一种宏基因组序列组装方法
文献发布时间:2023-06-19 13:45:04
技术领域
本发明涉及宏基因组学技术领域,尤其涉及一种宏基因组序列组装方法。
背景技术
宏基因组学(Metagenomics)也称为微生物环境基因组学,是由Handelsman等于1998年提出的,利用序列分析和功能基因筛选的研究方法以微生物的多样性、群落结构、进化关系、功能活性以及物种与环境之间的关系作为研究目的的一门新兴学科。宏基因组学避开了传统的微生物分离培养方法直接从环境样品中提取总DNA,借助于大规模测序技术和生物信息学工具发现大量过去无法获得的未知微生物、新基因或新的基因簇,让人们能够对大自然里99%的微生物(这些都还是没能在实验室里成功培养的微生物)进行分析和研究,还可以对整个大环境里的微生物进行研究。
目前,由于新一代高通量低成本测序技术的广泛应用,科学家们可以对环境中的全基因组进行测序,特别是下一代测序技术(NGS)的发展,产生庞大的宏基因组序列数据。这些海量数据的存储以及分析给研究人员带来前所未有的挑战,为了分析这些宏基因组序列,通常将构成宏基因组序列的核苷酸序列(例如DNA或RNA序列)组装成重叠群的较大序列,但传统宏基因组序列组装方法基本上都是基于单台计算机运行,需要在单台机器上对所有测序数据同时进行处理,而来自下一代测序平台的测序数据数量通常非常大,一个数据集中往往包含多个微生物物种,这样的测序数据不仅数据量大,而且测序数据也非常复杂。受限于单台计算机的内存和计算资源,传统组装方法不仅效率低下,而且组装完整度较低,并且随着数据量的增大可能无法完成运算。
发明内容
有鉴于现有技术的上述缺陷,本发明提供了一种宏基因组序列组装方法,在进行组装之前先对宏基因组序列进行聚类,然后再对聚类之后得到的簇按类进行组装,有效提高了组装的精确度和完整度。
本发明公开的宏基因组序列组装方法,包括:
获取测序后待组装的微生物宏基因组序列片段;
对所述序列片段进行聚类,将来自同一基因组的所述序列片段归为一类,得到分类簇;
对所述每个分类簇进行组装。
在本发明的较佳实施方式中,所述聚类步骤为:
对所述序列片段进行预处理,利用所述序列片段两两之间重叠关系,构建一个无向网络图;
基于分布式聚类算法对所述无向网络图进行迭代聚类操作;
得到聚类后的分类簇。
在本发明的另一较佳实施方式中,所述分布式聚类算法为基于分布式平台的Louvain算法,所述Louvain算法并行化聚类过程为:
对所述无向网络图中的每一个节点计算得到使之模块度增益最大的邻居节点,将所述邻居节点分配给所述邻居节点所属社区,迭代直至更改所在社区的节点数少于某一值,结束本次迭代得到某个层次的新图;
计算每次迭代前后的模块度变化值,模块度变化值小于一定值时,表示社区划分已经达到最优,停止迭代;
或者,当迭代次数达到限定值后中止迭代;
对聚类后结果处理得到序列信息及其对应社区信息。
在本发明的另一较佳实施方式中,所述分布式平台为Apache Spark平台。
在本发明的较佳实施方式中,所述组装步骤为:
使用MEGAHIT组装工具对所述每个分类簇中的序列片段进行组装;
对组装之后的结果进行对比和格式转换;
使用MetaBAT分箱工具对所述格式转换后的结果进行分箱。
由于宏基因组序列数据的数据量极其庞大、物种类别非常广泛,直接对宏基因组序列数据进行组装具有很大的难度。本发明公开的宏基因组序列组装方法首先对序列片段聚类,然后再进行组装,是处理宏基因组数据的一种新的方式,可有效地降低宏基因组数据的总体组装的复杂度。此外,基于分布式平台的并行化处理方法可以有效缓解单机内存和计算资源的不足,在海量复杂宏基因组序列数据下仍然可以实现快速和准确的基因组装。
以下将结合附图对本发明的构思、具体结构及产生的技术效果作进一步说明,以充分地了解本发明的目的、特征和效果。
附图说明
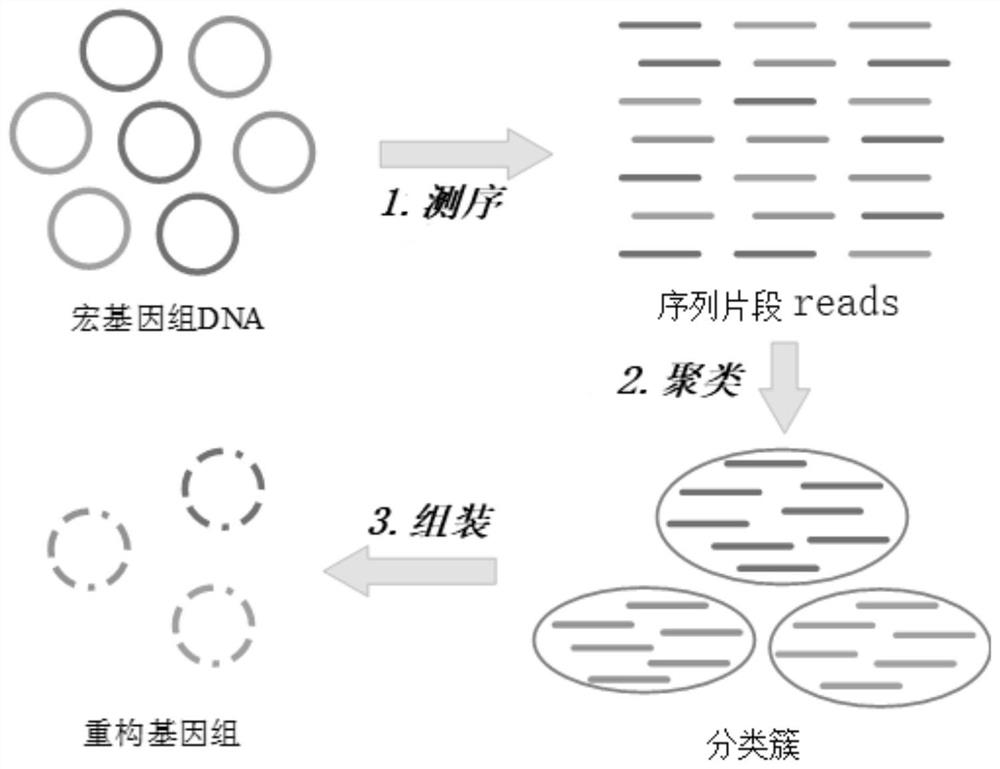
