一种检测基因SNV的方法
文献发布时间:2023-06-19 13:49:36
技术领域
本发明涉及生物技术领域,具体地,涉及一种检测基因SNV的方法。
背景技术
先天性心脏病(congenital heart defects,CHDs)是人类最常见的出生缺陷。在CHDs中,主动脉弓和流出道畸形约占40%,间室隔缺损约占30%。心脏神经嵴细胞(cardiacneural crest cells,NCCs)是一类与间室隔、主动脉发育息息相关的细胞。
miRNA广泛存在于生物体中,参与基因转录后表达的调控,与mRNA结合抑制其翻译效率,从而降低基因的表达水平,影响着细胞增殖、分化、迁移以及凋亡等活动,影响了生物体的发育和疾病的发生和发展。现有技术表明miRNA介导的基因表达失调很可能是先天性心脏病的重要致病原因之一,研究表明在小鼠模型中敲除NCCs的miRNAs表达,会导致小鼠CHDs的产生(Huang,Zhan-Peng et al.Loss of microRNAs in neural crest leads tocardiovascular syndromes resembling human congenital heartdefects.Arteriosclerosis,thrombosis,and vascular biology vol.30,12(2010):2575-86.)。
miRNA常常通过碱基互补配对与对应的基因的3’UTR结合,并降低靶标基因mRNA的稳定性或抑制其翻译,从而实现沉默靶标基因表达的分子功能。miRNA结合区域往往很短,关键的结合位点集中在miRNA的2~8bp,因此若基因的DNA突变发生在关键的区域将直接影响靶标基因正常的受miRNA的调控。单核苷酸突变(single-nucleotide variants,SNVs)则是最常见的DNA突变方式。
本研究团队曾报道过关于miRNA影响CHDs的研究,鉴定了多个基因中的3’UTR的SNVs与CHDs相关,证明了CHDs患者中,基因的SNVs干扰了miRNA介导的基因表达失调,从而导致CHDs的发生(Liu,Wangkai et al.Identification of Novel Single-NucleotideVariants With Potential of Mediating Malfunction of MicroRNA in CongenitalHeart Disease.Frontiers in cardiovascular medicine vol.8 739598.10 Sep.2021,doi:10.3389/fcvm.2021.739598)。
目前的SNV的检测往往需要依赖高通量测序技术,由于SNVs的变化水平极低,给后期测序数据的准确精细化处理带来了挑战,所以当下SNVs的检测成本和技术门槛都很高,亟需开发简单、快捷的检测方法。
发明内容
为了快速检测基因中是否存在单核苷酸突变,本发明的目的是提供一种检测基因单核苷酸突变的引物的设计方法。
本发明的第二个目的是提供一种检测基因单核苷酸突变的引物。
本发明的第三个目的是提供一种检测基因单核苷酸突变的方法。
为了实现上述目的,本发明是通过以下方案予以实现的:
一种检测基因单核苷酸突变的引物的设计方法,所述引物包含上游引物和下游引物1和2;
所述上游引物从5’端到3’端依次为:核苷酸序列如SEQ ID NO:1所示的通用引物序列1和上游扩增序列;所述上游扩增序列为与单核苷酸突变位点上游+144bp~+114bp相同的长度为25~27bp的片段;
所述下游引物1和2从5’端到3’端依次均为:通用引物序列2、下游扩增序列及单个碱基;所述下游引物1和2的通用引物序列2分别为序列(1)或(2)的不同序列,序列(1)为核苷酸序列如SEQ ID NO:2所示的序列;序列(2)为核苷酸序列如SEQ ID NO:3所示的序列;
所述下游扩增序列为与单核苷酸突变位点下游-1bp~-26bp反向互补的长度为24~26bp的片段;
所述单个碱基为碱基(1)或(2)的不同碱基,碱基(1)为与单核苷酸突变前碱基互补的碱基;碱基(2)为与单核苷酸突变后碱基互补的碱基。
优选地,所述单核苷酸突变为SNV。
