药物粉末的干燥
文献发布时间:2024-04-18 19:44:28
发明领域
本发明的技术领域涉及药物粉末的间歇和连续干燥。
发明的现有技术
本发明涉及将药物粉末干燥至极低残留量的工艺溶剂。供人服用的药物粉末中的工艺溶剂的残留量受到严格监管[1],并且根据溶剂的毒性,可以在百万分之几百(几百ppm)至0.5wt%的范围。然而,使用既定方法进行干燥是缓慢的过程,其需要花费大量时间[2],通常是工艺瓶颈并且因此与整体工艺生产率直接相关。
用于干燥这些药物粉末的现有技术主要基于间歇工艺,并且包括烘箱和盘式干燥器、流化床干燥器、单滚筒干燥器、真空干燥器和冷冻干燥器[3]。由于满足所需的溶剂水平(典型地超过6小时并且最高达几天)需要很长的干燥时间,因此大多数这些技术无法容易地整合到连续操作中。
此外,由于连续处理设备内的停留时间与干燥时间直接相关,并且停留时间与通量的乘积与设备内的产品总量成正比,因此连续设备相对于间歇设备的占用空间几乎没有差异。
流化床干燥器是最高效的干燥器之一,可以连续运行或间歇运行,并且已经成功地被用于中颗粒和粗颗粒材料的工业中[4]。然而,由于此类粉末的不良流化特性,这些干燥器不适合用于细且粘着性的产品,尤其是尺寸在100微米以下的颗粒。由于不良的流化质量和形成稳定喷动床,Geldart等人将这些粉末列入C组和D组[10]。另外,由于低斯托克斯数,这些细颗粒可以容易地被气体拽引,导致产品通过粉尘控制系统损失和/或最终导致堵塞。它们还可以聚集,改变产品尺寸分布,降低干燥速率并且导致不均匀的产品品质。
真空接触干燥是其中在真空条件下干燥材料的方法,降低快速干燥所需的温度[5]。在该方法中,热主要由通过传导来传递,但也可以通过使用蒸汽或惰性气体诸如氮气或氩气的对流来支持[5][6]。使用各种类型的真空干燥器用于干燥药物产品,包括双锥干燥器、具有螺杆混合器的锥形干燥器、桨式干燥器、真空带式干燥器和过滤器干燥器。达到药物粉末的低残留水平的干燥时间通常很长,通常为约12至48小时[5]。对于这些应用,由于非常大的占用空间要求以允许所需的停留时间,真空接触技术的连续形式是不合适的。
之前前解决了缩短干燥时间并且最终实现连续加工的挑战。Walter Kuelling描述了“连续流化床”(美国专利第3475832A号),其使用高流速的干燥气体来悬浮或流化竖直上升流中的小固体颗粒,使得气体和固体得以紧密接触。后来,Len.W.Hallen公开了具有强烈对流的锥形螺杆型混合器/干燥器(美国专利第5709036A号)。本发明是专门针对难以干燥的材料开发的,提供了在搅拌盘式干燥器中干燥的增加量的材料。这通过在干燥器内产生湍流来实现,特别是在产品的表面,通过迫使加压干燥气体经过喷嘴以高速进入干燥器。D.Dobry等人意识到从无定形材料中去除残留溶剂的困难,提出将颗粒暴露于挥发性流动性增强剂(专利第EP2043610A1号)。作者发现,将药物无定形材料暴露于试剂诸如水或乙醇会降低颗粒的玻璃化转变温度(Tg),导致残留溶剂的扩散速率增加并且因此导致更快的干燥速率。R.Ray等人已经提出了将真空、搅拌和汽提气体结合作为改善具有低于10wt%的残留溶剂含量的无定形材料的干燥时间的方法(专利第CA2594694A1号)。实施了流化床干燥器的许多其他解决方案和派生方案以进一步干燥喷雾干燥的材料。例如,在WO9513864中,提出了一种配置,其中在喷雾干燥单元的下游使用振动流化床,或者在US20030230004A1中,发明人要求保护对流化床内部腔室设计的改变以提高低批量大小的性能。
