460079; PMCID:PMC323873. 43.Rahmouni AR. Z-DNA as a probe for localized supercoiling in vivo.Mol Microbiol. 1992;6(5):569-72. PubMed PMID: 1552856. 44.Jovin TMS, D. M.; McIntosh, L. P. The Transition Between B-DNA andZ-DNA. Ann Rev Phys Chem. 1987;38:521-60. 45.Kim DH, Im H, Jee JG, Jang SB, Yoon HJ, Kwon AR, Kang SM, Lee BJ.beta-Arm flexibility of HU from Staphylococcus aureus dictates the DNA-binding and recognition mechanism. Acta Crystallogr D Biol Crystallogr. 2014;70(Pt 12):3273-89. doi: 10.1107/S1399004714023931. PubMed PMID: 25478845. 46.Chaires JB, Norcum MT. Structure and stability of Z* DNA. J BiomolStruct Dyn. 1988;5(6):1187-207.doi: 10.1080/07391102.1988.10506463. PubMedPMID: 3271507. 47.Thomas TJ, Bloomfield VA. Chain flexibility and hydrodynamics ofthe B and Z forms of poly(dGdC). poly(dG-dC). Nucleic Acids Res. 1983;11(6):1919-30. PubMed PMID: 6835843; PMCID: PMC325846. 48.Athanasiadis A. Zalpha-domains: at the intersection between RNAediting and innate immunity. Semin Cell Dev Biol. 2012;23(3):275-80. doi:10.1016/j.semcdb.2011.11.001. PubMed PMID: 22085847. 49.Bergen HR, 3rd, Losman MJ, O'Connor T, Zacharias W, Larson JE,Accavitti MA, Wells RD, Koopman WJ. Specificity of monoclonal anti-Z-DNAantibodies from unimmunized MRL/Mp-lpr/lpr mice. J Immunol. 1987;139(3):743-8. PubMed PMID: 3496387. 50.Herbert A, Rich A. The biology of left-handed Z-DNA. J Biol Chem.1996;271(20):11595-8. PubMed PMID: 8662853. 51.Gustave JE, Jurcisek JA, McCoy KS, Goodman SD, Bakaletz LO.Targeting bacterial integration host factor to disrupt biofilms associatedwith cystic fibrosis. J Cyst Fibros. 2013;12(4):384-9. doi: 10.1016/j.jcf.2012.10.011. PubMed PMID: 23168017; PMCID: PMC3582735. 52.Novotny LA, Jurcisek JA, Goodman SD, Bakaletz LO. Monoclonalantibodies against DNA-binding tips of DNABII proteins disrupt biofilms invitro and induce bacterial clearance in vivo. EBioMedicine. 2016;10:33-44.doi: 10.1016/j.ebiom.2016.06.022. PubMed PMID: 27342872; PMCID: PMC5006588. 53.Freire MO, Devaraj A, Young A, Navarro JB, Downey JS, Chen C,Bakaletz LO, Zadeh HH, Goodman SD. A bacteriuml-biofilm-induced oralosteolytic infection can be successfully treated by immuno-targeting anextracellular nucleoid-associated protein. Mol Oral Microbiol. 2017;32(1):74-88. doi: 10.1111/omi.12155. PubMed PMID: 26931773; PMCID: PMC5010536. 54.Bakaletz LO. Chinchilla as a robust, reproducible andpolymicrobial model of otitis media and its prevention. Expert Rev Vaccines.2009;8(8):1063-82. doi: 10.1586/erv.09.63. PubMed PMID: 19627188. 55.Iacomino G, Picariello G, Sbrana F, Di Luccia A, Raiteri R, D'Agostino L. DNA is wrapped by the nuclear aggregates of polyamines: theimaging evidence. Biomacromolecules. 2011;12(4):1178-86. doi: 10.1021/bm101478j. PubMed PMID: 21401020. 56.Paulin L, Lindberg LA, Poso H. Reversible inhibition of flagellaformation after specific inhibition of spermidine synthesis bydicyclohexylamine in Pseudomonas aeruginosa. Antonie Van Leeuwenhoek. 1986;52(6):483-90. PubMed PMID: 3101591. 57.Pegg AE, Bitonti AJ, McCann PP, Coward JK. Inhibition of bacterialaminopropyltransferases by Sadenosyl- 1, 8-diamino-3-thiooctane and bydicyclohexylamine. FEBS Lett. 1983;155(2):192-6. PubMed PMID: 6406265. 58.Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, ErsbollBK, Molin S. Quantification of biofilm structures by the novel computerprogram COMSTAT. Microbiology. 2000;146 ( Pt 10):2395-407. doi: 10.1099/00221287-146-10-2395. PubMed PMID: 11021916. 59.Yang CL, Chilvers M, Montgomery M, Nolan SJ. Dornase alfa forcystic fibrosis. Paediatr Respir Rev. 2017;21:65-7. doi: 10.1016/j.prrv.2016.09.001. PubMed PMID: 27769785. 60.Jang YJ, Lee C, Kim SK. Formation of Poly[d(A-T)2] Specific Z-DNAby a Cationic Porphyrin. Sci Rep. 2015;5:9943. doi: 10.1038/srep09943. PubMedPMID: 25943171; PMCID: PMC4421867. 61.Segall AM, Goodman SD, Nash HA. Architectural elements innucleoprotein complexes: interchangeability of specific and non-specific DNAbinding proteins. EMBO J. 1994;13(19):4536-48. PubMed PMID: 7925295; PMCID:PMC395386. 62.Tjokro NO, Rocco CJ, Priyadarshini R, Davey ME, Goodman SD. Abiochemical analysis of the interaction of Porphyromonas gingivalis HU PG0121protein with DNA. PLoS One. 2014;9(3):e93266. doi: 10.1371/journal.pone.0093266. PubMed PMID: 24681691; PMCID: PMC3969353. 63.Ayala E, Downey JS, Mashburn-Warren L, Senadheera DB, CvitkovitchDG, Goodman SD. A biochemical characterization of the DNA binding activity ofthe response regulator VicR from Streptococcus mutans. PLoS One. 2014;9(9):e108027. doi: 10.1371/journal.pone.0108027. PubMed PMID: 25229632; PMCID:PMC4168254. 64.Hung DC, Downey JS, Ayala EA, Kreth J, Mair R, Senadheera DB, QiF, Cvitkovitch DG, Shi W, Goodman SD. Characterization of DNA binding sitesof the ComE response regulator from Streptococcus mutans. J Bacteriol. 2011;193(14):3642-52. doi: 10.1128/JB.00155-11. PubMed PMID: 21602345; PMCID:PMC3133340. 65.Idicula WK, Jurcisek JA, Cass ND, Ali S, Goodman SD, Elmaraghy CA,Jatana KR, Bakaletz LO. Identification of biofilms in post-tympanostomy tubeotorrhea. Laryngoscope. 2016;126(8):1946-51. doi: 10.1002/lary.25826. PubMedPMID: 27426942; PMCID: PMC4955598. 66.Brandstetter KA, Jurcisek JA, Goodman SD, Bakaletz LO, Das S.Antibodies directed against integration host factor mediate biofilm clearancefrom Nasopore. Laryngoscope. 2013;123(11):2626-32. doi: 10.1002/lary.24183.PubMed PMID: 23670606; PMCID: PMC4060527. 