优选地,根据待测基因的3’UTR区序列设计引物。
一种检测基因单核苷酸突变的引物也应在本发明的保护范围之内,所述引物为通过上述的设计方法获得的引物,包含上游引物和下游引物1和2。
本发明还提供了一种快速检测基因单核苷酸突变的方法,所述方法使用上述的引物。具体包含以下步骤:使用上述的引物对待测样本基因组DNA进行PCR扩增;琼脂糖凝胶电泳检测。
优选地,下游引物1从5’端到3’端依次为核苷酸序列如SEQ ID NO:2所示的序列、下游扩增序列及单核苷酸突变前碱基互补的碱基;下游引物2从5’端到3’端依次为核苷酸序列如SEQ ID NO:3所示的序列下游扩增序列及单核苷酸突变后碱基互补的碱基。
进一步地,由上游引物及下游引物1扩增出的目的条带较长,表示为无单核苷酸突变;由上游引物及下游引物2扩增出的目的条带较短,表示为有单核苷酸突变。
更优选地,判定是否存在单核苷酸突变的标准为:有且仅有一条较长的目的条带,则无单核苷酸突变;有两条长短不一的目的条带,则仅一条染色体存在单核苷酸突变;有且仅有一条较短的目的条带,则两条染色体都存在单核苷酸突变;其余情况表示PCR体系或程序有问题,无法判定是否存在单核苷酸突变,需要重做。
优选地,所述PCR扩增的反应体系中上游引物的终浓度为0.4~0.6μM,下游引物1或下游引物2的终浓度为0.1~0.3μM。
更优选地,所述PCR扩增的反应体系中上游引物的终浓度为0.5μM,下游引物1或下游引物2的终浓度为0.2μM。
与普通PCR反应相比,本发明在PCR反应中共设计了两个循环步骤。第一个循环目的是通过提高退火温度来减少非特性条带的含量,增加目的条带的含量,从而到达最终产物中无或者尽量少非特异性条带的目的;第二个循环的主要目的是为了扩增目的条带达到凝胶电泳可观察到的量。
优选地,所述PCR的反应程序为98℃2min;98℃10s,64~75℃30s,72℃30s,11个循环;98℃10s,60℃30s,72℃30s,27个循环;72℃,3min。
更优选地,所述PCR的反应程序为98℃2min;98℃10s,67℃或73℃30s,72℃30s,11个循环;98℃10s,60℃30s,72℃30s,27个循环;72℃,3min。
本发明通过凝胶电泳检测结果显示的条带数量即可判定基因中是否存在SNV。
优选地,所述凝胶电泳检测的方法为:配制3.5%的琼脂糖凝胶,上样量为5μL,120V电泳150min,拍照观察。
优选地,判定是否存在单核苷酸突变的标准为:有且仅有一条较长的目的条带,则无单核苷酸突变;有两条长短不一的目的条带,则仅一条染色体存在单核苷酸突变;有且仅有一条较短的目的条带,则两条染色体都存在单核苷酸突变;其余情况表示PCR体系或程序有问题,无法判定是否存在单核苷酸突变,需要重做。
与现有技术相比,本发明具有以下有益效果:
本发明提供了一种低成本、快速检测基因中单核苷酸突变的方法,通过针对目标基因设计引物,利用普通的PCR技术就能甄别出基因中是否带有特定的单核苷酸突变,大大降低了对检测仪器的要求,节省了检测成本,加快了检测速度。
附图说明
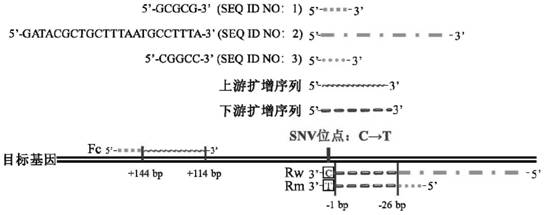
图1为检测基因SNV引物的设计方法示意图。
图2为基因NFATC13’UTR区的SNV检测结果;图A为不同退火温度下NFATC1的a、b两组检测引物的检测结果;图B为NFATC1的b组检测引物在不同质粒浓度、不同退火温度下的检测结果;图C为NFATC1的b组检测引物在最佳PCR反应体系及反应程序下的检测结果;M表示100bp DNA marker,WT表示人基因组DNA+plasmid-NTAFC1-WT组,MUT表示人基因组DNA+plasmid-NTAFC1-MUT组。
图3为基因FGFRL13’UTR区的SNV检测结果;图A为不同退火温度下FGFRL1的a、b两组检测引物的检测结果;图B为FGFRL1的b组检测引物在不同质粒浓度、不同退火温度下的检测结果;图C为FGFRL1的b组检测引物在最佳PCR反应体系及反应程序下的检测结果;M表示100bp DNA marker,WT表示人基因组DNA+plasmid-FGFRL1-WT组,MUT表示人基因组DNA+plasmid-FGFRL1-MUT组。