尽管有多种方法可用于将药物产品干燥至干燥产品中所需溶剂含量,但就简单性、干燥时间和整体生产率而言,它们仍然无法完全令人满意。为了从干燥技术中寻求更高的性能,也出现了微波的使用。J.Hradecky等人首次公开了用于“微波干燥药物明胶胶囊”的设备,旨在减少硬明胶胶囊的生产所涉及的时间并且降低实现胶囊中最佳水分含量所需的能量(专利第US4720924A号)。迄今为止,开发了多种基于微波的干燥器并且在新方法中添加了微波作为替代性能源。最近,L.Bohle等人公开了使用微波作为加热源的“用于干燥药物颗粒、球丸或类似物的方法和设备”(WO专利第WO2003027590A3号)。将产品在旋转驱动的透明管道中干燥,在其中干燥气体将蒸发的水分排出。另外,I.Ghebre-Sellassie等人提出了用于生产药物造粒产品的方法和装置,其考虑将射频或微波视作为加热源(专利第WO2001089679A2号)。关于微波干燥技术,尽管它是非常快速且高效的干燥方法,但设备还不完善,并且其构造也远非简单。另外,在干燥操作中使用微波辐射带来了与常规干燥操作不同的若干限制。使用微波作为辐射源的干燥工艺面临着一些关键挑战,即:对药物产品的品质和稳定性的影响;干燥室内的不均匀辐射分布;需要密封容器以避免微波辐射泄漏。
由于每种干燥方法的上述优点和缺点,对于细且粘着性的产品,诸如通过喷雾干燥所产生的无定形固体,二次干燥步骤通常在真空(典型地低于0.1巴)干燥器中进行,诸如盘式干燥器、双锥形干燥器或搅拌锥形干燥器。目前这些技术的现有技术并不能克服对于长干燥期的需要,这一方面常常使它们成为整个干燥方法的瓶颈,并且另一方面阻碍它们转变成半连续或连续制造。
对于细且粘着性的药物粉末(诸如由喷雾干燥方法产生的那些)所观察到的延长的干燥时间主要是由于在干燥的后期所观察到的极缓慢的干燥动力学(通常从1-3%w/w下降),通常被称为“降速”或“受阻”干燥[7]。由颗粒内质量和热传递所控制的干燥阶段通常是干燥结束时的缓慢干燥速率的原因[7,8,9],并且因此难以通过最常见的手段如增强对流来加速。加速颗粒内热传递的方式尤其包括使用微波和其他形式的能量,而颗粒内质量传递可以通过压力总体波动来加速,如由发明人所测试的,通过向任何单个颗粒的多孔网络中发生的扩散中添加对流。
为了寻找将会显著加速干燥动力学的替代性干燥方法,发明人出人意料地发现,通过在待干燥的粉末材料床上以高速(高对流)施加干燥气体流,可以显著减少达到目标溶剂水平所需的时间。这最初通过向下施加干燥气体,同时通过多孔膜支撑粉末床来测试。通过在干燥器中通过低孔径的可渗透元件(诸如过滤器/膜)支撑粉末床,可以使用高干燥气体流速而无粉末损失或产生优势通道。高流速不仅大幅缩短了干燥时间,而且还产生了均匀的产品,没有轻微的聚集,损失最小并且没有过滤器/膜堵塞。此外,发现干燥时间可以在一定程度上通过干燥气体流速的变化来操纵。减少的干燥时间是有利的,因为它们可以转化为半连续和连续制造。
与现有技术方法相比,本申请描述了一种方法,其允许快速干燥细且粘着性的药物粉末,使其能够用于间歇、半连续和连续操作。
发明目的
本发明的目的是提供一种间歇、半连续或连续干燥方法,其适用于基于对流干燥将细且粘着性的药物粉末干燥至所需的溶剂水平。
本发明的另一目的是提供一种更快的干燥方法以将残留溶剂减少至药物产品可接受的规格限度,以确保产品的安全和质量符合国际标准。
本发明的另一目的是提供一种使用高流速的干燥气体且不引起产品损失的间歇、半连续或连续对流干燥操作。
本发明的另一目的是提供一种用于干燥呈具有改善的均匀性的粉末形式的药物产品的残留溶剂含量的方法。
发明概述
本发明公开了一种用于干燥细且粘着性的药物粉末(D
本发明公开了一种用于将细且粘着性的药物粉末的残留溶剂含量干燥至可接受水平的方法,其包括施加穿过由可渗透元件支撑的干燥室中的粉末床的气体流,所述元件对所述气体流可渗透而保留产品。