67.Subashchandrabose S, Mobley HL. Virulence and Fitness Determinantsof Uropathogenic Escherichia coli. Microbiol Spectr. 2015;3(4). doi: 10.1128/microbiolspec.UTI-0015-2012. PubMed PMID: 26350328; PMCID: PMC4566162. 68.Subashchandrabose S, Mobley HL. Virulence and Fitness Determinantsof Uropathogenic Escherichia coli. Microbiol Spectr. 2015;3(4). doi: 10.1128/microbiolspec.UTI-0015-2012. PubMed PMID: 26350328; PMCID: PMC4566162. 69.Justice SS, Li B, Downey JS, Dabdoub SM, Brockson ME, Probst GD,Ray WC, Goodman SD. Aberrant community architecture and attenuatedpersistence of uropathogenic Escherichia coli in the absence of individualIHF subunits. PLoS One. 2012;7(10):e48349. doi: 10.1371/journal.pone.0048349.PubMed PMID: 23133584; PMCID: PMC3485042. 70.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, DatsenkoKA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol.2006;2:2006 0008. doi: 10.1038/msb4100050. PubMed PMID: 16738554; PMCID:PMC1681482. 71.Karperien A. FracLac for ImageJ 1999-2013. Available from:rsb.info.nih.gov/ij/plugins/fraclac/FLHelp/Introduction.htm. 72.Kashiwagi K, Miyamoto S, Suzuki F, Kobayashi H, Igarashi K.Excretion of putrescine by the putrescine-ornithine antiporter encoded by thepotE gene of Escherichia coli. Proc Natl Acad Sci U S A. 1992;89(10):4529-33.PubMed PMID: 1584788; PMCID: PMC49116. 73.Soksawatmaekhin W, Kuraishi A, Sakata K, Kashiwagi K, Igarashi K.Excretion and uptake of cadaverine by CadB and its physiological functions inEscherichia coli. Mol Microbiol. 2004;51(5):1401-12. doi: 10.1046/j.1365-2958.2003.03913.x. PubMed PMID: 14982633. 74.Higashi K, Ishigure H, Demizu R, Uemura T, Nishino K, Yamaguchi A,Kashiwagi K, Igarashi K. Identification of a spermidine excretion proteincomplex (MdtJI) in Escherichia coli. J Bacteriol. 2008;190(3):872-8. doi:10.1128/JB.01505-07. PubMed PMID: 18039771; PMCID: PMC2223573. 75.Sugiyama Y, Nakamura A, Matsumoto M, Kanbe A, Sakanaka M, HigashiK, Igarashi K, Katayama T, Suzuki H, Kurihara S. A Novel Putrescine ExporterSapBCDF of Escherichia coli. J Biol Chem. 2016;291(51):26343-51. doi:10.1074/jbc.M116.762450. PubMed PMID: 27803167; PMCID: PMC5159496. 76.Sabate Bresco M, Harris LG, Thompson K, Stanic B, Morgenstern M,O'Mahony L, Richards RG, Moriarty TF. Pathogenic Mechanisms and HostInteractions in Staphylococcus epidermidis Device-Related Infection. FrontMicrobiol. 2017;8:1401. doi: 10.3389/fmicb.2017.01401. PubMed PMID: 28824556;PMCID: PMC5539136. 77.Hakkinen MR, Roine A, Auriola S, Tuokko A, Veskimae E, KeinanenTA, Lehtimaki T, Oksala N, Vepsalainen J. Analysis of free, mono- anddiacetylated polyamines from human urine by LC-MS/MS. J Chromatogr B AnalytTechnol Biomed Life Sci. 2013;941:81-9. doi: 10.1016/j.jchromb.2013.10.009.PubMed PMID: 24185098. 78.Liu R, Li Q, Ma R, Lin X, Xu H, Bi K. Determination of polyaminemetabolome in plasma and urine by ultrahigh performance liquidchromatography-tandem mass spectrometry method: application to identifypotential markers for human hepatic cancer. Anal Chim Acta. 2013;791:36-45.doi: 10.1016/j.aca.2013.06.044. PubMed PMID: 23890604. 79.Xu H, Liu R, He B, Bi CW, Bi K, Li Q. Polyamine MetabolitesProfiling for Characterization of Lung and Liver Cancer Using an LC-Tandem MSMethod with Multiple Statistical Data Mining Strategies: DiscoveringPotential Cancer Biomarkers in Human Plasma and Urine. Molecules. 2016;21(8).doi: 10.3390/molecules21081040. PubMed PMID: 27517900. 80.Kabir A, Suresh Kumar G. Binding of the biogenic polyamines todeoxyribonucleic acids of varying base composition: base specificity and theassociated energetics of the interaction. PLoS One. 2013;8(7):e70510. doi:10.1371/journal.pone.0070510. PubMed PMID: 23894663; PMCID: PMC3722294. 81.Di Martino ML, Campilongo R, Casalino M, Micheli G, Colonna B,Prosseda G. Polyamines: emerging players in bacteria-host interactions. Int JMed Microbiol. 2013;303(8):484-91. doi: 10.1016/j.ijmm.2013.06.008. PubMedPMID: 23871215. 82.Karatan E, Michael AJ. A wider role for polyamines in biofilmformation. Biotechnol Lett. 2013;35(11):1715-7. doi: 10.1007/s10529-013-1286-3. PubMed PMID: 23881324. 83.Kim YG, Kim PS, Herbert A, Rich A. Construction of a Z-DNA-specific restriction endonuclease. Proc Natl Acad Sci U S A. 1997;94(24):12875-9. PubMed PMID: 9371768; PMCID: PMC24231. 84.Shin SI, Ham S, Park J, Seo SH, Lim CH, Jeon H, Huh J, Roh TY. Z-DNA-forming sites identified by ChIP-Seq are associated with activelytranscribed regions in the human genome. DNA Res. 2016. doi: 10.1093/dnares/dsw031. PubMed PMID: 27374614; PMCID: PMC5066173. 85.Kim J, Yang C, DasSarma S. Analysis of left-handed Z-DNA formationin short d(CG)n sequences in Escherichia coli and Halobacterium halobiumplasmids. Stabilization by increasing repeat length and DNA supercoiling butnot salinity. J Biol Chem. 1996;271(16):9340-6. PubMed PMID: 8621598. 86.Kwakye-Berko F, Meshnick S. Sequence preference of chloroquinebinding to DNA and prevention of Z-DNA formation. Mol Biochem Parasitol.1990;39(2):275-8. PubMed PMID: 2320060. 87.Shafer RH, Brown SC, Delbarre A, Wade D. Binding of ethidium andbis(methidium)spermine to Z DNA by intercalation. Nucleic Acids Res. 1984;12(11):4679-90. PubMed PMID: 6739293; PMCID: PMC318867. 88.Kim SK, Eriksson S, Norden B. Z-->B transition in poly[d(G-m5C)2]induced by interaction with 4',6-diamidino-2-phenylindole. Biopolymers. 