近红外发光双自由基材料及其制备和应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及发光材料与生物成像领域,具体涉及小分子发光材料技术领域。
背景技术
自由基材料因其具有未配对或弱成键电子,展现出独特的光、电和磁学性质,使其引起了有机电子学、非线性光学、自旋电子学和能量存储器件等领域学者的广泛关注。由于受到自旋选律的限制,传统的荧光分子只能产生25%的单线态激子能够进行发光;而发光自由基分子的基态与激发态均为双线态,在发光中不存在跃迁禁阻的问题,理论上有利于激子辐射跃迁产生高效率的荧光。但大多数有机自由基由于其光激发态中存在强烈的非辐射弛豫途径,通常是不发光的。近年来,三(2,4,6-三氯苯基)甲基(TTM)型自由基为代表的发光有机自由基逐渐涌现,这类材料克服了第一代基于传统闭壳发光体的有机发光二极管(OLED)75%的能量损失,成为新一代分子发光体。这种在基态和激发态具有双态的单极体可以在发光二极管中实现100%的激子利用率,激发了吸引了研究人员探索下一代自由基基发光体的兴趣。
最近,基于自旋约束中心(A·)周围多氯芳基的位阻设计的三芳基甲基自由基的重大进展,相关的结构修饰和研究主要集中在多氯三苯基甲基自由基核的去对称,即C
发明内容
为解决现有技术问题,本发明的首要目的在于提供了一种近红外发光双自由基材料(本发明也简称为自由基分子材料),旨在提供一种兼顾优异稳定性以及发光特性和效果的新材料。
本发明第二目的在于,提供所述的近红外发光双自由基材料的制备方法,成功制备所述的全新化合物。
本发明第三目的在于,提供所述的近红外发光双自由基材料在生物成像中的应用。
自由基化合物通常稳定性较差、且不具备发光特点,针对该问题,本发明经过深入研究,提供以下创新方案:
一种近红外发光双自由基材料,具有式1结构:
式1中,所述的R
或者,R
所述的Ar为苯环、五元杂芳环、六元杂芳环、或者由苯环、五元杂芳环、六元杂芳环中的两个及以上的芳香环并合形成的稠芳香环;
所述的Ar的芳香环上带有取代基或者不带取代基;所述的取代基为烷氧基、硝基、卤素、苯基、环烷基、-CN、-OTf中的至少一种。
本发明提供了一种全新结构的材料,其具有式1的互变异构结构。研究表明,本发明所述的全新的自由基化合物,其基于分子结构、基团的协同,能够实现协同,能够显著改善其空气稳定性、光稳定性,此外,还能够赋予化合物优异的发光特性,特别是能够使其在少见的近红外区发光。因此,由于本发明化合物优异的稳定性以及独特的发光特性,其在光动力治疗和发光器件开发领域具有广泛的应用前景。
本发明研究发现,化合物的结构以及砜基的引入是协同提高自由基材料稳定性以及发光效率的关键。所述的分子结构能够协同削弱自旋电子的离域,提高结构刚性,一定程度上抑制光激发态的非辐射弛豫途径,提高发光效率;并且可以调节激发态电子结构和发射波长。
本发明中,所述的R
优选地,R
优选地,所述的环状结构(包含环合的苯环)可以为芴环、萘环或硫杂芴环;
优选地,R
进一步优选,所述的R
本发明中,所述的Ar为苯基或蒽基;或卤素、C
进一步地,
本发明中,所述的近红外发光双自由基材料,其为具有以下结构的化合物:
本发明还提供了所述的近红外发光双自由基材料的制备方法,通过式2的化合物和氧化剂进行氧化偶联制得;
式2中,所述的R
