无金属石墨型氮化碳材料及其制备方法、污水处理方法
文献发布时间:2024-04-18 19:52:40
技术领域
本发明涉及环保领域中的一种氮化碳材料及其制备方法,具体涉及一种无金属石墨型氮化碳材料及其制备方法、采用所述无金属石墨型氮化碳材料的具有非光催化活性的活性催化剂、采用所述活性催化剂的降解废水中有机污染物的污水处理方法。
背景技术
随着工业化和人口的快速增长,有机污染物导致的水污染成为严重威胁公众健康和安全的全球性问题。各种合成有机物,如染料、杀虫剂、药品和个人护理产品等,在水体中具有较高的溶解性、稳定性和耐久性,因而容易进入到湖泊、河流、水库等自然水体中并不断被富集,造成严重的水污染问题。常规的污水处理技术,比如物理处理方法(过滤、吸附和絮凝)和生物处理方法(生物膜法、活性污泥法)无法实现废水的深度净化和完全处理,因此发展清洁高效、成本低廉的高级氧化水处理方法具有极为重要的意义。
高级氧化工艺是一种快速且有效去除难降解有机污染物的水处理技术。通过活化过氧化物氧化剂,获得具有更高氧化能力的自由基(如羟基自由基、硫酸根自由基)或非自由基(如单线态氧)来攻击稳定的有机污染物,实现污染物的完全降解。这种水处理工艺具有成本低廉、反应稳定性高、水质适应性广、降解速率快等优点。过氧化物氧化剂处理有机污染物时需要添加特殊的催化剂对其进行活化;石墨型氮化碳材料因其制备简单、成本低廉、化学稳定性高、具有可见光响应等特点,被广泛用于光催化领域,同时在水污染控制领域的应用包括有机污染物的降解以及过氧化物氧化剂的活化。值得注意的是,石墨型氮化碳本身固有的缺陷例如光生电子-空穴复合率高、以及可见光吸收范围窄等,严重限制了其光催化活性。此外,常规石墨型氮化碳材料的电子传输能力差,因此在没有光照的条件下该材料通常不具备催化活性。
此外,受限于自然光的不稳定性,外加光源也需投入更多的能源消耗。因此,为了拓展石墨型氮化碳的实际应用前景,学者们尝试了多种改性方法来赋予其非光催化活性。常用的方法是在石墨型氮化碳中掺入过渡金属元素(铁、钴、铈、锰、铜等)。但当前过渡金属化合物改性剂的用量过高,且石墨型氮化碳对金属的奥和能力不高,在长期使用中会导致过度金属的浸出,造成另一种污染;此外,一些金属改性方法为提升催化效率,使用复杂且成本高昂的制备方法,这类催化剂因制备成本过高而无法量产,极大地限制了材料本身的实际应用能力。
因此,如何提升石墨型氮化碳非光催化性能并避免过渡金属的使用,对于提高石墨型氮化碳在污水处理领域的实用价值具有重要意义,但是现有技术中仍然没有很好的,可以推广应用的技术方案。
发明内容
为解决现有石墨型氮化碳非光催化性能低并无法避免过渡金属使用的技术问题,本发明提供一种无金属石墨型氮化碳材料及其制备方法、采用所述无金属石墨型氮化碳材料的具有非光催化活性的活性催化剂、采用所述活性催化剂的降解废水中有机污染物的污水处理方法。
本发明采用以下技术方案实现:一种无金属石墨型氮化碳材料,其为通过将苯二甲腈类配体中富含碳的苯环引入到氮化碳前驱体的庚嗪环中,二者在氮化碳产物高温热聚合过程中得到的固形煅烧产物,所述固形煅烧产物是按照20:(0.1-2)的质量比准备氮化碳前驱体、苯二甲腈类配体作为原料,将两种原料以500-700℃的煅烧温度,保温煅烧1-3h后得到的块状固形煅烧产物。
作为上述方案的进一步改进,所述氮化碳前驱体包括硫脲、双氰胺、三聚氰胺、5-氨基四氮唑和尿素中的至少一种。
作为上述方案的进一步改进,所述苯二甲腈类配体包括邻苯二甲腈、间苯二甲腈、对苯二甲腈或4,4’-联苯甲腈中的至少一种。
本发明还提供一种无金属石墨型氮化碳材料的制备方法,其包括以下步骤:
按照20:(0.1-2)的质量比准备氮化碳前驱体、苯二甲腈类配体作为原料;
将二类原料充分混合均匀后,以500-700℃的煅烧温度,保温煅烧1-3h,得到的固形煅烧产物即为所需的无金属石墨型氮化碳材料。
作为上述方案的进一步改进,所述氮化碳前驱体包括硫脲、双氰胺、三聚氰胺、5-氨基四氮唑和尿素中的至少一种。
作为上述方案的进一步改进,所述苯二甲腈类配体包括邻苯二甲腈、间苯二甲腈、对苯二甲腈或4,4’-联苯甲腈中的至少一种。
作为上述方案的进一步改进,在煅烧过程中,将混合均匀后的二类原料先送入到马弗炉内,然后对马弗炉进行匀速升温,升温速率控制为2-10℃/min;炉温达到煅烧温度后,保温煅烧1-3h,煅烧结束后,将产物从马弗炉内取出并自然冷却至室温。