1993;33(11):1677-86. doi: 10.1002/bip.360331105. PubMed PMID: 8241427. 89.Muramatsu A, Shimizu Y, Yoshikawa Y, Fukuda W, Umezawa N, Horai Y,Higuchi T, Fujiwara S, Imanaka T, Yoshikawa K. Naturally occurring branched-chain polyamines induce a crosslinked meshwork structure in a giant DNA. JChem Phys. 2016;145(23):235103. doi: 10.1063/1.4972066. PubMed PMID:28010109. 90.Zimmer C, Marck C, Guschlbauer W. Z-DNA and other non-B-DNAstructures are reversed to B-DNA by interaction with netropsin. FEBS Lett.1983;154(1):156-60. PubMed PMID: 6299791. 91.Temiz NA, Donohue DE, Bacolla A, Luke BT, Collins JR. The role ofmethylation in the intrinsic dynamics of B- and Z-DNA. PLoS One. 2012;7(4):e35558. doi: 10.1371/journal.pone.0035558. PubMed PMID: 22530050; PMCID:PMC3328458. 92.Goodman SD, Nash HA. Functional replacement of a protein-inducedbend in a DNA recombination site. Nature. 1989;341(6239):251-4. doi: 10.1038/341251a0. PubMed PMID: 2528697. 93.Herbert AG, Rich A. A method to identify and characterize Z-DNAbinding proteins using a linear oligodeoxynucleotide. Nucleic Acids Res.1993;21(11):2669-72. PubMed PMID: 8332463; PMCID: PMC309597. 94.Shishido K, Sakaguchi R, Nosoh Y. Enhancement of S1 nuclease-susceptibility of negatively superhelical DNA by netropsin. Biochem BiophysRes Commun. 1984;124(2):388-92. PubMed PMID: 6093797. 95.Mattila T, Honkanen-Buzalski T, Poso H. Reversible inhibition ofbacterial growth after specific inhibition of spermidine synthase bydicyclohexylamine. Biochem J. 1984;223(3):823 30. PubMed PMID: 6508744;PMCID: PMC1144368. 96.Muth A, Madan M, Archer JJ, Ocampo N, Rodriguez L, Phanstiel Ot.Polyamine transport inhibitors: design, synthesis, and combination therapieswith difluoromethylornithine. J Med Chem. 2014;57(2):348-63. doi: 10.1021/jm401174a. PubMed PMID: 24405276. 97.Takaji S, Hibasami H, Imoto I, Hashimoto Y, Nakao K, Ikemura N,Taguchi Y, Nakashima K. Growth inhibition of Helicobacter pylori by apolyamine synthesis inhibitor, methylglyoxal bis(cyclopentylamidinohydrazone). Lett Appl Microbiol. 1997;25(3):177-80. PubMedPMID: 9351259. 98.Weinstein MP. Comparative evaluation of penicillin, ampicillin,and imipenem MICs and susceptibility breakpoints for vancomycin-susceptibleand vancomycin-resistant Enterococcus faecalis and Enterococcus faecium. JClin Microbiol. 2001;39(7):2729-31. doi: 10.1128/JCM.39.7.2729-2731.2001.PubMed PMID: 11427608; PMCID: PMC88224. 99.LaPlante KL, Leonard SN, Andes DR, Craig WA, Rybak MJ. Activitiesof clindamycin, daptomycin, doxycycline, linezolid, trimethoprim-sulfamethoxazole, and vancomycin against community-associated methicillin-resistant Staphylococcus aureus with inducible clindamycin resistance inmurine thigh infection and in vitro pharmacodynamic models. Antimicrob AgentsChemother. 2008;52(6):2156-62. doi: 10.1128/AAC.01046-07. PubMed PMID:18411321; PMCID: PMC2415789. 100.Pinheiro L, Brito CI, Pereira VC, Oliveira A, Bartolomeu AR,Camargo CH, Cunha ML. Susceptibility Profile of Staphylococcus epidermidisand Staphylococcus haemolyticus Isolated from Blood Cultures to Vancomycinand Novel Antimicrobial Drugs over a Period of 12 Years. Microb Drug Resist.2016;22(4):283-93. doi: 10.1089/mdr.2015.0064. PubMed PMID: 26623676. 101.Sader HS, Mendes RE, Streit JM, Flamm RK. AntimicrobialSusceptibility Trends among Staphylococcus aureus Isolates from U.S.Hospitals: Results from 7 Years of the Ceftaroline (AWARE) SurveillanceProgram, 2010 to 2016. Antimicrob Agents Chemother. 2017;61(9). doi: 10.1128/AAC.01043-17. PubMed PMID: 28630196; PMCID: PMC5571371. 102.Liang-Yu C, Kuo SC, Liu CY, Luo BS, Huang LJ, Lee YT, Chen CP,Chen TL, Fung CP. Difference in imipenem, meropenem, sulbactam, and colistinnonsusceptibility trends among three phenotypically undifferentiatedAcinetobacter baumannii complex in a medical center in Taiwan, 1997-2007. JMicrobiol Immunol Infect. 2011;44(5):358-63. doi: 10.1016/j.jmii.2011.01.032.PubMed PMID: 21524973. 103.Hindler JA, Humphries RM. Colistin MIC variability by method forcontemporary clinical isolates of multidrug-resistant Gram-negative bacilli.J Clin Microbiol. 2013;51(6):1678-84. doi: 10.1128/JCM.03385-12. PubMed PMID:23486719; PMCID: PMC3716094. 104.Rivera CG, Narayanan PP, Patel R, Estes LL. Impact of CefepimeSusceptible-Dose-Dependent MIC for Enterobacteriaceae on Reporting andPrescribing. Antimicrob Agents Chemother. 2016;60(6):3854-5. doi: 10.1128/AAC.00442-16. PubMed PMID: 27067319; PMCID: PMC4879407. 105.Dervan PB, Becker, MM. Molecular recognition of DNA by smallmolecules. Synthesis of bis(methidium)spermine, a DNA polyintercalatingmolecule. 1978;100(6):1968-1970 106.Rajecky, M (2015) Interaction of benzo[ 107.Le TN, et al., A Versatile Total Synthesis of Benzo[ 108.Michael AJ. Polyamines in Eukaryotes, Bacteria, and Archaea. JBiol Chem. 2016;291(29):14896-903. doi: 10.1074/jbc.R116.734780. PubMed PMID:27268252; PMCID: PMC4946907. 109.D'Agostino L, di Pietro M, Di Luccia A. Nuclear aggregates ofpolyamines are supramolecular structures that play a crucial role in genomicDNA protection and conformation. FEBS J. 2005;272(15):3777-87. doi: 10.1111/j.1742-4658.2005.04782.x. PubMed PMID: 16045750. 110.Miller-Fleming L, Olin-Sandoval V, Campbell K, Ralser M.Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell.J Mol Biol. 2015;427(21):3389-406. doi: 10.1016/j.jmb.2015.06.020. PubMedPMID: 26156863. 