本发明中,作为可列举的实施方式,所述的氧化剂可以为三氯化铁、DDQ、四氯对苯醌中的至少一种;
优选地,氧化偶联中,式2和氧化剂的摩尔比为1:1.5~2.5;
优选地,氧化偶联的反应温度为15~40℃;
优选地,氧化偶联的溶剂为THF、Et
优选地,氧化偶联阶段,预先将式2和有机碱混合,再加入氧化剂进行氧化偶联反应,随后通过柱层析分离得到式1。
所述的有机碱可以为C
本发明中,所述的式2化合物通过式3氧化反应制备得到:
式3中,所述的R
氧化反应阶段选用的氧化剂a包括但不限于过氧化物、过硫酸盐中的至少一种,作为一种可列举的方案,其具体可以为间氯过氧苯甲酸;
优选地,氧化反应中,式3和氧化剂a的摩尔比为1:2~4;
作为优选,氧化反应的溶剂为THF、Et
作为优选,氧化反应的温度为15℃~40℃。
本发明中,所述的式3通过式4化合物、烷基锂和表达式为Ar-X的卤代芳化合物进行反应制备:
式4中,所述的R
优选地,所述的X为卤素,优选为Br;
优选地,所述的卤代芳化合物和式4的摩尔比为0.8~1:1;
优选地,所述的烷基锂为C
优选地,烷基锂和式4的摩尔比为1~1.2:1;
优选地,反应的溶剂为THF、Et
优选地,反应的温度为-10℃以下,优选为-40℃以下。
本发明提供了一种所述的近红外发光双自由基材料的应用,用于制备半导体电子器件、生物成像、自旋材料、近红外光激发的光动力治疗纳米药物等领域。
进一步优选,将所述的自由基分子材料用于有机发光二极管、生物荧光探针、光动力治疗等。
本发明还提供了一种近红外发光器件,包含所述的近红外发光双自由基材料。
本发明还提供了一种近红外光激发的光动力治疗纳米药物,其包含药学有效量的所述的近红外发光双自由基材料。
本发明所述的药物,可以基于已知的制剂手段以及辅料,将本发明所述的所述的近红外发光双自由基材料制备任意能够实现光治疗的给药制剂。
本发明技术方案具有以下有益效果:
本发明提供了一种全新结构的材料,其基于分子结构、基团的协同,能够实现协同,能够显著改善该自由基化合物的空气稳定性、光稳定性,此外,还具有优异的发光特性,特别是能够在少见的近红外区发光。
本发明所述的自由基发光材料具有深近红外发射和大斯托克斯位移特性,并且具有超氧自由基阴离子产生能力,在光动力治疗中存在巨大的应用潜力。
附图说明
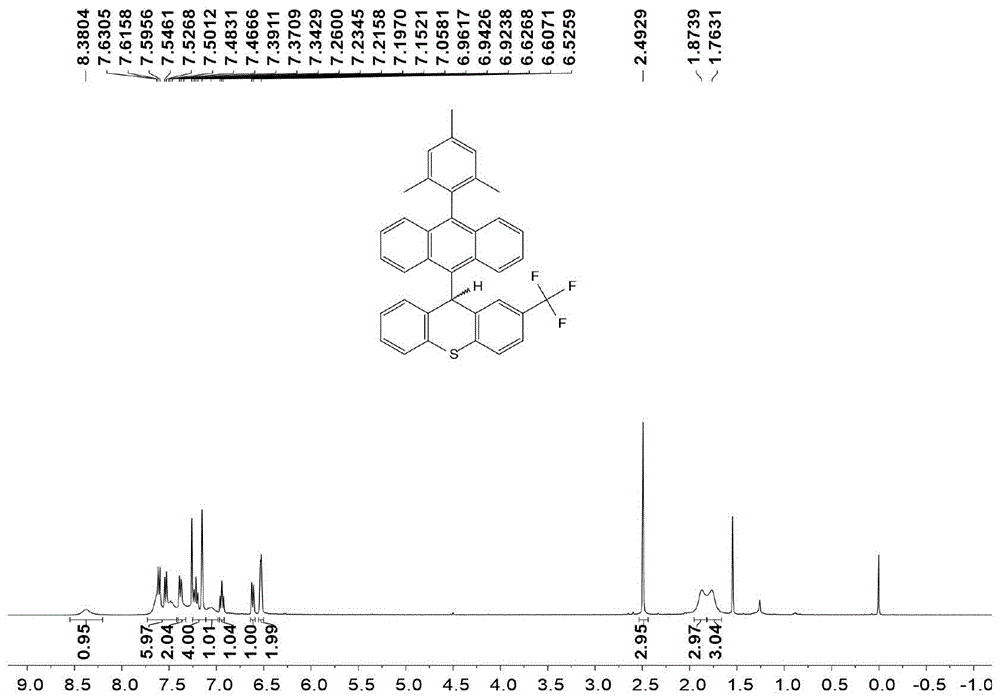
图1是实施例2制得的化合物5的核磁共振氢谱。
图2是实施例2制得的化合物5的核磁共振碳谱。
图3是实施例2制得的化合物5的质谱(Mw为560.7)。
图4是实施例2制得的化合物6的核磁共振氢谱。
图5是实施例2制得的化合物6的核磁共振碳谱。
图6是实施例2制得的化合物6的质谱(Mw为554.7)。
图7是实施例2制得的化合物SD-2的变温核磁共振氢谱。
图8是实施例2制得的化合物SD-2的质谱(Mw为1181.3)。
图9是实施例1~3制得的化合物SD-1~SD-3在甲苯中的UV-vis-NIR吸收谱图(A)和光致发光光谱(B)。
图10是实施例1~3制得的化合物SD-1~SD-3的单晶堆积结构。
图11为实施例1~3制得的SD-1~SD-3在空气环境、甲苯溶液态下的稳定性监测性质。
图12为实施例1~3制得的SD-1~SD-3在空气环境、甲苯溶液态且持续光照下的稳定性监测性质。