已经发现,通过增加干燥气体流速和温度来提高残留溶剂的去除速率。发现通过固定床向下送入干燥气体比在流化床配置中示出更好的结果。通过在低孔径的可渗透元件(诸如过滤器/膜)上支撑粉末,可以使用高干燥气体流速而无需使颗粒被拖出干燥器,如在流化床中所发生的。此外,通过各种实验发现,未观察到优势通道的典型表现,诸如干燥粉末中的不均匀性。
优选地,将可渗透元件诸如过滤器或膜支撑在干燥室的底部的烧结多孔板上,允许干燥气体离开装置,将干燥产物保持在内部。膜可以是多孔的、有机械阻力的(mechanically resistant)并且对干燥产品几乎没有/没有亲和力。可渗透元件有助于将颗粒锁定在适当位置,从而能够使用高干燥气体流速,这转而又增加了气流与颗粒之间的速度,导致更快的干燥。
将先前加热至目标干燥温度的干燥气体向下送入干燥室中。干燥室表面也优选地被加热至目标干燥温度。
另外,可以将振动系统引入干燥装置中用于振动或搅动装置。施加振动可以避免在干燥器内形成粉末的聚集体并且允许将产品从容器壁上脱离。
在本发明的一个方面,干燥产品/粉末的均匀性也可以通过对干燥器中的粉末床施加振动、搅动或其他等效方法来改进。另外,通过对粉末床施加振动、搅动或其他等效方法,也可以减轻可渗透元件中颗粒的堵塞。
本申请中的实验的研究结果是,允许干燥时间大幅度减少的主要区分因素是使气体流穿过由可渗透元件支撑的粉末床,这增加了气体与粉末颗粒之间的相对速度。这样的高相对速度不出现在常用的对流干燥器中,诸如在流化床干燥器中,因为存在一个极限,超过该极限时颗粒将通过气体流气动地输送的。因此,本方法提供了穿过由可渗透膜支撑的粉末床的更高速度的气体流。考虑到高速,细颗粒(诸如低于100微米的颗粒)可以更快地干燥,这与流化床干燥器不同,流化床干燥器不支持穿过流化床的气体流之间的高速。
优选地,干燥气体流基本上垂直于可渗透元件。
在本发明的一个方面,气体流被向下引导,但是干燥方法也可以采用向上或径向流动来应用,只要颗粒被可渗透元件保持在适当位置,这可实现气体与粉末颗粒之间的高相对气体速度。例如,在通过从底部送入气体流的向上流动方法中,其中渗透元件将位于顶部,流动速度可以受益于足够高,使得在干燥操作开始时颗粒上的空气动力学阻力将大于重力,并且因此颗粒将被拖入可渗透元件和粉末床中,在此处颗粒将被保持在适当位置。
因此,本发明提供了干燥细且粘着性产物诸如药物粉末的改进的方法,其包括以下步骤:
i)将药物粉末送入或装载到干燥室中;
ii)将气体流送入干燥室中的粉末床,和
iii)提供可渗透元件以支撑粉末床。可以在送入干燥室之前加热干燥气体,使得粉末在高流速下被推靠在可渗透元件上以使气体流与粉末颗粒之间的相对速度最大化。
然后将干燥的粉末颗粒或产品从干燥器中排出。
当送入或装载到干燥室的粉末沉降在膜/烧结板上时,可以形成粉末床。也可以将待干燥的粉末与干燥气体一起引入干燥室中。干燥气体可以拖动待干燥的粉末并且形成粉末床。
优选地,干燥气体流穿过粉末床和可渗透元件向下、向上或径向流动。
可以在将气体流送入干燥室中之前将其加热至目标干燥温度。优选地,加热干燥室表面以避免热损失,特别是在初始阶段期间。优选地,使用电加热器将初始温度设定至40℃至70℃的范围内,优选地设定至40℃、50℃、52℃、55℃和70℃。
气体的温度可以在室温与材料的熔融温度或玻璃化转变温度之间变化。优选地,典型地将温度选择为尽可能的最大值,而不损害产品的品质,即避免杂质的形成,多晶型特性、堆积密度等的改变。
可以将气体流以足以导致粉末床被推靠在可渗透元件上的流速送入,以使气体与粉末颗粒之间的相对速度最大化。
优选地,气体流速在约0.06kg/h至约3.3kg/h的范围内。优选地,气体流速是0.06kg/h、0.17kg/h、0.28kg/h、0.5kg/h或3.3kg/h。