111.Michael AJ. Biosynthesis of polyamines and polyamine-containingmolecules. Biochem J. 2016;473(15):2315-29. doi: 10.1042/BCJ20160185. PubMedPMID: 27470594. 112.D'Agostino L, di Pietro M, Di Luccia A. Nuclear aggregates ofpolyamines. IUBMB Life. 2006;58(2):75-82. doi: 10.1080/15216540600662525.PubMed PMID: 16608821. 113.Karatan E, Michael AJ. A wider role for polyamines in biofilmformation. Biotechnol Lett. 2013;35(11):1715-7. doi: 10.1007/s10529-013-1286-3. PubMed PMID: 23881324. 114.McGinnis MW, Parker ZM, Walter NE, Rutkovsky AC, Cartaya-Marin C,Karatan E. Spermidine regulates Vibrio cholerae biofilm formation viatransport and signaling pathways. FEMS Microbiol Lett. 2009;299(2):166-74.doi: 10.1111/j.1574-6968.2009.01744.x. PubMed PMID: 19694812. 115.Wortham BW, Oliveira MA, Fetherston JD, Perry RD. Polyamines arerequired for the expression of key Hms proteins important for Yersinia pestisbiofilm formation. Environ Microbiol. 2010;12(7):2034-47. doi: 10.1111/j.1462-2920.2010.02219.x. PubMed PMID: 20406298; PMCID: PMC3039482. 116.Goytia M, Dhulipala VL, Shafer WM. Spermine impairs biofilmformation by Neisseria gonorrhoeae. FEMS Microbiol Lett. 2013;343(1):64-9.doi: 10.1111/1574-6968.12130. PubMed PMID: 23506248; PMCID: PMC3651792. 117.Cardile AP, Woodbury RL, Sanchez CJ, Jr., Becerra SC, Garcia RA,Mende K, Wenke JC, Akers KS. Activity of Norspermidine on Bacterial Biofilmsof Multidrug-Resistant Clinical Isolates Associated with Persistent ExtremityWound Infections. Adv Exp Med Biol. 2017;973:53-70. doi: 10.1007/5584_2016_93. PubMed PMID: 27864804; PMCID: PMC5438303. 118.Wang Y, Kim SH, Natarajan R, Heindl JE, Bruger EL, Waters CM,Michael AJ, Fuqua C. Spermidine Inversely Influences Surface Interactions andPlanktonic Growth in Agrobacterium tumefaciens. J Bacteriol. 2016;198(19):2682-91. doi: 10.1128/JB.00265-16. PubMed PMID: 27402627; PMCID: PMC5019071. 119.Qu L, She P, Wang Y, Liu F, Zhang D, Chen L, Luo Z, Xu H, Qi Y,Wu Y. Effects of norspermidine on Pseudomonas aeruginosa biofilm formationand eradication. Microbiologyopen. 2016;5(3):402-12. doi: 10.1002/mbo3.338.PubMed PMID: 26817804; PMCID: PMC4905993. 120.Konai MM, Adhikary U, Samaddar S, Ghosh C, Haldar J. Structure-Activity Relationship of Amino Acid Tunable Lipidated NorspermidineConjugates: Disrupting Biofilms with Potent Activity against BacterialPersisters. Bioconjug Chem. 2015;26(12):2442-53. doi: 10.1021/acs.bioconjchem.5b00494. PubMed PMID: 26452096. 121.Si X, Quan X, Wu Y. A small-molecule norspermidine andnorspermidine-hosting polyelectrolyte coatings inhibit biofilm formation bymulti-species wastewater culture. Appl Microbiol Biotechnol. 2015;99(24):10861-70. doi: 10.1007/s00253-015-6943-0. PubMed PMID: 26350146. 122.Dewangan RP, Joshi S, Kumari S, Gautam H, Yar MS, Pasha S. N-terminally modified linear and branched spermine backbone dipeptidomimeticsagainst planktonic and sessile methicillin-resistant Staphylococcus aureus.Antimicrob Agents Chemother. 2014;58(9):5435-47. doi: 10.1128/AAC.03391-14.PubMed PMID: 24982082; PMCID: PMC4135887. 123.Ding Y, Peng N, Du Y, Ji L, Cao B. Disruption of putrescinebiosynthesis in Shewanella oneidensis enhances biofilm cohesiveness andperformance in Cr(VI) immobilization. Appl Environ Microbiol. 2014;80(4):1498-506. doi: 10.1128/AEM.03461-13. PubMed PMID: 24362428; PMCID:PMC3911039. 124.Planet PJ, LaRussa SJ, Dana A, Smith H, Xu A, Ryan C, UhlemannAC, Boundy S, Goldberg J, Narechania A, Kulkarni R, Ratner AJ, Geoghegan JA,Kolokotronis SO, Prince A. Emergence of the epidemic methicillin-resistantStaphylococcus aureus strain USA300 coincides with horizontal transfer of thearginine catabolic mobile element and speG-mediated adaptations for survivalon skin. MBio. 2013;4(6):e00889-13. doi: 10.1128/mBio.00889-13. PubMed PMID:24345744; PMCID: PMC3870260. 125.Hobley L, Li B, Wood JL, Kim SH, Naidoo J, Ferreira AS, KhomutovM, Khomutov A, Stanley-Wall NR, Michael AJ. Spermidine promotes Bacillussubtilis biofilm formation by activating expression of the matrix regulatorslrR. J Biol Chem. 2017;292(29):12041-53. doi: 10.1074/jbc.M117.789644.PubMed PMID: 28546427; PMCID: PMC5519356. 126.Ou M, Ling J. Norspermidine changes the basic structure of S.mutans biofilm. Mol Med Rep. 2017;15(1):210-20. doi: 10.3892/mmr.2016.5979.PubMed PMID: 27922663; PMCID: PMC5355703. 127.Nesse LL, Berg K, Vestby LK. Effects of norspermidine andspermidine on biofilm formation by potentially pathogenic Escherichia coliand Salmonella enterica wild-type strains. Appl Environ Microbiol. 2015;81(6):2226-32. doi: 10.1128/AEM.03518-14. PubMed PMID: 25595767; PMCID:PMC4345383. 128.Ramon-Perez ML, Diaz-Cedillo F, Contreras-Rodriguez A, Betanzos-Cabrera G, Peralta H, Rodriguez-Martinez S, Cancino-Diaz ME, Jan-Roblero J,Cancino Diaz JC. Different sensitivity levels to norspermidine on biofilmformation in clinical and commensal Staphylococcus epidermidis strains.Microb Pathog. 2015;79:8-16. doi: 10.1016/j.micpath.2014.12.004. PubMed PMID:25549879. 129.Hobley L, Kim SH, Maezato Y, Wyllie S, Fairlamb AH, Stanley-WallNR, Michael AJ. Norspermidine is not a self-produced trigger for biofilmdisassembly. Cell. 2014;156(4):844-54. doi: 10.1016/j.cell.2014.01.012.PubMed PMID: 24529384; PMCID: PMC3969229. 