图13为实施例5制得的SD-2@DSPE-PEG在水溶液中的吸收和发射光谱。
图14为实施例5制得的SD-2@DSPE-PEG在小鼠体内的共聚焦荧光图(a)和短波红外图(b)。
具体实施方式
下面通过具体实验方式来进一步说明本发明的技术方案。
下述实施例中所述实验方法,如无特殊说明,均为常规方法,所述试剂和材料,如无特殊说明,均可从商业途径获得。
实施例1 SD-1的合成
在干燥的200mL史莱克瓶中加入化合物1(595mg,3.00mmol),在氩气气氛下加入60mL干燥的四氢呋喃。将反应瓶冷却至-40℃,然后缓慢滴加正丁基锂的正己烷溶液(1.2mL,3.00mmol)。混合物在-40℃下搅拌1小时。然后,将9-溴-10-三甲基蒽(935mg,2.50mmol)溶解在30mL四氢呋喃中后,加入反应瓶中。将反应液缓慢恢复至室温,在室温下搅拌,用TLC监测反应。反应完成后,向反应瓶中加入10mL水淬灭反应,用40mL二氯甲烷萃取5次。合并有机层,用50mL水洗涤3次后,用无水硫酸钠干燥,过滤,滤液用旋转蒸发仪除去溶剂,得到米白色粗产物。粗产物用硅胶柱层析进一步分离纯化,洗脱剂为石油醚:二氯甲烷=10:1,得到788mg白色固体化合物2,产率为64%。
1
13
HRMS(MALDI-TOF)m/z:Calcd for C
向100mL单口瓶中加入化合物2(591mg,1.2mmol),加入30mL二氯甲烷溶解,完全溶解后向瓶中加入3-氯过氧苯甲酸(619mg,3.6mmol),在室温下搅拌4小时。向反应瓶中加入2M Na
1
13
HRMS(MALDI-TOF)m/z:Calcd for C
向50mL干燥的史莱克瓶中加入化合物3(262mg,0.50mmol),加入20mL无水四氢呋喃溶解。在氩气气氛下,向反应瓶中加入叔丁醇钾(112mg,1.00mmol),在室温搅拌,用紫外-可见吸收光谱监测反应情况。3小时后,所有起始物质消失,形成棕黑色的阴离子溶液。然后,在氩气保护下,向反应瓶中加入对氯胺(185mg,0.75mmol),溶液变为深蓝色,用TLC监测反应。反应完成后,将反应液转移至50mL单口瓶中,用旋转蒸发仪除去溶剂,得到蓝色粗产物。粗产物用硅胶柱层析进一步分离纯化,洗脱剂为石油醚:四氢呋喃=5:1,得到371mg白色固体化合物SD-1,产率为71%。
HRMS(MALDI-TOF)m/z:Calcd for C
实施例2SD-2的合成
按照实施例1中的化合物2的方法合成化合物8,产率为69%。
1
13
HRMS(MALDI-TOF)m/z:Calcd for C
按照实施例1中的化合物3的方法合成化合物9,产率为84%。
1
13
HRMS(MALDI-TOF)m/z:Calcd for C
按照实施例1中的化合物SD-1的方法合成化合物SD-2,产率为62%。
HRMS(MALDI-TOF)m/z:Calcd for C
实施例3SD-3的合成
按照实施例1中的化合物2的方法合成化合物8,产率为63%。
1
13
HRMS(MALDI-TOF)m/z:Calcd for C
按照实施例1中的化合物3的方法合成化合物9,产率为87%。
1
13
HRMS(MALDI-TOF)m/z:Calcd for C
按照实施例1中的化合物SD-1的方法合成化合物SD-3,产率为35%。