在本发明的干燥方法中,干燥气体流速与产品质量之间的比率可以是每1kg产品至少0.4kg/h气体。优选地,干燥气体流速与产品质量之间的比率在每1kg产品约0.4至约36kg/h气体的范围内。
气体与粉末之间的相对速度可以根据所使用的产品和气体流速而变化。优选地,颗粒的相对速度可以是至少0.05cm/s。气体与粉末之间的相对速度优选地在约0.05至0.25cm/s的范围内。
本文所描述的方法可以使用单个单元以间歇方法操作。
本文所描述的方法可以通过组合几个干燥单元以半连续模式操作。例如,可以通过组合至少两个并行操作的干燥器来创建半连续方法,一个从上游工艺装载,而另一个干燥产品。
本文所描述的方法可以通过使用螺杆进料器(或等效技术)连续添加湿粉末并且通过使用螺杆单元连续排出干燥粉末来以完全连续模式操作。这样的进料和出料单元将位于干燥室的边界处。此外,通过将这样的干燥器与上游连续干燥步骤诸如喷雾干燥组合,可以创建完全连续的集成方法。
本发明的这些和其他特征从优选实施方案的以下描述和附图将变得更加明显。
附图简述
以下附图提供了优选实施方案以说明描述,但不限制本发明的范围。
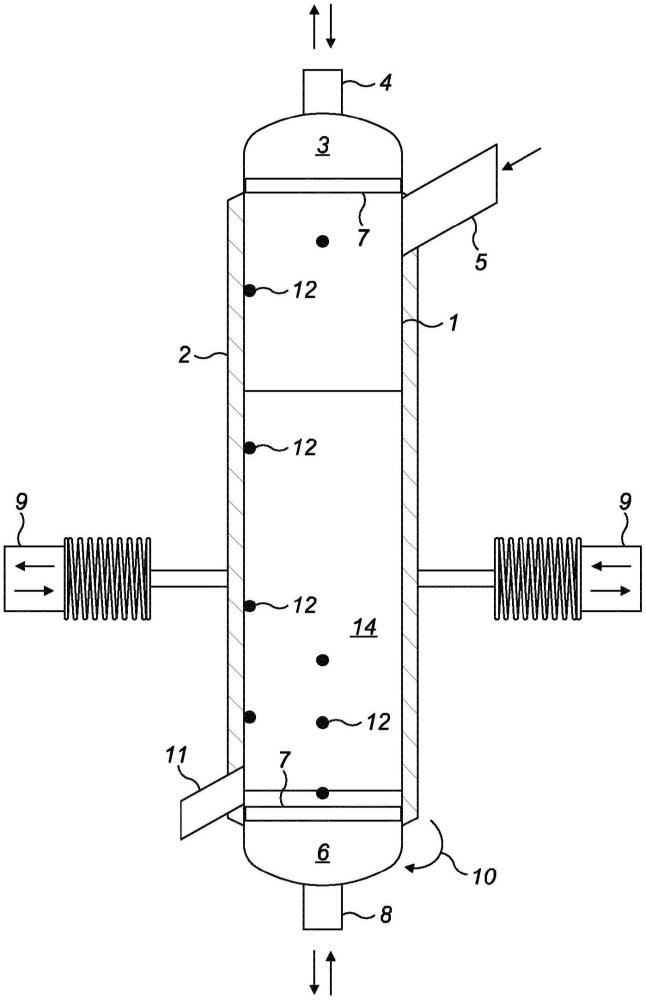
图1示出了用于以间歇方法干燥药物粉末产品的装置的简化示意图。
图2示出了用于以间歇方法干燥药物粉末产品的具有两个同心烧结管的装置的简化示意图。
发明详述
本发明公开了能够对具有低密度(<0.6g/ml)的细且粘着性的药物粉末产品(D
所公开的方法被开发用于干燥药物产品和中间体,诸如由喷雾干燥单元或类似技术产生的那些,其中产品中的溶剂含量高于由药物产品的国际推荐标准所定义的限度[1]。药物产品可以是通过喷雾干燥或类似技术所产生的无定形材料。药物产品也可以是纳米晶体。发明人所进行的实验证明,通过增加干燥气体流速和温度,提高了溶剂从产品中的去除速率。干燥气体与颗粒之间的相对速度的增加也改善了质量和热传输。
在本发明的一个方面,干燥气体流速与产物质量之间的比率至少是0.4(kg
另外,发现使用具有优选地≤1μm的孔径的可渗透元件或多孔膜(其是有机械阻力的并且对被干燥的产品几乎没有亲和力)可以有效地支撑粉末床并且避免堵塞问题。可渗透元件或多孔膜可以进一步被具有更高机械稳定性的烧结金属或聚合物板或载体支撑或替换。
可渗透元件的孔隙率应当使得其应当保留颗粒,但应当允许气体流动。
在选择可渗透元件时可以考虑其与被干燥的产品或粉末中的溶剂的化学相容性及其物理特性(最大操作温度和疏水性)。在选择可渗透元件孔隙率时可以考虑被干燥的产品或药物粉末的粒度。
优选地,可渗透元件诸如多孔膜或过滤器具有≤2μm、或≤1μm的孔尺寸或孔径。优选地,可渗透元件的孔隙率可以在0.02至2μm或0.02至1μm的范围内。
优选地,可渗透元件诸如多孔膜或过滤器由诸如特氟龙、PP、PVDF、PCTE或其混合物的材料制成,其具有机械阻力并且对被干燥的产品几乎没有亲和力。
为了有利于从颗粒到干燥气体的溶剂质量传递,温度必须尽可能高,但不影响最终产品的物理-化学特性。
干燥气体的性质也可以在溶剂的去除速率中起作用。可以使用任何合适的干燥气体或不促进与产品的不期望的反应的其他气体或气体混合物。优选地,干燥气体选自由以下组成的组:N2、CO2、空气及其组合或混合物。优选地,干燥气体是二氧化碳。在另外的研究中,发明人发现使用二氧化碳已经证明是有利的。用二氧化碳的干燥实验显示,当与氮气相比时,最终产品中的溶剂含量降低。二氧化碳具有更高的热容并且在一些情况下具有高于氮气的对颗粒表面的亲和力,并且因此可以更容易地置换溶剂。