130.Sakamoto A, Terui Y, Yamamoto T, Kasahara T, Nakamura M, TomitoriH, Yamamoto K, Ishihama A, Michael AJ, Igarashi K, Kashiwagi K. Enhancedbiofilm formation and/or cell viability by polyamines through stimulation ofresponse regulators UvrY and CpxR in the two-component signal transducingsystems, and ribosome recycling factor. Int J Biochem Cell Biol. 2012;44(11):1877-86. doi: 10.1016/j.biocel.2012.07.010. PubMed PMID: 22814172. 131.Burrell M, Hanfrey CC, Murray EJ, Stanley-Wall NR, Michael AJ.Evolution and multiplicity of arginine decarboxylases in polyaminebiosynthesis and essential role in Bacillus subtilis biofilm formation. JBiol Chem. 2010;285(50):39224-38. doi: 10.1074/jbc.M110.163154. PubMed PMID:20876533; PMCID: PMC2998088. 132.Lee J, Sperandio V, Frantz DE, Longgood J, Camilli A, PhillipsMA, Michael AJ. An alternative polyamine biosynthetic pathway is widespreadin bacteria and essential for biofilm formation in Vibrio cholerae. J BiolChem. 2009;284(15):9899-907. doi: 10.1074/jbc.M900110200. PubMed PMID:19196710; PMCID: PMC2665113. 133.Patel CN, Wortham BW, Lines JL, Fetherston JD, Perry RD, OliveiraMA. Polyamines are essential for the formation of plague biofilm. JBacteriol. 2006;188(7):2355-63. doi: 10.1128/JB.188.7.2355-2363.2006. PubMedPMID: 16547021; PMCID: PMC1428407. 134.Chen X, Wu H, Cao Y, Yao X, Zhao L, Wang T, Yang Y, Lv D, Chai Y,Cao Y, Zhu Z. Ion-pairing chromatography on a porous graphitic carbon columncoupled with time-of-flight mass spectrometry for targeted and untargetedprofiling of amino acid biomarkers involved in Candida albicans biofilmformation. Mol Biosyst. 2014;10(1):74-85. doi: 10.1039/c3mb70240e. PubMedPMID: 24150280. 135.Liao Z, ZhangGuan X, Zhu Z, Yao X, Yang Y, Jiang Y, Cao Y.Enhancement of the antibiofilm activity of amphotericin B by polyaminebiosynthesis inhibitors. Int J Antimicrob Agents. 2015;46(1):45-52. doi:10.1016/j.ijantimicag.2015.02.021. PubMed PMID: 25937097. 136.Visick KL, Schembri MA, Yildiz F, Ghigo JM. Biofilms 2015:Multidisciplinary Approaches Shed Light into Microbial Life on Surfaces. JBacteriol. 2016;198(19):2553-63. doi: 10.1128/JB.00156-16. PubMed PMID:26977109; PMCID: PMC5019050. 137.Hoiby N. A short history of microbial biofilms and biofilminfections. APMIS. 2017;125(4):272-5. doi: 10.1111/apm.12686. PubMed PMID:28407426. 138.Gunn JS, Bakaletz LO, Wozniak DJ. What's on the Outside Matters:The Role of the Extracellular Polymeric Substance of Gram-negative Biofilmsin Evading Host Immunity and as a Target for Therapeutic Intervention. J BiolChem. 2016;291(24):12538-46. doi: 10.1074/jbc.R115.707547. PubMed PMID:27129225; PMCID: PMC4933457. 139.Martins M, Uppuluri P, Thomas DP, Cleary IA, Henriques M, Lopez-Ribot JL, Oliveira R. Presence of extracellular DNA in the Candida albicansbiofilm matrix and its contribution to biofilms. Mycopathologia. 2010;169(5):323-31. doi: 10.1007/s11046-009-9264-y. PubMed PMID: 20012895; PMCID:PMC3973130. 140.Brandstetter KA, Jurcisek JA, Goodman SD, Bakaletz LO, Das S.Antibodies directed against integration host factor mediate biofilm clearancefrom Nasopore. Laryngoscope. 2013;123(11):2626-32. doi: 10.1002/lary.24183.PubMed PMID: 23670606; PMCID: PMC4060527. 141.Brockson ME, Novotny LA, Mokrzan EM, Malhotra S, Jurcisek JA,Akbar R, Devaraj A, Goodman SD, Bakaletz LO. Evaluation of the kinetics andmechanism of action of anti-integration host factor-mediated disruption ofbacterial biofilms. Mol Microbiol. 2014;93(6):1246-58. doi: 10.1111/mmi.12735. PubMed PMID: 25069521; PMCID: PMC4160410. 142.Devaraj A, Justice SS, Bakaletz LO, Goodman SD. DNABII proteinsplay a central role in UPEC biofilm structure. Mol Microbiol. 2015;96(6):1119-35. doi: 10.1111/mmi.12994. PubMed PMID: 25757804; PMCID: PMC4464964. 143.Freire MO, Devaraj A, Young A, Navarro JB, Downey JS, Chen C,Bakaletz LO, Zadeh HH, Goodman SD. A bacteriuml-biofilm-induced oralosteolytic infection can be successfully treated by immuno-targeting anextracellular nucleoid-associated protein. Mol Oral Microbiol. 2017;32(1):74-88. doi: 10.1111/omi.12155. PubMed PMID: 26931773; PMCID: PMC5010536. 144.Gustave JE, Jurcisek JA, McCoy KS, Goodman SD, Bakaletz LO.Targeting bacterial integration host factor to disrupt biofilms associatedwith cystic fibrosis. J Cyst Fibros. 2013;12(4):384-9. doi: 10.1016/j.jcf.2012.10.011. PubMed PMID: 23168017; PMCID: PMC3582735. 145.Novotny LA, Clements JD, Goodman SD, Bakaletz LO. TranscutaneousImmunization with a Band-Aid Prevents Experimental Otitis Media in aPolymicrobial Model. Clin Vaccine Immunol. 2017;24(6). doi: 10.1128/CVI.00563-16. PubMed PMID: 28381402; PMCID: PMC5461379. 146.Novotny LA, Jurcisek JA, Goodman SD, Bakaletz LO. Monoclonalantibodies against DNA-binding tips of DNABII proteins disrupt biofilms invitro and induce bacterial clearance in vivo. EBioMedicine. 2016;10:33-44.doi: 10.1016/j.ebiom.2016.06.022. PubMed PMID: 27342872; PMCID: PMC5006588. 147.Novotny LA, Jurcisek JA, Ward MO, Jr., Jordan ZB, Goodman SD,Bakaletz LO. Antibodies against the majority subunit of type IV Pili dispersenontypeable Haemophilus influenzae biofilms in a LuxS-dependent manner andconfer therapeutic resolution of experimental otitis media. Mol Microbiol.2015;96(2):276-92. doi: 10.1111/mmi.12934. PubMed PMID: 25597921; PMCID:PMC4423401. 148.Rocco CJ, Davey ME, Bakaletz LO, Goodman SD. Natural antigenicdifferences in the functionally equivalent extracellular DNABII proteins ofbacterial biofilms provide a means for targeted biofilm therapeutics. MolOral Microbiol. 2017;32(2):118-30. doi: 10.1111/omi.12157. PubMed PMID:26988714; PMCID: PMC5023461. 