具体实施方式
下面结合说明书附图及具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
以下实施例所用材料如下:
实验试剂:Ex Taq(RR902A)、Premix Ex Taq
实验仪器:4℃冰箱(海尔)、-20℃低温冰箱(海尔)、-80℃超低温冰箱(Thermo)、PCR仪(TC1000-G,DLAB)、超净工作台(HDL)、凝胶电泳仪(DYY-6D型,北京市六一仪器厂)、凝胶成像系统(TANON)、37℃恒温CO
实施例1检测基因SNV引物的设计方法
根据目的基因中存在SNV的序列设计引物,设计原理如图1所示。具体方法如下:
在SNV上游的+144bp~+114bp处,选择其中长度为25~27bp的一段序列作为上游扩增序列,在其5’端加上固定序列5’-GCGCG-3’(SEQ ID NO:1),即构成上游引物Fc。
在SNV下游的-1bp~-26bp处,选择与其中长度为24~26bp的一段序列反向互补的序列作为下游扩增序列,在其5’端加上固定序列5’-GATACGCTGCTTTAATGCCTTTA-3’(SEQ IDNO:2),在其3’端加上与SNV突变前碱基互补的碱基,即构成下游引物Rw。
在SNV下游的-1bp~-26bp处,选择与其中长度为24~26bp的一段序列反向互补的序列作为下游扩增序列,在其5’端加上固定序列5’-CGGCC-3’(SEQ ID NO:3),在其3’端加上与SNV突变后碱基互补的碱基,即构成下游引物Rm。
实施例2检测基因SNV的方法
(1)引物的设计及筛选
使用实施例1所述的方法,根据目的基因中存在SNV的序列设计多组引物Fc、Rw、Rm,经对比扩增效果,筛选到特异性最好的一组引物。
(2)样本基因组DNA提取
采用商业化的试剂盒提取待测样本的基因组DNA。
(3)PCR鉴定
1.配制PCR的反应体系,其中Fc的终浓度为0.5μM,Rw及Rm的终浓度各为0.2μM。
2.设定PCR反应程序为98℃2min;98℃10s,67℃或73℃30s,72℃30s,11个循环;98℃10s,60℃30s,72℃30s,27个循环;72℃,3min。
3.配制3.5%的琼脂糖凝胶,上样量为5μL,120V电泳150min,拍照观察。
4.判定是否存在单核苷酸突变的标准为:有且仅有一条较长的目的条带,则无单核苷酸突变;有两条长短不一的目的条带,则仅一条染色体存在单核苷酸突变;有且仅有一条较短的目的条带,则两条染色体都存在单核苷酸突变;其余情况表示PCR体系或程序有问题,无法判定是否存在单核苷酸突变,需要重做。
实施例3检测基因NFATC1 3’UTR区的SNV
一、实验方法
(1)引物的设计及筛选
1.使用实施例1所述的方法,根据NFATC1(NM_172387.3)的3’UTR区序列以及文献(DOI:10.3389/fcvm.2021.739598)中公开的SNV位点,设计a、b两组检测引物如表1所示,划线标注的碱基为固定序列,方框标注的碱基为SNV位点。
表1 检测基因NFATC1 3’UTR区SNV的引物
2.如图2A所示,经筛选鉴定,与a组相比,用b组引物扩增产生的条带特异性更好,故选用b组引物进行后续试验。
(2)人基因组DNA提取
采集健康成年人的血液,利用试剂盒(TIANGEN,DP304-03)对人外周血进行基因组DNA的提取,详细步骤如下:
1.取200μL血液加入离心管中。
2.加入20μL proteinase K溶液混匀。
3.加入200μL GB缓冲液,充分颠倒混匀,70℃放置10min。
4.加入200μL无水乙醇,充分混匀后,加入吸附柱CB3中。
5.12000rpm,30s离心弃掉废液,加入500μL GD缓冲液。
6.12000rpm,30s离心弃掉废液,加入600μL PW漂洗液。
7.重复步骤6一遍。
8.12000rpm,30s离心弃掉废液,12000rpm,2min离心。
9.将吸附柱转移至新的离心管中,室温静置2min;加入50μL TE洗脱液;室温静置2min。
10.12000rpm,2min离心,即获得人基因组DNA。
(3)pMD19T-NTAFC1-WT及pMD19T-NTAFC1-MUT质粒构建