在本发明的一个方面,干燥方法是间歇方法,在圆柱形装置中进行,如图1中所示。装置包含圆柱形干燥器(1)、电加热系统(2)、顶盖(3)、干燥气体入口阀(4)、重力进料阀(5)、底盖(6)、多孔烧结金属板上的可渗透元件(7)、干燥气体出口阀(8)、振动单元(9)、底盖开启机构(10)、样品阀(11)和温度和/或压力测量点(12)。将待干燥的粉末或产品从顶部通过进料门(5)装载到装置中,直到其达到根据粉末的干燥特性的预定体积。粉末沉淀在由烧结板支撑的可渗透元件上,该烧结板充当载体并且避免小颗粒被拖出。通过开启围绕整个装置分布的电加热器(2)并且通过引入干燥气体(4)来启动干燥过程。在预定的加热升温之后,温度随着干燥时间持续升高。数个热电偶(12)沿着装置分布以获得精确的读数,以便控制温度。将先前加热的干燥气体向下供应至装置。干燥气体流速与产品质量之间的比率在约0.4至约38(kg
优选地,气体压力从真空至最高达10巴的压力、优选地在2-4巴之间周期性地变化。本申请的装置可以在2-4巴之间以60s的压力循环操作。
在本发明的另一方面,干燥方法是半连续方法。在半连续方法中,装置包含两个干燥单元,并且当其中所述单元之一被用通过上游干燥技术(诸如喷雾干燥器)所产生的产品装载时,另一个单元正在干燥。装载和干燥过程是同步的,以这样的方式使得一旦第一单元的装载步骤结束,第二单元中的干燥步骤也结束;第一单元跟随干燥步骤,并且第二单元在卸载到接收器(16)中之后启动装载步骤。
在本发明的另一方面,干燥方法是间歇方法,在具有两个同心烧结管(20,21)的圆柱形装置中进行,如图2中所示。装置包含圆柱形干燥器(1)、电加热系统(2)、顶盖(3)、干燥气体入口阀(4)、底盖(6)、覆盖有膜的外部烧结管(20)和覆盖有膜的内部烧结管(21)、干燥气体出口阀(8)、旋转单元(22)和数个温度和/或压力测量点(12)。
将粉末装载到位于这两个同心烧结管(20,21)之间的装置中,直到其达到根据粉末的干燥特性的预定体积。
通过开启围绕整个装置分布的电加热器(2)并且通过引入干燥气体(4)来启动干燥过程。数个热电偶(12)沿着装置分布以控制温度并且获得精确的读数。将先前加热的干燥气体供应至装置。气体方向是径向的并且可以假设为向内的方向。优选地,干燥气体流速与产品质量之间的比率在约1至约10(kg
在本发明的另一方面,提供了如本文以上所描述的可渗透元件(诸如过滤器或多孔膜)用于将药物产品的残留溶剂含量干燥至根据行业标准的可接受水平的用途,所述可渗透元件在干燥室中用于支撑在干燥器中的药物产品的粉末床。
在本发明的另一方面,提供了已知干燥装置(其可以与喷雾干燥器、过滤器干燥器、真空干燥器或任何其他已知干燥器组合使用)通过引入可渗透元件以支撑将干燥气体流施加通过其的粉末床,用于将药物产品的残留溶剂含量干燥至根据行业标准的可接受水平的用途。
在本发明的另一方面,提供了通过本文所描述的干燥方法所获得的颗粒或药物产品。
图1中所示出的干燥单元使用共聚维酮(制药工业中且特别是在喷雾干燥的分散体中常用的聚合物)进行测试,其中溶剂(乙醇)含量为大约5wt%。目的是根据既定指南(即低于5000ppm)[1]并且在几小时内将产品干燥至可接受的残留乙醇水平。通过重力进料阀(5)引入100g的共聚维酮质量;粉末沉降在膜/烧结板(7)上。膜是具有0.2μm的孔径的特氟龙膜。使用由PID控制器控制的电加热器(2)将温度设置至50℃,其中温度信息由靠近圆柱壁的热电偶(12)提供。向下引入0.28kg/h的气体流速;在该情况下为CO2。系统在2-4巴之间以60s的压力循环操作。每小时收集干燥粉末的样品以评价干燥方法;通过顶空气相色谱法分析样品。将该方法与在真空(50毫巴)下以2h
表1-干燥方法50℃和70℃的比较。
结果表明,在使用CO2作为干燥气体干燥3小时之后,使用本发明的方法(本发明)的乙醇含量是参考盘式干燥器的五分之一。