HRMS(MALDI-TOF)m/z:Calcd for C
实施例4基于SD-1~SD-3的性质验证
本发明实例通过变温核磁共振氢谱、质谱、紫外-可见-近红外吸收光谱、单晶衍射等手段确认了SD-1~SD-3结构,并对其相关性质进行了研究。
图9为化合物SD-1~SD-3的甲苯溶液的紫外-可见-近红外吸收光谱(A)和激发光致发光光谱(B),相关的光物理性质结果如表1所示。电子吸收光谱结果表明,化合物SD-1和SD-1的最大吸收波长为625nm,而化合物SD-3的吸收谱带红移大约42nm,最大吸收波长为657nm。通过计算可得化合物SD-1~SD-3的光致发光量子产率(PLQY)分别为5.2%、2.4%和2.0%,表明这些砜功能化的二自由基类化合物的供体/受体取代基在其辐射衰变过程中发挥了重要作用。
表1 SD-1~3在甲苯中的光物理性质
图10为SD-1~SD-3的单晶结构图,化合物SD-1~SD-3都是由两个三芳基甲基段组成,由三个正交排列的部分组成,包括嵌入砜的π-段、蒽基段和均三甲苯基段。SD-1、SD-2和SD-3连接两个半分子段的C-C键的键长分别为1.418、1.424和
空气稳定性鉴定:
将SD-1~SD-3分子溶解在甲苯中,得到均相溶液(溶质的浓度均为10
光稳定性鉴定:
将SD-1~SD-3分子溶解在甲苯中,得到均相溶液(溶质的浓度均为0.1M),将该均相溶液在365nm波段的光下辐照;并通过紫外测试SD-1~SD-3分子分解情况。
溶液态下SD-1~SD-3分子在空气环境以及光照条件下的稳定性可由其紫外吸收监测图证明。如图11所示,在空气环境中三个双自由基分子的半衰期最高达到70天;如图12所示,在光照下其半衰期则都达到10
实施例5基于SD-2@DSPE-PEG的合成以及性质验证
本发明实例基于SD-2@DSPE-PEG的合成以及性质研究,具体合成方法如下:
(1)SD-2@DSPE-PEG纳米粒子(也简称为SD-2NPS)的制备:
基于SD-2@DSPE-PEG纳米粒子直接采用纳米共沉淀法合成,首先准备包含SD-2(100μg)和二硬脂酰基磷脂酰乙醇胺-聚乙二醇5000(DSPE-mPEG 5k)(2.5mg)的THF母液1mL,取9mLH
(2)基于SD-2@DSPE-PEG的性质研究
图13为实施例5制得的SD-2@DSPE-PEG在水溶液中的吸收(SD-2NPS-Abs)和发射(SD-2NPS-PL)光谱。根据图13可以发现SD-2@DSPE-PEG在901nm处有一个明显的发射峰。
图14为660nm光照射下SD-2@DSPE-PEG(10μg/mL)处理的HeLa细胞在DHE存在下的共聚焦荧光图像(a)和SD-2@DSPE-PEG(右)和空白(左)在660nm激发下小鼠皮下的短波红外图像及相应的荧光强度(b)。SD-2@DSPE-PEG在光照射下表现出优异的细胞内活性氧生成能力,HeLa细胞内的2',7'-二氯二氢荧光素二乙酸酯(DCFH-DA,ROS)指示剂22和二氢乙二酸(DHE,超氧化物)23进一步证实了SD-2@DSPE-PEG的存在,可应用于660nm激发下的短波长红外。
综上,本发明所述的式1结构的化合物,具有深近红外发射和大斯托克斯位移特性,并且具有超氧自由基阴离子产生能力,在光动力治疗中存在巨大的应用潜力。