一种阴离子交换膜及其制备方法和应用
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及燃料电池技术领域,具体涉及一种阴离子交换膜及其制备方法和应用。
背景技术
燃料电池作为一种清洁能源转换方式已在汽车领域取得了巨大的成就,但商业化的质子膜燃料电池(PEMFC)由于在使用过程中使用贵重金属铂等极大地限制了它的广泛应用。阴离子交换膜燃料电池(AEMFC)因在使用过程中使用非贵重金属Fe、Ni等作为催化剂大大降低了运行成本,被视为PEMFC的替代品。
阴离子交换膜(AEM)是AEMFC的核心组件,可以在电池使用期间作为隔绝燃料的屏障有效地减少燃料交叉,但是其传导OH
发明内容
本发明的目的在于提供一种阴离子交换膜及其制备方法和应用。本发明提供的阴离子交换膜电导率高、耐碱性好且结构稳定性好。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种阴离子交换膜的制备方法,包括以下步骤:
1)将1-甲基哌啶、二溴代烷烃与乙酸乙酯混合进行季铵化反应,得到季铵化的哌啶阳离子;
2)将2,3-二酮二氢吲哚、联苯和有机溶剂混合后,滴加催化剂进行傅克酰基化反应,得到聚吲哚芳烃;
3)将所述步骤1)得到的季铵化的哌啶阳离子、所述步骤2)得到的聚吲哚芳烃与二甲亚砜和带羟基的溴化物混合后加入碳酸钾,然后进行门舒特金反应,得到阴离子交换膜基质;
4)将所述步骤3)得到的阴离子交换膜基质浸渍于碱性溶液中进行离子交换,得到阴离子交换膜;
所述步骤1)和步骤2)不分时间先后顺序。
优选地,所述步骤1)中1-甲基哌啶与二溴代烷烃物质的量的比为1:(5~10)。
优选地,所述步骤1)中的二溴代烷烃包括1,4二溴丁烷、1,6二溴己烷、1,8二溴辛烷、1,10-二溴癸烷和1,12-二溴十二烷中的一种。
优选地,所述步骤2)中2,3-二酮二氢吲哚与联苯的物质的量的比为(1~1.3):1。
优选地,所述步骤2)中的联苯包括联苯、三联苯和四联苯中的一种或两种。
优选地,所述步骤3)中季铵化的哌啶阳离子与聚吲哚芳烃的物质的量的比为(0.6~0.9):1。
优选地,所述步骤3)中带羟基的溴化物与聚吲哚芳烃的物质的量的比为(0.1~0.4):1。
优选地,所述步骤3)中带羟基的溴化物包括溴代醇。
优选地,所述步骤3)中溴代醇包括2-溴乙醇、4-溴丁醇、6-溴己醇和8-溴辛醇中的一种。
本发明还提供了上述技术方案所述的制备方法值得的阴离子交换膜,包括聚吲哚芳烃和接枝于聚吲哚芳烃上的带季铵的哌啶阳离子和带羟基的溴代烷烃。
本发明还提供了上述技术方案所述的制备方法制得的阴离子交换膜在阴离子交换膜燃料电池中的应用。