使用实验设计(DOE)研究了干燥产物质量、温度与流速之间的关系。DOE考虑了三级参数全因子(具有中心点),并且使用氮气作为干燥气体。因此,在类似于图1中所示出的干燥单元中进行实验,并且考虑三种温度(40℃、55℃和70℃)、三种流速(0.06kg/h、0.17kg/h、0.50kg/h)和三种质量的共聚维酮(13.3g、40g和120g),均具有约5wt.%的初始溶剂(乙醇)含量。表2中公开了实验条件和所获得的主要结果。
表2-DOE实验和主要结果。
结果表明,对于更大量的产品,流速对干燥方法具有影响并且可以显著改善它。此外,产品量的增加几乎可以通过增加气体流速线性地补偿;该观察结果对所有研究的温度都是有效的。另外,观察到温度对干燥速率的影响很大;在该情况下,质量与温度之间的关系在所研究的范围内几乎是线性的。
此外,观察到高于一定的流速-质量比率时,干燥速率没有显著变化并且达到平台期。
从所进行的实验中观察到,总体干燥速率直接随着气体速度的增加而响应,达到一定的水平。然后,干燥速率稳定,表明内部阻力控制剩余的总体干燥过程,这可以通过增加温度来改善。
使用先前所提及的干燥单元(图1)评价了具有不同孔隙率的不同类型的膜对干燥方法的影响。这些测试还允许理解膜和烧结板中的压降和堵塞现象。选择膜时考虑其与溶剂的化学相容性及其物理特性(最大操作温度和疏水性)。所选择的膜是PTFE、PCTE、PP和PVDF。膜孔隙率是0.2μm和1μm;选择这些孔隙率时考虑共聚维酮粒度范围在2.5μm至63μm(普遍粒度是约20μm)。使用120g的共聚维酮,在55℃下以0.5kg/h的N
干燥过程期间的乙醇含量不受膜的类型的影响。观察到的差异很小并且可以归因于取样和样品暴露过程。
表3-使用不同类型的膜的干燥结果。
另外,在实验之前、期间和之后测量膜/烧结板中的压降(表4)。结果表明,具有0.2μm的孔径的膜不仅在测试之前具有高于具有1μm的孔径的膜的压降,而且在干燥测试期间也具有更高的压降增加。尽管如此,在数个干燥循环期间,未观察到任何测试膜的显著堵塞。考虑到测试膜的实验结果和物理-化学特征,具有1μm孔隙率的非层压PTFE更适用于该特定应用。
表4-干燥实验之前、干燥期间和干燥之后膜/烧结板中的压降。
使用之前提到的干燥单元(图1)评价了柱高对干燥方法、粉末压实和压降的影响(表5)。实验在55℃下在N
表5-压降、床压实和干燥结果。
使用共聚维酮测试图2中所示出的干燥单元,其中溶剂(乙醇)含量为约5wt%。在设备中在竖直位置中引入890g的共聚维酮质量,沉降在这两个同心的烧结管之间。装载和关闭之后,将设备水平放置。设备具有致动器以旋转<270°(两个方向)。使用由PID控制器控制的电加热器(2)将温度设置至52℃,其中温度信息由靠近圆柱壁的热电偶(12)提供。向内送入3.3kg/h的气体流速。测试期间的压降是约0.71巴(仅0.08巴由粉末引起)。3小时之后在三个不同的点中收集干燥粉末的样品以评价干燥方法;通过顶空气相色谱法分析样品。表6中呈现的结果表明,有可能在3小时内达到5000ppm,并且未观察到比较这三个取样点的严重不均匀性。此外,在实验期间,未观察到期间的堵塞或压力增加,确证了该方法的可行性。
表6-在52℃下的干燥结果。
文献目录
专利文献
US3475832A-Continuous fluid bed dryer,1986年11月。
US5709036A-Aggressive convective drying in a conical screw typemixer/dryer,1998年1月。
EP2043610A1-Drying of drug-containing particles,2007年3月US4720924A-Microwave drying of pharmaceutical gelatin capsules,1988年1月。