图1是宏基因组序列经典处理流程示意图;
图2是本发明所述的宏基因组序列片段组装方法流程示意图;
图3是本发明一较佳实施例中聚类算法流程示意图;
图4是本发明一较佳实施例中并行化Louvain算法流程示意图;
图5是本发明一较佳实施例中组装流程示意图;
图6(a)是本发明所述组装方法的覆盖度和完整度之间关系图;
图6(b)是传统组装方法的覆盖度和完整度之间关系图。
具体实施方式
下面对本发明的较佳实施例进行详细阐述,以使本发明的优点和特征能更易于被本领域技术人员理解,从而对本发明的保护范围做出更为清楚明确的界定。
序列组装(Assembly)和分箱(Binning)是宏基因组分析中的常见方法,同时也是关键步骤。图1为宏基因组序列分析的经典流程,在图1所示的流程中,序列组装作为重建宏基因组的第一步,往往会面临如计算量大、复杂度高等诸多问题。
序列组装指利用测序得到的序列片段(reads)之间的重叠关系,对reads进行拼接,得到较长连续序列重叠群contigs的过程。随着测序精度及其序列长度的改变,宏基因组序列组装方法也在随之不断发展。但是,当下用于宏基因组序列组装的方法不足以生成高质量的contigs和处理大规模数据,特别是对于万亿级宏基因组学项目。
通常,由于下一代测序(Next Generation Sequencing,NGS)的高通量特性以及组装软件对计算机磁盘和内存的高性能要求,现有的组装工具往往缺乏在所得contigs噪声与序列长度增益之间取得良好折衷的能力,无法在大规模宏基因组序列项目中进行更好的推广使用。
本发明在组装之前先对序列片段reads进行聚类,具体流程如图2所示,首先根据reads之间重叠关系进行聚类,然后将聚类得到的分类簇进行组装,重构基因组。
对于测序后的序列片段,本发明实施例采用分布式聚类方法,具体聚类流程如图3所示:
①将原始的FASTQ文件转化为序列文件,同时为每条read加上编号,以降低后续处理对内存资源的占用;
②对得到的reads片段进行处理,根据预定义k-mer长度将所有reads切割成固定长度的k-mer,并统计k-mer在reasd中的分布信息;
③根据得到的k-mer在reads中的分布信息,以reads为顶点,reads之间共享k-mer数为边来生成无向网络图;
④通过图聚类算法对生成的无向网络图进行划分,将来自同一基因组的reads归为一分类簇。
当reads长度比较短时,还可以对得到的分类簇进行再聚类。
Louvain算法是一种基于模块度的社区发现算法。模块度常用于描述网络中的各个社区内部节点之间联系的紧密程度,可定义为各社区内部的总边数和网络中总边数的比例减去一个固定值,该固定值是网络为随机网络时各社区内部的总边数和网络中总边数的比例。如果一个网络模块度值越高,则网络中各个社区内部的节点交互较为紧密,社区之间的节点交互较为稀疏。因此,将模块度作为一个优化函数,求得其全局最优解,即可得到一个网络的最优社区划分。
本发明实施例中对无向网络图进行图聚类,可以通过并行化Louvain算法对所述无向网络图进行迭代运算,得到模块度最优的层次化聚类结果。将社区结构与已知信息进行比对,对聚类过程中重要参数进行调整。
Apache Spark分布式框架基于弹性分布式数据集,数据被缓存在工作节点的内存中,适合进行机器学习及智能优化算法中的迭代运算,因此,在并行Louvain算法的框架选择上,优选用Apache Spark框架提升其性能。为了在分布式集群上实现Louvain算法,需要对串行Louvain算法进行改进。分布式集群中,所有顶点不再按顺序进行节点更新,而是根据上一轮节点的信息来同步地更新节点信息,并在每一轮更改后更新图形状态。这种同步的操作可能会导致消息滞后,并出现“互换社区”或“社区归属延迟”的问题。针对以上问题,本发明具体实施方式中首先对得到的结果构建逻辑图,求解逻辑图中的连通区域,将同一连通区域内的点都归为一个社区。该方法充分考虑的图的特性,能有效地保证并行化算法的稳定性。针对图整体的模块度计算,充分考虑到图中各个节点的内部边数以及外部边数。首先在集群节点上针对各个节点按照模块度计算公式进行并行计算,然后先在集群节点上集合节点数据,最后将各集群节点数据相加可得到整体的模块度。