149.Novotny LA, Amer AO, Brockson ME, Goodman SD, Bakaletz LO.Structural stability of Burkholderia cenocepacia biofilms is reliant on eDNAstructure and presence of a bacteriuml nucleic acid binding protein. PLoSOne. 2013;8(6):e67629. doi: 10.1371/journal.pone.0067629. PubMed PMID:23799151; PMCID: PMC3682984. 150.Baelo A, Levato R, Julian E, Crespo A, Astola J, Gavalda J, EngelE, Mateos-Timoneda MA, Torrents E. Disassembling bacterial extracellularmatrix with DNase-coated nanoparticles to enhance antibiotic delivery inbiofilm infections. J Control Release. 2015;209:150-8. doi: 10.1016/j.jconrel.2015.04.028. PubMed PMID: 25913364. 151.Brown HL, Hanman K, Reuter M, Betts RP, van Vliet AH.Campylobacter jejuni biofilms contain extracellular DNA and are sensitive toDNase I treatment. Front Microbiol. 2015;6:699. doi: 10.3389/fmicb.2015.00699. PubMed PMID: 26217328; PMCID: PMC4498105. 152.Frederiksen B, Pressler T, Hansen A, Koch C, Hoiby N. Effect ofaerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients withcystic fibrosis. Acta Paediatr. 2006;95(9):1070-4. doi: 10.1080/08035250600752466. PubMed PMID: 16938752. 153.Martins M, Henriques M, Lopez-Ribot JL, Oliveira R. Addition ofDNase improves the in vitro activity of antifungal drugs against Candidaalbicans biofilms. Mycoses. 2012;55(1):80-5. doi: 10.1111/j.1439-0507.2011.02047.x. PubMed PMID: 21668524; PMCID: PMC3175262. 154.Goodman SD, Obergfell KP, Jurcisek JA, Novotny LA, Downey JS,Ayala EA, Tjokro N, Li B, Justice SS, Bakaletz LO. Biofilms can be dispersedby focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol. 2011;4(6):625-37. doi: 10.1038/mi.2011.27. PubMed PMID: 21716265. 155.Pasini A, Caldarera CM, Giordano E. Chromatin remodeling bypolyamines and polyamine analogs. Amino Acids. 2014;46(3):595-603. doi:10.1007/s00726-013-1550-9. PubMed PMID: 23836422. 156.Baeza I, Ibanez M, Wong C, Chavez P, Gariglio P, Oro J. Possibleprebiotic significance of polyamines in the condensation, protection,encapsulation, and biological properties of DNA. Orig Life Evol Biosph. 1991;21(4):225-42. PubMed PMID: 1668678. 157.Nayvelt I, Hyvonen MT, Alhonen L, Pandya I, Thomas T, KhomutovAR, Vepsalainen J, Patel R, Keinanen TA, Thomas TJ. DNA condensation bychiral alpha-methylated polyamine analogues and protection of cellular DNAfrom oxidative damage. Biomacromolecules. 2010;11(1):97-105. doi: 10.1021/bm900958c. PubMed PMID: 19919070. 158.Cavaliere R., Ball J. L., Turnbull L., and Whitchurch C. (2014).The biofilm matri destabilizers, EDTA, and DNAseI, enhance biofilms totreatment with ampicillin and ciprofloxacin. Microbiology Open. 3(4):557-567. 159.Chmielewski R.A.N. and Frank J.F. (2003). Biofilm Formation andControl in Food Processing Facilities. Comprehensive Reviews in Food Scienceand Food Safety. Vol. 2. 22-32. 160.Devaraj A., Justice S.S., Bakaletz L.O. , and Goodman S.D.(2015). DNABII Proteins play a central role in UPEC biofilm structure.Molecular Microbiology. 96 (6): 1119-1135. 161.Dongari-Bagtzoglou A. (2008). Pathogenesis of mucosal biofilminfections: challenges and progress. Expert Rev Anti Infect Ther.6(2):201-8.Donlan R.M. (2001). Biofilms and device-associated infections. EmergingInfectious Diseases. 7(2):277-281. 162.Flemming H. and Wingender J. (2010). The biofilm matrix. NatureReviews Microbiology. 8:623-633 163.Goodman S.D., Obergfell K.P., Jurcisek J.A., Novotny L.A., DowneyJ.S., Ayala E.A., Tjokro N., Li B., Justice S.S., and Bakaletz L.O. (2011).Mucosal Immunology. 4 (6): 625-637. 164.Gueroult M., Boittin O., Mauffret O., Etchebest C., and HartmannB. (2012). Mg 2+ in the Major Groove Modulates B-DNA Structure and Dynamics.PLOS ONE. (7)-7-e41704. 165.Hackl E. V., Kornilova S.V., and Blagoi Y. P. (2005) DNAstructural transitions induced by divalent metals ions in aqueous solutions.International Journal of Biological Macromolecules 35:175-191. 166.Hobley L., Li B., Wood J.L., Kim S.H., Naidoo J., Ferreira A.S.,Khomutov M., Khomutov A., Stanley-Wall N.R., and Michael A.J. (2017).Spermidine promotoes Bacillus subtilis biofilm formation by activatingexpression of the matrix regulator slrR. Journal of Biological Chemistry. 292(29): 12041-12053. 167.Nash H. A., Robertson C. A., Flamm E., Weisberg R. A., and MillerH. I. (1987). Overproduction of Escherichia coli Integration Host Factor, aProtein with Nonidentical Subunits. Journal of Bacteriology. 4124-4127. 168.Novotny L.A., Jucisek J.A., Goodman S. D., and Bakaletz L.O.(2016). Monoclonal antibodies against DNA-binding tips of DNABII proteinsdisrupt biofilms in vitro and induce bacterial clearance in vivo.EBioMedicine. (10) :33-44. 169.Patel C.N., Wortham B.W., Lines J.L., Fetherston J.D., PerryR.D., and Oliveira M.A. (2006). Polyamines Are Essential for the Formation ofPlague Biofilm. Journal of Bacteriology. 2355-2363. 170.Tan Z-J. and Chen S-J. (2006). Nucleic Acid Helix Stability:Effects of Salt Concentration, Cation Valence and Size, and Chain Length.Biophysical Journal. (90): 1175-1190. 171.Vorgias C. E., and Wilson K. S. (1991). A rapid purificationprocedure of recombinant integration host factor from Escherichia coli.Protein Expression and Purifcation. 2(5-6):317-20. 172.Wood T.L. , Guha R., Tang. L., Geitner M., Kumar M., and WoodT.K. (2016). Living biofouling-resistant membranes as a model for thebeneficial use of engineered biofilms. PNAS. E2802-E2811. 