本发明还提供一种具有非光催化活性的活性催化剂,所述活性催化剂由上述任意无金属石墨型氮化碳材料制成。
本发明还提供一种降解废水中有机污染物的污水处理方法,其采用了上述的具有非光催化活性的活性催化剂,所述活性催化剂作为利用过氧化物氧化剂降解废水中有机污染物时使用。
作为上述方案的进一步改进,所述污水处理方法的过程如下:
在含有目标污染物的污水中,根据处理不同污染物时不同药品的目标添加量,向污水中投入最佳目标用量的过氧化物氧化剂和活性催化剂,对污水进行混匀搅拌;直到目标污染物的浓度降低至污水中目标污染物的允许值以下。
作为上述方案的进一步改进,所述过氧化物氧化剂包括过一硫酸氢钾、过硫酸钾、过硫酸钠和过氧乙酸。
作为上述方案的进一步改进,所述有机污染物包括对氯苯酚、2,4-二氯苯酚、对乙酰氨基酚、诺氟沙星和磺胺甲恶唑。
本发明的制备方法可以得到一种改进型的无金属石墨型氮化碳材料,经过对配方的改良,使得材料的获得了非光催化活性。因此在使用本发明提供的产物作为过氧化物氧化剂降解有机物污染物时的活性催化剂使用时,可以不受光照强度的限制,保持稳定的有机污染物降解效率。其中本发明的无金属石墨型氮化碳材,其材料的性能提升是通过将苯二甲腈类配体中富含碳的苯环引入到庚嗪环中实现的。二者在氮化碳产物高温热聚合过程中,改变了材料的电子排布和电子离域能力,有效发挥出最终产物的非光催化活性。此外,本发明提供的无金属石墨型氮化碳材料彻底避免了过渡金属的使用,无任何金属浸出的风险。
附图说明
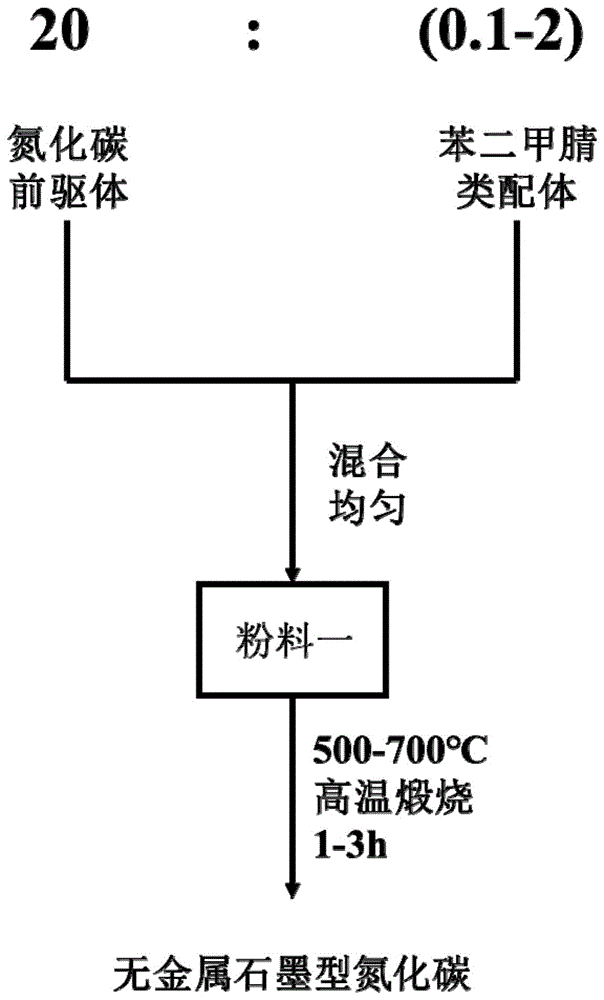
图1为本发明的无金属石墨型氮化碳材料的制备方法的工艺流程图。
图2为图1中制备方法制备的产品CN1和对比例的产品CN17之间的外观对比图。
图3为图2中CN7和CN17样品的XRD谱图。
图4为图2中CN1和CN17的样品的X射线光电子能谱图。
图5为图2中CN1和CN17的样品的固体核磁共振(SSNMR)
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
本实施例介绍的无金属石墨型氮化碳材料为通过将苯二甲腈类配体中富含碳的苯环引入到氮化碳前驱体的庚嗪环中,二者在氮化碳产物高温热聚合过程中得到的固形煅烧产物。所述氮化碳前驱体可包括硫脲、双氰胺、三聚氰胺、5-氨基四氮唑和尿素中的至少一种。所述苯二甲腈类配体可包括邻苯二甲腈、间苯二甲腈、对苯二甲腈或4,4’-联苯甲腈中的至少一种。
无金属石墨型氮化碳材料的性能提升是通过将苯二甲腈类配体中富含碳的苯环引入到庚嗪环中实现的。二者在氮化碳产物高温热聚合过程中,改变了材料的电子排布和电子离域能力,有效发挥出最终产物的非光催化活性。此外,本发明提供的无金属石墨型氮化碳材料彻底避免了过渡金属的使用,无任何金属浸出的风险。
无金属石墨型氮化碳材料可作为利用过氧化物氧化剂降解废水中有机污染物时使用的活性催化剂。本发明的活性催化剂具有非光催化活性,可应用在降解废水中有机污染物的污水处理中。降解废水中有机污染物的污水处理方法的过程如下:在含有目标污染物的污水中,根据处理不同污染物时不同药品的目标添加量(如专家经验值),向污水中投入最佳目标用量(也可以是相应的专家经验值)的过氧化物氧化剂和活性催化剂,对污水进行混匀搅拌;直到目标污染物的浓度降低至污水中目标污染物的允许值以下。在本实施例中,在含有目标污染物的污水中,根据处理不同污染物时不同药品的添加量的专家经验值,向污水中投入最佳用量的过硫酸盐和活性催化剂,对污水进行混匀搅拌;直到目标污染物的浓度降低至污水中目标污染物的允许值以下。其中,所述目标污染物包括对氯苯酚、2,4-二氯苯酚、对乙酰氨基酚、诺氟沙星、磺胺甲恶唑和磺胺吡啶。所述过氧化物氧化剂用于对目标污染物进行降解,所述过氧化物氧化剂包括过一硫酸氢钾、过硫酸钾、过硫酸钠和过氧乙酸。所述活性催化剂对过氧化物氧化剂进行催化活化以降解有机污染物。本发明的污水处理方法提升有机污染物的降解速率和去除率。投入药品之后,有机污染物的降解过程在光照和非光照条件下均可以完成。
请参阅图1,图1为本发明的无金属石墨型氮化碳材料的制备方法的工艺流程图。无金属石墨型氮化碳材料在制备时,其制备方法包括以下步骤:按照20:(0.1-2)的质量比准备氮化碳前驱体、苯二甲腈类配体作为原料;将二类原料充分混合均匀后,以500-700℃的煅烧温度,保温煅烧1-3h,得到的固形煅烧产物即为所需的无金属石墨型氮化碳材料。