CA2594694A1-Drying of drug-containing particles,2006年1月WO9513864-AProcess and a Spray Drying Apparatus for Producing An Agglomerated Powder
US20030230004A1-Batch Fluid Bed Processor
WO2001089679A2-Continuous production of pharmaceutical granulates,2001年11月。
WO2003027590A3-Method and device for drying pharmaceuticalgranulates,pellets or similar,2003年4月。
其他参考文献
[1]“IMPURITIES:GUIDELINE FOR RESIDUAL SOLVENTS Q3C(R6),第4版”,ICHHARMONISED GUIDELINE,第4版,2016.
[2]K.R.Morris,S.L.Nail,G.E.Peck,S.R.Byrn,U.J.Griesser,J.G.Stowell,S.-J.Hwang和K.Park,“Advances in pharmaceutical material and procesing,”Pharmaceutical science&technology today,第235-245页,1998.
[3]F.J.Muzzio,T.Shinbrot和B.J.Glasser,“Powder technology in thepharmaceutical industry:the need to catch up fast,”Powder Technology,第1-7页,2002.
[4]W.R.W.Daud,“Fluidized Bed Dryers-Recent Advances,”Advanced PowderTechology,第403-418页,2008.
[5]D.Parikh,“Vacuum drying:basics and applications,”ChemicalEngineering,第48-54页,2015.
[6]C.L.Hii,C.L.Law,M.Cloke和S.Suzannah,“Thin layer drying kinetics ofcocoa and dried product quality,”Biosystems Engineering,第102卷,第153-161页,2009.
[7]I.T.Administration,“2016Top Markets Report-Pharmaceuticals,”U.S.Department of Commerce,2016.
[8]F.X.McConville,“Tips for Drying Active PharmaceuticalIngredients,”process HEATING,2007.
[9]H.Leuenberger,“New trends in the production of pharmaceticalgranules:batch versus continuous processing,”European Journal ofPharmaceutics and Biopharmaceutics,第289-296页,2001.
[10]D.Geldart,“Types of Gas Fluidization,”Powder Technology,第7卷,第285-292页,1973.