本发明提供一种阴离子交换膜的制备方法,包括以下步骤:1)将1-甲基哌啶、二溴代烷烃与乙酸乙酯混合进行季铵化反应,得到季铵化的哌啶阳离子;2)将2,3-二酮二氢吲哚、联苯和有机溶剂混合后,滴加催化剂进行傅-克酰基化反应,得到聚吲哚芳烃;3)将所述步骤1)得到的季铵化的哌啶阳离子、所述步骤2)得到的聚吲哚芳烃与二甲亚砜和带羟基的溴化物混合后加入碳酸钾,然后进行门舒特金反应,得到阴离子交换膜基质;4)将所述步骤3)得到的阴离子交换膜基质浸渍于碱性溶液中进行离子交换,得到阴离子交换膜。本发明通过将1-甲基哌啶季铵化得到季铵化的哌啶阳离子,使其易与聚吲哚芳烃发生门舒特金反应;通过采用带羟基的溴化物与聚吲哚芳烃发生门舒特金反应,对聚吲哚芳烃进行接枝,从而减少季铵化哌啶阳离子与聚吲哚芳烃主链发生门舒特金反应,间接减少膜的吸水率,提高结构稳定性;带羟基的溴化物中羟基自身的亲水特性可在一定程度上减少因失去部分哌啶阳离子而损失的离子电导率,羟基的存在促使以哌啶阳离子为头基的侧链发生自聚集进而形成高效的离子传输通道,提高电导率;且带羟基的溴化物与哌啶阳离子基团的静电吸引形成较大的离子团簇,提高电导率、耐碱性和结构稳定性。实施例结果显示,本发明提供的制备方法制得的阴离子交换膜离子电导率可达11.37mS·cm
附图说明
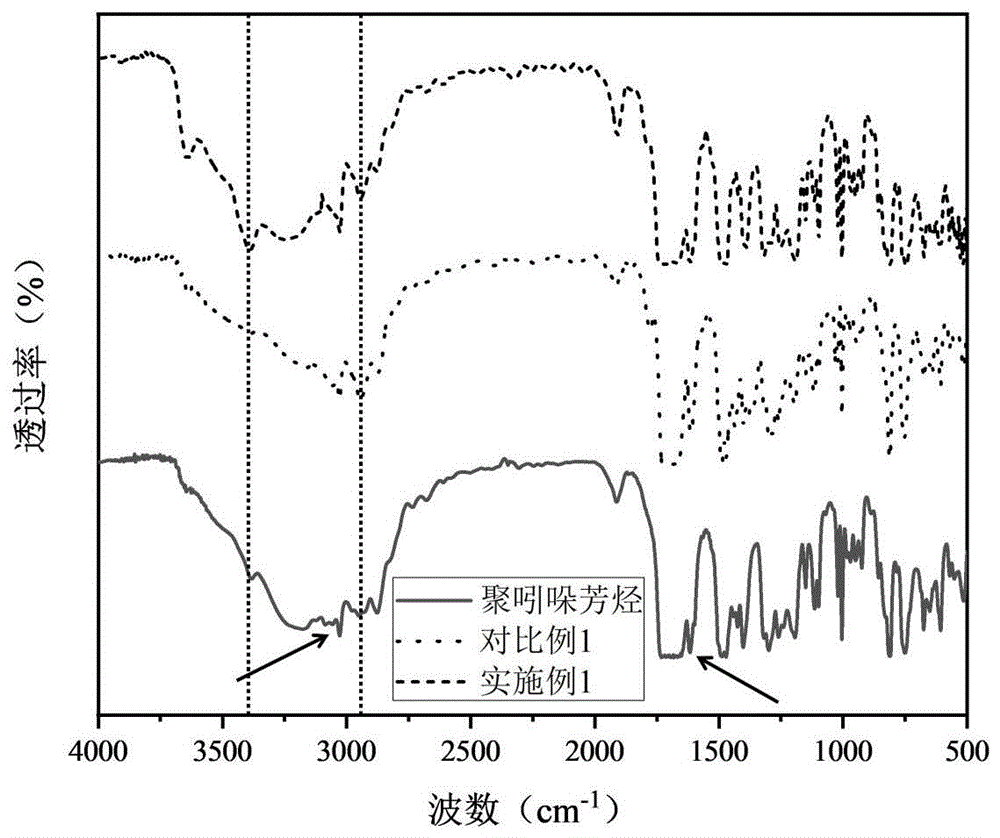
图1为实施例1和对比例1制得的阴离子交换膜的FT-IR图;
图2为实施例1制得的阴离子交换膜的TEM图;
图3为实施例2制得的阴离子交换膜的TEM图;
图4为对比例1制得的阴离子交换膜的TEM图;
图5为实施例2制得的阴离子交换膜的SEM图;
图6为实施例2制得的阴离子交换膜的断面SEM图。
具体实施方式
本发明提供了一种阴离子交换膜的制备方法,包括以下步骤:
1)将1-甲基哌啶、二溴代烷烃与乙酸乙酯混合进行季铵化反应,得到季铵化的哌啶阳离子;
2)将2,3-二酮二氢吲哚、联苯和有机溶剂混合后,滴加催化剂进行傅克酰基化反应,得到聚吲哚芳烃;
3)将所述步骤1)得到的季铵化的哌啶阳离子、所述步骤2)得到的聚吲哚芳烃与二甲亚砜和带羟基的溴化物混合后加入碳酸钾,然后进行门舒特金反应,得到阴离子交换膜基质;
4)将所述步骤3)得到的阴离子交换膜基质浸渍于碱性溶液中进行离子交换,得到阴离子交换膜。
本发明将1-甲基哌啶、二溴代烷烃与乙酸乙酯混合进行季铵化反应,得到季铵化的哌啶阳离子。在本发明中,所述季铵化反应的化学式为:
在本发明中,所述1-甲基哌啶与二溴代烷烃物质的量的比优选为1:(5~10),更优选为1:7。本发明通过限定1-甲基哌啶与二溴代烷烃物质的量的比以保证1-甲基哌啶的季铵化更为充分。在本发明中,所述二溴代烷烃优选包括1,4二溴丁烷、1,6二溴己烷、1,8二溴辛烷、1,10-二溴癸烷和1,12-二溴十二烷中的一种。
在本发明中,所述1-甲基哌啶与乙酸乙酯的体积比优选为1:(24~26)。本发明通过限定乙酸乙酯的用量以保证1-甲基哌啶和溴代烷烃充分混合。
本发明对所述1-甲基哌啶、二溴代烷烃与乙酸乙酯混合的方式没有特殊的限定,采用本领域熟知的混合方式即可。