并行Louvain算法具体流程如图4所示。在对基因序列数据进行预处理,构建原始无向网络图图之后,通过Louvain算法进行聚类,具体过程如下:
①在聚类迭代过程中首先对所述无向网络图中的每一个节点计算使之模块度增益最大的邻居节点,将所述邻居节点分配给该节点所属社区,直至更改所在社区的节点数少于某一值,结束本次迭代得到某一层次的新图;
②计算每次迭代前后的模块度变化值,模块度变化值小于一定值时,表示社区划分已经达到最优,可停止迭代;
③或者,为避免迭代层数过深导致社区过大,当迭代次数达到限定值后中止迭代;
④对聚类后结果处理得到序列信息及其对应社区信息。
根据聚类结果,对每一个分类簇中的reads进行组装,组装具体流程如图5所示,具体步骤如下;
首先,使用MEGAHIT组装工具对分类之后的原始FASTQ格式的序列进行组装,产生final.contigs.fa文件;
然后,使用BWA、Samtools对final.contigs.fa文件进行对比和格式转换;
最后,使用MetaBAT分箱工具进行分箱,将得到的Bins合并,进行后续的分析对比。
为了验证本发明方法的有效性,通过实验对本发明方法和传统组装方法进行对比。实验数据从一个含有791个基因组,1,024,000,000条reads,数据集大小为320GB的数据集中进行采样,在保持每个子采样数据集reads总数近似一致地情况下,使用50个样本数据集进行组装实验结果验证。采样之后的SEQ格式的数据集大小为25GB,原始的FASTQ格式的数据集大小约为50GB。测序片段的长度为150*2bp,测序片段的数量为83,029,581。组装结果采用纯度和完整度作为评价指标来进行分析
首先对比传统方法和本发明组装方法生成的最大10个Bin纯度。表1和表2分别为传统方法和基于本发明组装方法生成的最大10个Bin纯度。
表2 本发明组装方法最大10个Bin纯度
通过表1和表2可以看到,传统组装方法和本发明组装方法最大的10个Bin的纯度均在95%以上,但是本发明组装方法产生的最大十个Bins包含reads数量均要比传统组装方法的reads数量多。本发明组装方法在保证纯度的情况下能够得到reads数量更多的Bins。
然后测试了两种不同方法的高覆盖度情况下的组装结果,实验数据集中共包含79个不同覆盖度的基因组,其中覆盖度为100x以上的基因组共有20个。图6(a)和图6(b)分别给了本发明组装方法和传统组装方法产生的Bins完整度与其相对应覆盖度分布的对比,从两图中可以看出基于本发明组装方法在覆盖度在100x以下部分整体完整度较低,表现不如传统宏基因组装方法,在覆盖度为100x以上部分整体完整度基本在100%,表现较好;传统组装方法在覆盖度100x以下部分整体完整度比本发明的组装方法高,但是在覆盖度为100x以上的部分表现不如本发明组装方法。因此本发明组装方法在覆盖度为100x以上时表现较好。进一步通过实验测试得到在覆盖度大于100x以上的基因组,本发明的组装方法能够拼接出的纯度在95%以上和完整度在95%以上的基因组数量为15个,而传统组装方法能够拼接出的纯度在95%以上和完整度在95%以上的基因组数量为11个。在整体高覆盖度基因组数量为20的情况下,本发明的组装方法能够比传统的组装方法多组装产生4个基因组,能够有效提高传统宏基因组装方法拼接基因组的完整度可,起到了较好的优化效果。
以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。