173.Visick KL, Schembri MA, Yildiz F, Ghigo JM. Biofilms 2015:Multidisciplinary Approaches Shed Light into Microbial Life on Surfaces. JBacteriol. 2016;198(19):2553-63. doi: 10.1128/JB.00156-16. PubMed PMID:26977109; PMCID: PMC5019050. 174.Hoiby N. A short history of microbial biofilms and biofilminfections. APMIS. 2017;125(4):272-5. doi: 10.1111/apm.12686. PubMed PMID:28407426. 175.Gunn JS, Bakaletz LO, Wozniak DJ. What's on the Outside Matters:The Role of the Extracellular Polymeric Substance of Gram-negative Biofilmsin Evading Host Immunity and as a Target for Therapeutic Intervention. J BiolChem. 2016;291(24):12538-46. doi: 10.1074/jbc.R115.707547. PubMed PMID:27129225; PMCID: PMC4933457. 176.Okshevsky M, Meyer RL. The role of extracellular DNA in theestablishment, maintenance and perpetuation of bacterial biofilms. Crit RevMicrobiol. 2015;41(3):341-52. doi: 10.3109/1040841X.2013.841639. PubMed PMID:24303798. 177.Wilton M, Charron-Mazenod L, Moore R, Lewenza S. ExtracellularDNA Acidifies Biofilms and Induces Aminoglycoside Resistance in Pseudomonasaeruginosa. Antimicrob Agents Chemother. 2015;60(1):544-53. doi: 10.1128/AAC.01650-15. PubMed PMID: 26552982; PMCID: PMC4704225. 178.Brandstetter KA, Jurcisek JA, Goodman SD, Bakaletz LO, Das S.Antibodies directed against integration host factor mediate biofilm clearancefrom Nasopore. Laryngoscope. 2013;123(11):2626-32. doi: 10.1002/lary.24183.PubMed PMID: 23670606; PMCID: PMC4060527. 179.Brockson ME, Novotny LA, Mokrzan EM, Malhotra S, Jurcisek JA,Akbar R, Devaraj A, Goodman SD, Bakaletz LO. Evaluation of the kinetics andmechanism of action of anti-integration host factor-mediated disruption ofbacterial biofilms. Mol Microbiol. 2014;93(6):1246-58. doi: 10.1111/mmi.12735. PubMed PMID: 25069521; PMCID: PMC4160410. 180.Devaraj A, Justice SS, Bakaletz LO, Goodman SD. DNABII proteinsplay a central role in UPEC biofilm structure. Mol Microbiol. 2015;96(6):1119-35. doi: 10.1111/mmi.12994. PubMed PMID: 25757804; PMCID: PMC4464964. 181.Freire MO, Devaraj A, Young A, Navarro JB, Downey JS, Chen C,Bakaletz LO, Zadeh HH, Goodman SD. A bacteriuml-biofilm-induced oralosteolytic infection can be successfully treated by immuno-targeting anextracellular nucleoid-associated protein. Mol Oral Microbiol. 2017;32(1):74-88. doi: 10.1111/omi.12155. PubMed PMID: 26931773; PMCID: PMC5010536. 182.Gustave JE, Jurcisek JA, McCoy KS, Goodman SD, Bakaletz LO.Targeting bacterial integration host factor to disrupt biofilms associatedwith cystic fibrosis. J Cyst Fibros. 2013;12(4):384-9. doi: 10.1016/j.jcf.2012.10.011. PubMed PMID: 23168017; PMCID: PMC3582735. 183.Novotny LA, Clements JD, Goodman SD, Bakaletz LO. TranscutaneousImmunization with a Band-Aid Prevents Experimental Otitis Media in aPolymicrobial Model. Clin Vaccine Immunol. 2017;24(6). doi: 10.1128/CVI.00563-16. PubMed PMID: 28381402; PMCID: PMC5461379. 184.Novotny LA, Jurcisek JA, Goodman SD, Bakaletz LO. Monoclonalantibodies against DNA-binding tips of DNABII proteins disrupt biofilms invitro and induce bacterial clearance in vivo. EBioMedicine. 2016;10:33-44.doi: 10.1016/j.ebiom.2016.06.022. PubMed PMID: 27342872; PMCID: PMC5006588. 185.Rocco CJ, Davey ME, Bakaletz LO, Goodman SD. Natural antigenicdifferences in the functionally equivalent extracellular DNABII proteins ofbacterial biofilms provide a means for targeted biofilm therapeutics. MolOral Microbiol. 2017;32(2):118-30. doi: 10.1111/omi.12157. PubMed PMID:26988714; PMCID: PMC5023461. 186.Novotny LA, Amer AO, Brockson ME, Goodman SD, Bakaletz LO.Structural stability of Burkholderia cenocepacia biofilms is reliant on eDNAstructure and presence of a bacteriuml nucleic acid binding protein. PLoSOne. 2013;8(6):e67629. doi: 10.1371/journal.pone.0067629. PubMed PMID:23799151; PMCID: PMC3682984. 187.Baelo A, Levato R, Julian E, Crespo A, Astola J, Gavalda J, EngelE, Mateos-Timoneda MA, Torrents E. Disassembling bacterial extracellularmatrix with DNase-coated nanoparticles to enhance antibiotic delivery inbiofilm infections. J Control Release. 2015;209:150-8. doi: 10.1016/j.jconrel.2015.04.028. PubMed PMID: 25913364. 188.Brown HL, Hanman K, Reuter M, Betts RP, van Vliet AH.Campylobacter jejuni biofilms contain extracellular DNA and are sensitive toDNase I treatment. Front Microbiol. 2015;6:699. doi: 10.3389/fmicb.2015.00699. PubMed PMID: 26217328; PMCID: PMC4498105. 189.Frederiksen B, Pressler T, Hansen A, Koch C, Hoiby N. Effect ofaerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients withcystic fibrosis. Acta Paediatr. 2006;95(9):1070-4. doi: 10.1080/08035250600752466. PubMed PMID: 16938752. 190.Martins M, Henriques M, Lopez-Ribot JL, Oliveira R. Addition ofDNase improves the in vitro activity of antifungal drugs against Candidaalbicans biofilms. Mycoses. 2012;55(1):80-5. doi: 10.1111/j.1439-0507.2011.02047.x. PubMed PMID: 21668524; PMCID: PMC3175262. 191.Goodman SD, Obergfell KP, Jurcisek JA, Novotny LA, Downey JS,Ayala EA, Tjokro N, Li B, Justice SS, Bakaletz LO. Biofilms can be dispersedby focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol. 2011;4(6):625-37. doi: 10.1038/mi.2011.27. PubMed PMID: 21716265. 192.Cho C, Chande A, Gakhar L, Bakaletz LO, Jurcisek JA, Ketterer M,Shao J, Gotoh K, Foster E, Hunt J, O'Brien E, Apicella MA. Role of thenuclease of nontypeable Haemophilus influenzae in dispersal of organisms frombiofilms. Infect Immun. 2015;83(3):950-7. doi: 10.1128/IAI.02601-14. PubMedPMID: 25547799; PMCID: PMC4333478. 