其中,在煅烧过程中,将混合均匀后的二类原料先送入到马弗炉内,然后对马弗炉进行匀速升温,升温速率控制为2-10℃/min;炉温达到煅烧温度后,以500-700℃的煅烧温度,保温煅烧1-3h,煅烧结束后,将产物从马弗炉内取出并自然冷却至室温,得到的固形煅烧产物即为所需的石墨型氮化碳材料。
氮化碳前驱体作为提供富含碳氮元素的小分子,在高温条件下通过热缩聚反应生成规则的庚嗪结构。由于氮化碳前驱体可以选择包括硫脲、双氰胺、三聚氰胺、5-氨基四氮唑和尿素中的一种或任意多种,以上氮化碳前驱体材料的制备方法简单,成本低廉,性状稳定,原料简单易得。具体使用时可以根据需要进行选择,除以上列举出的的氮化碳前驱体材料之外,其它可以通过高温热缩聚反应生成的稳定的庚嗪环结构的小分子材料,也可以作为本发明中所需的氮化碳前驱体原料。本发明中的苯二甲腈类配体原料在反应过程中,苯环作为富碳结构被引入庚嗪环结构中,甲腈基团作为碳-氮官能团,可以与氮化碳前驱体原料共同热缩聚,将苯环引入庚嗪环中。当苯环被引入庚嗪环环结构后,改变了原本碳-氮结构的电子分布和电子离域能力,赋予了其非光催化的能力。本发明解决了现有石墨型氮化碳材料不具备非光催化活性、材料中掺入金属的问题,即解决现有石墨型氮化碳材料在污水处理过程中不具有非光催化活性,以及现有金属改性的石墨型氮化碳非光催化剂中,金属浸出且成本高昂的问题,避免了大量金属浸出带来的二次污染,提高了催化剂的可重复使用性。
以下通过具体的生产实例对提供的制备方法生产出的产品及其性能进行进一步的说明。
实施例1
称取20g尿素,1.0g对苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至550℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN1。
实施例2
称取20g双氰胺,1.0g对苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至550℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN2。
实施例3
称取20g三聚氰胺,2.0g对苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以2.5℃/min的升温速率由室温升至550℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN3。
实施例4
称取20g硫脲,2.0g对苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至600℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN4。
实施例5
称取20g5-氨基四氮唑,0.1g4,4’-联苯甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至700℃,并且保温1小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN5。
实施例6
称取20g三聚氰胺、双氰胺和尿素三者的等质量比混合物,2g间苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以10℃/min的升温速率由室温升至650℃,并且保温1小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN6。
实施例7
称取20g尿素和5-氨基四氮唑的等质量比混合物,0.5g对苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以2℃/min的升温速率由室温升至600℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN7。
实施例8
称取20g硫脲和5-氨基四氮唑的等质量比混合物,0.5g邻苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以10℃/min的升温速率由室温升至600℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN8。
实施例9