在本发明中,所述季铵化反应的温度优选为常温;所述季铵化反应的时间优选为24~48h。本发明通过限定季铵化反应的温度和时间以保证1-甲基哌啶的季铵化更为充分。在本发明中,所述季铵化反应中1-甲基哌啶上的氮把二溴代烷烃中的溴取代得到季铵化的哌啶阳离子,易与聚吲哚芳烃主链发生门舒特金反应,有利于提高制得的阴离子交换膜的离子电导率和碱稳定性。
季铵化反应完成后,本发明优选将所述季铵化的产物依次进行抽滤和乙醚洗涤,得到季铵化的哌啶阳离子。本发明对所述抽滤的仪器没有特殊的限定,采用本领域熟知的抽滤仪器将产物和溶剂分离即可。在本发明中,所述乙醚洗涤的次数优选为4~5次。本发明通过限定乙醚洗涤的次数以去除季铵化的哌啶阳离子上的杂质。
本发明将2,3-二酮二氢吲哚、联苯和有机溶剂混合后,滴加催化剂进行傅克酰基化反应,得到聚吲哚芳烃。在本发明中,所述傅克酰基化反应的化学式为:
在本发明中,所述2,3-二酮二氢吲哚与联苯的物质的量的比优选为(1~1.3):1,更优选为1.2:1。本发明通过限定2,3-二酮二氢吲哚与联苯的物质的量以保证傅克酰基化反应更充分。
在本发明中,所述联苯优选包括联苯、三联苯和四联苯中的一种或两种。
在本发明中,所述有机溶剂优选包括二氯甲烷和/或三氟乙酸,更优选为二氯甲烷和三氟乙酸。
在本发明中,当所述有机溶剂包括二氯甲烷时,所述2,3-二酮二氢吲哚与二氯甲烷的质量比优选为1:(11.7~17.58)。本发明通过限定2,3-二酮二氢吲哚与二氯甲烷的质量比以保证2,3-二酮二氢吲哚与联苯更加充分溶解。
在本发明中,当所述有机溶剂包括三氟乙酸时,所述2,3-二酮二氢吲哚与三氟乙酸的质量比优选为1:(7.6~20.35)。在本发明中,所述三氟乙酸一方面作为溶剂起助溶解的作用,使制得的聚吲哚芳烃呈粉末状,有利于下一步门舒特金反应时更好的溶解,参与反应;另一方面可以促进傅-克酰基化反应的进行起助催化的作用。
本发明对所述2,3-二酮二氢吲哚、联苯和有机溶剂的混合方式没有特殊的限定,采用本领域熟知的混合方式即可。
在本发明中,所述2,3-二酮二氢吲哚、联苯和有机溶剂的混合优选为将2,3-二酮二氢吲哚、联苯和二氯甲烷混合后,在冰浴下滴加三氟乙酸,并进行搅拌。在本发明中,所述搅拌的时间优选为0.5~1h。
本发明对所述催化剂的种类没有特殊限定,采用本领域熟知的催化剂可催化傅克酰基化反应即可。在本发明中,所述催化剂优选为三氟甲烷磺酸。在本发明中,所述2,3-二酮二氢吲哚与三氟甲烷磺酸的质量比优选为1:(9.45~18.9)。在本发明中,所述三氟甲烷磺酸优选在冰浴下进行滴加。在本发明中,所述滴加速率优选为15~20min。本发明通过限定滴加速率使2,3-二酮二氢吲哚与联苯反应更充分。本发明通过添加三氟甲烷磺酸,利用其具有超强酸性,作为催化剂,促进傅克酰基化酰基化~反应的进行。本发明对所述滴加方式没有特殊限定,采用本领域熟知的滴加方式即可。