193.Kiedrowski MR, Kavanaugh JS, Malone CL, Mootz JM, Voyich JM,Smeltzer MS, Bayles KW, Horswill AR. Nuclease modulates biofilm formation incommunity-associated methicillin-resistant Staphylococcus aureus. PLoS One.2011;6(11):e26714. doi: 10.1371/journal.pone.0026714. PubMed PMID: 22096493;PMCID: PMC3214024. 194.Liu J, Sun L, Liu W, Guo L, Liu Z, Wei X, Ling J. A Nuclease fromStreptococcus mutans Facilitates Biofilm Dispersal and Escape from Killing byNeutrophil Extracellular Traps. Front Cell Infect Microbiol. 2017;7:97. doi:10.3389/fcimb.2017.00097. PubMed PMID: 28401067; PMCID: PMC5368189. 195.Steichen CT, Cho C, Shao JQ, Apicella MA. The Neisseriagonorrhoeae biofilm matrix contains DNA, and an endogenous nuclease controlsits incorporation. Infect Immun. 2011;79(4):1504-11. doi: 10.1128/IAI.01162-10. PubMed PMID: 21300774; PMCID: PMC3067525. 196.Hall-Stoodley L, Nistico L, Sambanthamoorthy K, Dice B, Nguyen D,Mershon WJ, Johnson C, Hu FZ, Stoodley P, Ehrlich GD, Post JC.Characterization of biofilm matrix, degradation by DNase treatment andevidence of capsule downregulation in Streptococcus pneumoniae clinicalisolates. BMC Microbiol. 2008;8:173. doi: 10.1186/1471-2180-8-173. PubMedPMID: 18842140; PMCID: PMC2600794. 197.Izano EA, Shah SM, Kaplan JB. Intercellular adhesion and biocideresistance in nontypeable Haemophilus influenzae biofilms. Microb Pathog.2009;46(4):207-13. doi: 10.1016/j.micpath.2009.01.004. PubMed PMID: 19490830;PMCID: PMC2691864. 198.Kaplan JB, LoVetri K, Cardona ST, Madhyastha S, Sadovskaya I,Jabbouri S, Izano EA. Recombinant human DNase I decreases biofilm andincreases antimicrobial susceptibility in staphylococci. J Antibiot (Tokyo).2012;65(2):73-7. doi: 10.1038/ja.2011.113. PubMed PMID: 22167157; PMCID:PMC3288126. 199.Tetz VV, Tetz GV. Effect of extracellular DNA destruction byDNase I on characteristics of forming biofilms. DNA Cell Biol. 2010;29(8):399-405. doi: 10.1089/dna.2009.1011. PubMed PMID: 20491577. 200.Wang G, Vasquez KM. Z-DNA, an active element in the genome. FrontBiosci. 2007;12:4424-38. PubMed PMID: 17485386. 201.Barraud P, Allain FH. ADAR proteins: double-stranded RNA and Z-DNA binding domains. Curr Top Microbiol Immunol. 2012;353:35-60. doi:10.1007/82_2011_145. PubMed PMID: 21728134; PMCID: PMC3226063. 202.Choi J, Majima T. Conformational changes of non-B DNA. Chem SocRev. 2011;40(12):5893-909. doi: 10.1039/c1cs15153c. PubMed PMID: 21901191. 203.Yang L, Wang S, Tian T, Zhou X. Advancements in Z-DNA:development of inducers and stabilizers for B to Z transition. Curr Med Chem.2012;19(4):557-68. PubMed PMID: 22204344. 204.D'Agostino L, di Pietro M, Di Luccia A. Nuclear aggregates ofpolyamines. IUBMB Life. 2006;58(2):75-82. doi: 10.1080/15216540600662525.PubMed PMID: 16608821. 205.Balasundaram D, Tyagi AK. Polyamine--DNA nexus: structuralramifications and biological implications. Mol Cell Biochem. 1991;100(2):129-40. PubMed PMID: 2008175. 206.Thomas TJ, Gunnia UB, Thomas T. Polyamine-induced B-DNA to Z-DNAconformational transition of a plasmid DNA with (dG-dC)n insert. J Biol Chem.1991;266(10):6137-41. PubMed PMID: 1848849. 207.Kim SK, Eriksson S, Norden B. Z-->B transition in poly[d(G-m5C)2]induced by interaction with 4',6-diamidino-2-phenylindole. Biopolymers. 1993;33(11):1677-86. doi: 10.1002/bip.360331105. PubMed PMID: 8241427. 208.Mirau PA, Kearns DR. The effect of intercalating drugs on thekinetics of the B to Z transition of poly(dG-dC). Nucleic Acids Res. 1983;11(6):1931-41. PubMed PMID: 6835844; PMCID: PMC325847. 209.Waga S, Mizuno S, Yoshida M. Nonhistone protein HMG1 removes thetranscriptional block caused by left-handed Z-form segment in a supercoiledDNA. Biochem Biophys Res Commun. 1988;153(1):334-9. PubMed PMID: 3377790. 210.Pasini A, Caldarera CM, Giordano E. Chromatin remodeling bypolyamines and polyamine analogs. Amino Acids. 2014;46(3):595-603. doi:10.1007/s00726-013-1550-9. PubMed PMID: 23836422. 211.Baeza I, Ibanez M, Wong C, Chavez P, Gariglio P, Oro J. Possibleprebiotic significance of polyamines in the condensation, protection,encapsulation, and biological properties of DNA. Orig Life Evol Biosph. 1991;21(4):225-42. PubMed PMID: 1668678. 212.D'Agostino L, di Pietro M, Di Luccia A. Nuclear aggregates ofpolyamines are supramolecular structures that play a crucial role in genomicDNA protection and conformation. FEBS J. 2005;272(15):3777-87. doi: 10.1111/j.1742-4658.2005.04782.x. PubMed PMID: 16045750. 213.Nayvelt I, Hyvonen MT, Alhonen L, Pandya I, Thomas T, KhomutovAR, Vepsalainen J, Patel R, Keinanen TA, Thomas TJ. DNA condensation bychiral alpha-methylated polyamine analogues and protection of cellular DNAfrom oxidative damage. Biomacromolecules. 2010;11(1):97-105. doi: 10.1021/bm900958c. PubMed PMID: 19919070. 214.Duell BL, Su YC, Riesbeck K. Host-pathogen interactions ofnontypeable Haemophilus influenzae: from commensal to pathogen. FEBS Lett.2016;590(21):3840-53. doi: 10.1002/1873-3468.12351. PubMed PMID: 27508518. 215.Blango MG, Mulvey MA. Persistence of uropathogenic Escherichiacoli in the face of multiple antibiotics. Antimicrob Agents Chemother. 2010;54(5):1855-63. doi: 10.1128/AAC.00014-10. PubMed PMID: 20231390; PMCID:PMC2863638. 216.Jang YJ, Lee C, Kim SK. Formation of Poly[d(A-T)2] Specific Z-DNAby a Cationic Porphyrin. Sci Rep. 2015;5:9943. doi: 10.1038/srep09943. PubMedPMID: 25943171; PMCID: PMC4421867。
- 用于介导EPS的组合物和方法
- 用于EP4介导的骨相关疾病和病症的二氟内酰胺组合物