称取20g双氰胺和三聚氰胺等质量比混合物,0.5g对苯二甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以10℃/min的升温速率由室温升至700℃,并且保温1小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN9。
实施例10
称取20g硫脲和尿素的等质量比混合物,0.4g4,4’-联苯甲腈;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至500℃,并且保温3小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN10。
实施例11
称取20g三聚氰胺,2.0g对苯二甲腈和间苯二甲腈的等质量比混合物,混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至650℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN11。
实施例12
称取20g双氰胺,2g对苯二甲腈和邻苯二甲腈的等质量比混合物;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至600℃,并且保温3小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN12。
实施例13
称取20g尿素,2g对苯二甲腈、邻苯二甲腈和间苯二甲腈的等质量比混合物;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至550℃,并且保温3小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN13。
实施例14
称取20g5-氨基四氮唑,0.5g对苯二甲腈和4,4’-联苯甲腈等质量比混合物;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至550℃,并且保温3小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN14。
实施例15
称取20g硫脲,1g对苯二甲腈和间苯二甲腈等质量比混合物;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以10℃/min的升温速率由室温升至550℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN15。
实施例16
称取20g尿素和5-氨基四氮唑的等比混合物,1g对苯二甲腈和间苯二甲腈等质量比混合物;混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以2.5℃/min的升温速率由室温升至550℃,并且保温3小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN16。
为了确定本案提供的石墨型氮化碳材料中各原料与最终产物理化性能间的关系,本案还以实施例1~6为对照组,采用控制变量的原则制备六组产品,并以该六组样品为对比例。具体地,对比例的制备方法如下:
对比例1
称取20g尿素,装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至550℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN17。
对比例2
称取20g双氰胺,装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至550℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN18。
对比例3
称取20g三聚氰胺,装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以2.5℃/min的升温速率由室温升至550℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN19。
对比例4
称取20g硫脲,装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至600℃,并且保温2小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN20。