在本发明中,所述傅克酰基化反应优选在搅拌条件下进行。本发明对所述搅拌的方式和速率没有特殊的限定,采用本领域技术人员熟知的搅拌方式和速率即可。在本发明中,所述搅拌方式优选为磁力搅拌。在本发明中,所述傅-克酰基化反应中,联苯上的氢键被2,3-二酮二氢吲哚中的二酮取代,发生缩聚,得到聚吲哚芳烃。在本发明中,所述傅-克酰基化反应的温度和时间优选为先在冰浴下反应0.5~1h,然后在20~35℃下反应2.5~4h。本发明在冰浴下进行反应,防止一开始反应过热发生副反应,然后进行常温反应提高反应速率。
傅-克酰基化反应完成后,本发明优选对所述傅-克酰基化反应的产物依次进行抽滤、洗涤再抽滤和干燥,得到聚吲哚芳烃。
本发明对所述抽滤、洗涤再抽滤和干燥的方式没有特殊限定,采用本领域熟知的技术方案对反应产物进行分离提纯即可。在本发明中,所述洗涤再抽滤优选为一个组合。在本发明中,所述洗涤再抽滤优选为:将所述抽滤得到的产物与预热后的甲醇混合后再抽滤。在本发明中,所述甲醇与2,3-二酮二氢吲哚的质量比优选为(35.39~70.79):1。在本发明中,所述预热的温度优选为50℃~55℃;所述预热方式优选为水浴预热。在本发明中,所述混合优选在搅拌下进行;所述搅拌的方式优选为机械搅拌。在本发明中,所述混合的时间优选为20~30min。本发明优选重复进行洗涤再抽滤至再抽滤得到的滤液澄清时停止。
在本发明中,所述干燥的温度优选为50~70℃;所述干燥的时间优选为24~48h;所述干燥的设备优选为真空干燥箱。
得到季铵化的哌啶阳离子和聚吲哚芳烃后,本发明将所述季铵化的哌啶阳离子、聚吲哚芳烃与二甲亚砜和带羟基的溴化物混合后加入碳酸钾,然后进行门舒特金反应,得到阴离子交换膜基质。在本发明中,所述门舒特金反应的化学式为:
在本发明中,所述季铵化的哌啶阳离子与聚吲哚芳烃的物质的量的比优选为(0.6~0.9):1,更优选为0.7:1。本发明通过限定季铵化的哌啶阳离子与聚吲哚芳烃的物质的量的比以保证季铵化的哌啶阳离子与聚吲哚芳烃充分反应,提高阴离子交换膜的离子电导率和碱稳定性。
在本发明中,所述带羟基的溴化物与聚吲哚芳烃的物质的量的比优选为(0.1~0.4):1,更优选为0.3:1。本发明通过限定带羟基的溴化物与聚吲哚芳烃的物质的量的比以保证带羟基的溴化物与聚吲哚芳烃充分反应,以减少后续季铵化哌啶阳离子与聚吲哚芳烃主链的接枝位点,间接减少阴离子交换膜的吸水率。
在本发明中,所述带羟基的溴化物优选包括溴代醇,更优选包括2-溴乙醇、4-溴丁醇、6-溴己醇和8-溴辛醇中的一种,最优选为6-溴正己醇。本发明通过限定带羟基的溴化物与聚吲哚芳烃主链发生门舒特金反应,对聚吲哚芳烃进行接枝,从而减少季铵化哌啶阳离子与聚吲哚芳烃主链发生门舒特金反应,间接减少膜的吸水率,提高结构稳定性;减少因失去部分哌啶阳离子而损失的离子电导率,羟基的存在促使以哌啶阳离子为头基的侧链发生自聚集进而形成高效的离子传输通道,提高电导率;与哌啶阳离子基团的静电吸引形成较大的离子团簇,提高电导率、耐碱性和结构稳定性。
在本发明中,所述二甲亚砜与聚吲哚芳烃的质量比优选为(48.69~97.38):1,更优选为60:1。本发明通过限定二甲亚砜的添加量以保证季铵化的哌啶阳离子和聚吲哚芳烃更加充分的溶解。
在本发明中,所述混合优选为先将季铵化的哌啶阳离子、聚吲哚芳烃与二甲亚砜混合溶解,然后与带羟基的溴化物混合。在本发明中,所述混合和门舒特金反应均在搅拌下进行。
在本发明中,所述碳酸钾与季铵化的哌啶阳离子的物质的量的比优选为(1~1.2):1。本发明通过限定碳酸钾的用量以保证可以更好的拔去聚吲哚芳烃上的氢,使被带羟基的溴化物可以取代拔去的氢,得到阴离子交换膜基质。
在本发明中,所述碳酸钾添加后优选在混合液的上方覆一层保鲜膜以防止溶剂挥发过多。
在本发明中,所述门舒特金反应的温度优选为70~90℃;所述门舒特金反应的时间优选为48~72h。。在本发明中,所述门舒特反应中碳酸钾拔去聚吲哚芳烃上的氢,然后被带羟基的溴化物取代;季铵化的哌啶阳离子取代剩余的氢,得到阴离子交换膜基质。本发明通过限定门舒特金反应的温度和时间以保证反应更加充分。
门舒特金反应完成后,本发明优选将所述门舒特金反应的产物依次进行乙酸乙酯洗涤抽滤、蒸馏水洗涤抽滤、干燥、溶解和再干燥,得到阴离子交换膜基质。
本发明对所述乙酸乙酯洗涤抽滤、蒸馏水洗涤抽滤和干燥的方式没有特殊限定,采用本领域熟知的技术方案对反应产物进行分离提纯即可。
在本发明中,所述乙酸乙酯洗涤抽滤优选为一个组合。在本发明中,所述乙酸乙酯洗涤抽滤优选为将所述门舒特金反应的产物与乙酸乙酯混合,然后抽滤。在本发明中,所述乙酸乙酯与季铵化的哌啶阳离子的质量比优选为(135.2~180):1。在本发明中,所述混合优选在搅拌下进行;所述搅拌的方式优选为机械搅拌。在本发明中,所述混合的时间优选为20~30min。在本发明中,所述乙酸乙酯洗涤抽滤的次数优选为2~3次。
在本发明中,所述蒸馏水洗涤抽滤优选为将乙酸乙酯洗涤抽滤后的产物与蒸馏水混合,然后抽滤。在本发明中,所述蒸馏水与季铵化的哌啶阳离子的质量比优选为(253~300):1。在本发明中,所述混合优选在搅拌下进行;搅拌的方式优选为机械搅拌。在本发明中,所述混合的时间优选为15~20min。本发明优选通过在抽滤过程中补加蒸馏水重复进行蒸馏水洗涤抽滤。在本发明中,所述蒸馏水与季铵化的哌啶阳离子的质量比优选为(93~100):1。在本发明中,所述蒸馏水的补加次数优选为2~3次。
在本发明中,所述干燥的温度优选为50~70℃;所述干燥的时间优选为24~48h。本发明对所述干燥的设备没有特殊的限定,采用本发明熟知的干燥设备即可。在本发明中,所述干燥的设备优选为真空干燥箱。
在本发明中,所述溶解优选为将所述干燥后的产物溶解在N-甲基吡咯烷酮中。在本发明中,所述溶解的作用是得到质量分数为4~10wt%的均质溶液。在本发明中,所述溶解优选在搅拌下进行。在本发明中,所述搅拌的方式优选为机械搅拌。在本发明中,所述搅拌的时间优选为48h。在本发明中,所述均质溶液中干燥后的产物的质量分数优选为8wt%。
在本发明中,所述均质溶液在再干燥之前优选倒入玻璃铸膜板中。在本发明中,所述玻璃铸膜板的规格优选为10×10cm