对比例5
称取20g5-氨基四氮唑,装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以5℃/min的升温速率由室温升至700℃,并且保温1小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN21。
对比例6
称取20g三聚氰胺、双氰胺和尿素三者的等质量比混合物,混合均匀装入带盖的陶瓷坩埚中,将坩埚置于马弗炉中;将马弗炉以10℃/min的升温速率由室温升至650℃,并且保温1小时,煅烧结束后自然冷却至室温;收集所得固体在玛瑙研钵中研磨成粉末,记为CN22。
观察上述产物的物理性状,本案与对比例的产品均为均匀的粉末状产物。其中,图2显示了本案的产品(CN1,右边图示区域)和对比例的产品(CN17,左边图示区域)之间的外观对比图。本案的产品颜色较深呈深棕色,且不同实施例中原料类型比例不同颜色的深浅存在差异;而对比例中产品的颜色较浅呈乳黄色。
接着,设计如下的测试试验对制备出的CN1~21的产品进行检测,确定各组样本的理化性质。具体的测试试验包括:
测试试验1:
CN17产品是采用市场上常规的石墨型氮化碳材料的生产原料,并按照本案提供的生产工艺制备而成的。CN1是按照本案的提供的原料和制备工艺制备而成的。本试验以CN1和CN17的产品为样本,使用X射线衍射(XRD)技术进行分析;根据分析结果绘制如图3所示的CN7和CN17样品的XRD谱图。
分析XDR谱图中的数据可以发现:CN17具有典型的石墨型氮化碳(100)面和(002)面的特征衍射峰。而CN1和CN17谱图相似,表明苯二甲腈类配体的改性保持了与对比例1中类似的催化剂的周期性堆叠结构。
测试试验2
接下来,继续以CN1和CN17的产品为样本,使用X射线光电子能谱(XPS)仪进行分析,并根据试验数据绘制如图4所示的CN1和CN17的样品的X射线光电子能谱图。
分析X射线光电子能谱图中的数据可以发现:CN17具有典型石墨型氮化碳的N-C=N(288.2eV)、C-NH
测试试验3
同样以CN1和CN17的产品为样本,使用固体核磁共振技术进行分析,并根据试验数据绘制如图5所示的CN1和CN17的样品的固体核磁共振(SSNMR)
测试试验4
称取前述实施例制备的CN1-17中的石墨型氮化碳材料作为活性催化剂,并选择过一硫酸氢钾作为污染物降解药品。将二者加入50mL有机污染物水溶液中,进行污染物降解试验。
污染物降解试验中选择的有机污染物分别为:
污染物1:对氯苯酚。
污染物2:2,4-二氯苯酚。
污染物3:对乙酰氨基酚。
污染物4:诺氟沙星。
污染物5:磺胺甲恶唑。
在污染物降解试验的溶液体系中,加入的活性催化剂的浓度为0.2g/L,过硫酸氢钾或过硫酸氢钠的浓度为1mmol/L,有机污染物浓度为5mg/L。
在遮光条件下对溶液体系进行磁力搅拌。然后每隔一定时间反应后的溶液进行取样。并通过滤膜分离样品中的固体,通过高效液相色谱测定取样的溶液中剩余污染物的浓度,进而计算出各组活性催化剂对应的污染物降解的表观速率常数(min
表1:各组活性催化剂在污染物降解试验中的降解速率检测结果
分析表中数据可以发现,本实施例提供的制备方法生成的石墨型氮化碳材料(CN1~16)作为活性催化剂使用时,在遮光条件下相对常规的石墨型氮化碳产品(CN17~22)而言,污染物的催化降解速率提升了110-370倍。这证明,本实施例提供的材料催化活性显著增强,且具有明显的非光催化活性。
测试试验5
为了验证本案提供的无金属石墨型氮化碳材料降解有机污染物时,对多种过氧化物氧化剂均有催化能力,因此采用实施例中的CN1~5作为活性催化剂,对氯苯酚作为污染物,在50mL水溶液中进行污染物降解实验。
选用的过氧化物氧化剂为:
氧化剂1:过一硫酸氢钾。
氧化剂2:过硫酸钾。
氧化剂3:过硫酸钠。
氧化剂4:过氧乙酸。
表2:各组活性催化剂在活化不同过氧化物氧化剂降解对氯苯酚的降解效率
分析表2中数据可以发现,本实施例提供的制备方法生成的石墨型氮化碳材料(CN1~5)作为活性催化剂使用时,在遮光条件下均可以有效活化不同类型的过氧化物氧化剂,证明本例提供的制备方法生成的产品具有较广的应用范围。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。