得到阴离子交换膜基质后,本发明将所述阴离子交换膜基质浸渍于碱性溶液中进行离子交换,得到阴离子交换膜。
在本发明中,所述碱性溶液优选为氢氧化物溶液,更优选为KOH或NaOH的水溶液。在本发明中,所述碱性溶液的浓度优选为0.5~1mol/L。
在本发明中,所述离子交换的时间优选为24~48h。本发明通过限定离子交换的时间以保证离子交换进行完全。在本发明中,所述离子交换中阴离子膜中的溴转换为-OH。
离子交换完成后,本发明优选将所述离子交换的产物依次进行洗涤和浸渍,得到阴离子交换膜。
在本发明中,所述洗涤优选采用蒸馏水。本发明对所述蒸馏水的用量没有特殊限定,采用本领域熟知的能够将离子交换的产物表面碱洗去即可。在本发明中,所述浸渍优选为将洗涤后的离子交换基质浸渍于蒸馏水。在本发明中,所述浸渍的时间优选为24~48h。在本发明中,所述浸渍的条件优选为密封条件。
本发明通过将1-甲基哌啶季铵化得到季铵化的哌啶阳离子,使其易与聚吲哚芳烃发生门舒特金反应;采用带羟基的溴化物与聚吲哚芳烃主链进行接枝,从而减少后续季铵化哌啶阳离子与聚吲哚芳烃主链的接枝位点,间接减少膜的吸水率,提高结构稳定性;带羟基的溴化物侧链的引入加深了聚合物之间的化学不相容性,它们彼此之间的相互吸引增强了碱性阴离子交换膜的强度和韧性,进一步提高结构稳定性;带羟基的溴化物中羟基自身的亲水特性可在一定程度上减少因失去部分哌啶阳离子而损失的离子电导率,羟基的存在促使以哌啶阳离子为头基的侧链发生自聚集进而形成高效的离子传输通道,提高电导率高;且带羟基的溴化物与哌啶阳离子基团的静电吸引形成较大的离子团簇,提高电导率、耐碱性和结构稳定性。
本发明还提供了一种由上述技术方案所述的制备方法制得的阴离子交换膜,所述阴离子交换膜的主链是聚吲哚芳烃、接枝侧链是季铵化的哌啶阳离子与6-溴正己醇、季铵化哌啶阳离子的接枝率(60~100%)、带羟基的溴化物的接枝率(0~40%)。
本发明提供的阴离子交换膜采用带羟基的溴化物与聚吲哚芳烃主链进行接枝,从而减少后续季铵化哌啶阳离子与聚吲哚芳烃主链的接枝位点,间接减少膜的吸水率,提高结构稳定性;加深聚合物之间的化学不相容性,它们彼此之间的相互吸引增强了碱性阴离子交换膜的强度和韧性,进一步提高结构稳定性;利用自身的亲水特性在一定程度上减少因失去部分哌啶阳离子而损失的离子电导率,羟基的存在促使以哌啶阳离子为头基的侧链发生自聚集进而形成高效的离子传输通道,提高电导率高;且带羟基的溴化物与哌啶阳离子基团的静电吸引形成较大的离子团簇,提高电导率、耐碱性和结构稳定性。
本发明提供的制备方法制得的阴离子交换膜离子电导率可达16.24mS·cm
本申请还提供了一种由上述技术方案所述阴离子交换膜的应用。
以上所述仅是本发明的优选实施方式,并非对本发明作任何形式上的限制。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
实施例1
一种阴离子交换膜的制备方法,由以下步骤组成:
1)将0.02mol(2.5mL)1-甲基哌啶、0.1mol 1,6二溴己烷与60mL乙酸乙酯混合,在常温下进行季铵化反应,所述季铵化反应的时间为24h,并将季铵化反应的产物抽滤,用乙醚洗涤5次,得到季铵化的哌啶阳离子;所述1-甲基哌啶与溴代烷烃物质的量的比为1:5;所述1-甲基哌啶与乙酸乙酯的体积比为1:24;
2)将0.0077mol(1.13g)2,3-二酮二氢吲哚、0.007mol对三联苯烃和13.25g(10mL)二氯甲烷混合后,在冰浴下滴加8.6g(5.6mL)三氟乙酸磁力搅拌0.5h,继续滴加10.68g(6.3mL)三氟甲烷磺酸进行傅-克酰基化反应,所述反应的温度和时间为先在冰浴下磁力搅拌反应0.5h,然后在25℃下磁力搅拌反应3.5h,反应完成后,将反应后的产物抽滤,然后与35.39g(50mL)50℃预热的甲醇混合洗涤,磁力搅拌20min后抽滤,再洗涤,再抽滤,重复5次至用热甲醇洗涤后滤液澄清时停止洗涤,放入真空干燥箱70℃干燥24h,得到聚吲哚芳烃;所述2,3-二酮二氢吲哚与对三联苯的物质的量的比为1.1:1;所述2,3-二酮二氢吲哚与二氯甲烷的质量比为1:11.7;所述2,3-二酮二氢吲哚与三氟乙酸的质量比为1:7.6;所述2,3-二酮二氢吲哚与三氟甲烷磺酸的质量比为1:9.45;所述甲醇与2,3-二酮二氢吲哚的质量比为35.39:1;
3)将所述步骤1)得到的2.25mmol(0.592g)季铵化的哌啶阳离子、所述步骤2)得到的2.5mmol(0.9036g)聚吲哚芳烃与48.69g(40mL)二甲亚砜混合搅拌溶解,然后与0.25mmol(0.063mL)6-溴正己醇混合搅拌后加入2.5mmol(0.346g)碳酸钾,在混合液上方覆一层保鲜膜,然后搅拌进行门舒特金反应,所述门舒特金反应的温度为80℃,反应的时间为48h,门舒特金反应完成后,将反应后的混合物与150mL乙酸乙酯混合机械搅拌20min,进行洗涤,然后抽滤,再洗涤,再抽滤,重复3次之后,将抽滤后的产物与150mL水混合搅拌15~20min,进行洗涤,然后抽滤,在抽滤过程中再加50mL蒸馏水再抽滤,重复加蒸馏水抽滤3次后,真空烘箱70℃干燥24h,然后将0.6g干燥后的产物溶解在60mLN-甲基吡咯烷酮中,磁力搅拌48h,得到质量分数为10wt%的均质溶液,倒入10×10cm
4)将所述步骤3)得到的阴离子交换膜基质浸渍于0.5mol/LNaOH溶液中,进行离子交换,所述离子交换的时间优选为24h,离子交换完成后,用~蒸馏水洗涤,然后在密封条件下,浸渍于蒸馏水24h,得到阴离子交换膜。
实施例2
实施例2与实施例1的区别在于,步骤3)中季铵化的哌啶阳离子为2.22mmol(0.526g),六溴正己醇为0.5mmol(0.126mL),所述季铵化的哌啶阳离子与聚吲哚芳烃的物质的量的比优选为0.8:1;所述带羟基的溴化物与聚吲哚芳烃的物质的量的比优选为0.2:1;其他与实施例1相同。
实施例3
实施例3与实施例1的区别在于,步骤3)中季铵化的哌啶阳离子为1.94mmol(0.461g),六溴正己醇为0.75mmol(0.189mL),所述季铵化的哌啶阳离子与聚吲哚芳烃的物质的量的比优选为0.7:1;所述带羟基的溴化物与聚吲哚芳烃的物质的量的比优选为0.3:1;其他与实施例1相同。
实施例4
实施例4与实施例1的区别在于,步骤3)中季铵化的哌啶阳离子为1.67mmol(0.395g),六溴正己醇为1mmol(0.252mL),所述季铵化的哌啶阳离子与聚吲哚芳烃的物质的量的比优选为0.6:1;所述带羟基的溴化物与聚吲哚芳烃的物质的量的比优选为0.4:1;其他与实施例1相同。
对比例1
对比例1与实施例1的区别在于,步骤3)中未添加六溴正己醇,其他与实施例1相同。
本发明通过电子万能拉伸试验机,根据国标GB/T 1040对实施例1~4和对比例1制得的阴离子交换膜的机械性能进行了测试,如表1所示。
表1实施例1~4和对比例1制得的阴离子交换膜的机械性能
由表1可以看出:
对比例1制得的阴离子交换膜未添加6-溴正己醇,其拉伸强度与断裂伸长率分别为3.98MPa,1.86%;本发明实施例4制得的阴离子交换膜拉伸强度与断裂伸长率分别为17.57MPa,6.94%;由此可见,本发明添加6-溴正己醇制得的阴离子交换膜结构稳定性好;且随着6-溴正己醇添加量的增加,拉伸强度与断裂伸长率均有所提升。
本发明采用FT-IR对实施例1和对比例1制得的阴离子交换膜的结构进行了分析,如图1所示。由图可知:在3292cm-1处的宽峰是2,3-二酮二氢吲哚在聚吲哚芳烃中的N-H伸缩振动造成的;而在聚吲哚芳烃苯的C-H伸缩出现在约2988cm
本发明采用透射电子显微镜对实施例1~2和对比例1制得的阴离子交换膜进行了分析,如图2~4所示。由图可知:实施例1~2和对比例1制得的阴离子交换膜均有着良好的微相分离结构,这得益于侧链型的阴离子交换膜,并且,随着6-溴正己醇的含量增加,图中红色圈圈起来的暗区变得更加明显,表明离子团簇变大,这有利于OH
本发明通过采用采用交流阻抗法对实施例1~4和对比例1制得的阴离子交换膜的电导率进行测试,以及对实施例1~4和对比例1制得的阴离子交换膜在60℃1M KOH浸泡15d后的电导率进行测试,结果如表2所示。
表2实施例1~4和对比例1制得的阴离子交换膜的电导率测试
由表2可知:
对比例1制得的阴离子交换膜离子电导率为8.23mS·cm
本发明通过采用ZEISS扫描电子显微镜对实施例2制得的阴离子交换膜表面及断面形貌进行了分析,如图5~6所示。由图可知:阴离子交换膜表面平整,说明哌啶阳离子基团和带羟基的溴代烷烃与聚吲哚芳烃的骨架接枝程度较好,相容性较高;而且膜的断面非常紧密,这赋予了膜较高的机械强度,适用于碱性阴离子交换膜燃料电池。
以上所述仅是本发明的优选实施方式,并非对本发明作任何形式上的限制。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。