1.以上述所得人基因组DNA为模板,通过PCR扩增含有本实施例步骤(1)中SNV位点的目的片段,所用引物序列如表2所示,方框标注的碱基为SNV位点。
表2 扩增NFATC1目的片段的引物
PCR反应体系为:Ex Taq,25μL;NTAFC-TA-WT-F、NTAFC-TA-WT-R各1μL或NTAFC-TA-MUT-F、NTAFC-TA-MUT-R各1μL;基因组DNA 200ng以下;ddH
PCR反应程序为:预变性98℃,2min;变性98℃,10s;退火55℃,30s;延伸72℃,20s;29个循环。
2.PCR反应结束后进行1.5%核酸凝胶电泳,在目的大小位置观察到单一的条带证明扩增成功,进一步利用凝胶DNA回收试剂盒(DP209-03,TIANGEN)进行纯化回收,即可得到大量的目的片段。
3.将纯化好的目的基因片段连接到pMD19T载体(6013,TAKARA),方法参照相关产品说明书,即获得连接产物。
4.将获得连接产物转化至感受态细胞Ecoli.DH5α(KTSM101L,KT HEALTH)中,方法参照相关产品说明书。将转化后的Ecoli.DH5α接种于LB培养基,培养过夜。
5.挑取单克隆菌落于300μL含氨苄抗生素的LB培养基中,37℃,220rpm,培养5h。培养结束后进行菌落PCR鉴定阳性克隆,PCR所用引物、反应体系、反应程序同本实施例步骤(3)的第1步。
6.将所得阳性克隆送到华大基因公司测序,若序列正确无突变,对菌液进行扩大培养,用试剂盒(DP120-01,TIANGEN)提取质粒,详细步骤参考产品说明书,即获得pMD19T-NTAFC1-WT及pMD19T-NTAFC1-MUT质粒。
(4)pMD19T-NTAFC1-WT及pMD19T-NTAFC1-MUT质粒的线性化处理
为模拟人体所含SNV的基因组,对pMD19T-NTAFC1-WT质粒及pMD19T-NTAFC1-MUT质粒进行线性化处理。具体步骤如下:
1.使用BamHI-HF内切酶(R3136S,NEB)对pMD19T-NTAFC1-WT质粒及pMD19T-NTAFC1-MUT质粒进行单酶切,详细步骤以及反应体系可参照产品说明书,获得酶切产物。
2.将所得酶切产物分别用普通DNA产物纯化试剂盒(DP204-03,TIANGEN)进行回收,详细步骤参照产品说明书,即获得plasmid-NTAFC1-WT及plasmid-NTAFC1-MUT。
(5)PCR反应检测基因NFATC1 3’UTR区的SNV
1.使用表1所示的引物配制PCR体系如表3所示,其中基因组DNA为本实施例所得人基因组DNA。
表3 检测基因NFATC1 3’UTR区SNV的PCR体系
2.以表4所示反应程序进行PCR扩增。
表4 检测基因NFATC1 3’UTR区SNV的PCR反应程序
3.琼脂糖凝胶电泳
配制3.5%的琼脂糖凝胶,上样量为5μL,120V电泳150min,拍照观察。
二、实验结果
如图2B所示,plasmid-NTAFC1-WT组只有一条目的条带,而plasmid-NTAFC1-MUT组有两条大小不一的目的条带,表明已成功鉴定出NTAFC1的3’UTR区存在SNV。
退火温度和质粒模板的浓度也影响着PCR反应的扩增效果。当plasmid-NTAFC1-WT或plasmid-NTAFC1-MUT的模板量为60~70pg,且第一步循环的退火温度为67℃时,PCR的扩增效果最好,在该条件下NFATC1 3’UTR区的SNV的检测结果如图2C所示。
实施例4检测基因FGFRL1 3’UTR区的SNV
一、实验方法
(1)引物的设计
1.使用实施例1所述的方法,根据FGFRL1(NM_001004356.3)的3’UTR区序列以及文献(DOI:10.3389/fcvm.2021.739598)中公开的SNV位点,设计a、b两组检测引物如表5所示,划线标注的碱基为固定序列,方框标注的碱基为SNV位点。
表5 检测基因FGFRL1 3’UTR区SNV的引物
(2)人基因组DNA提取
方法同实施例3的步骤(2)。
(3)pMD19T-FGFRL1-WT及pMD19T-FGFRL1-MUT质粒构建
1.以上述所得人基因组DNA为模板,通过PCR扩增含有本实施例步骤(1)中SNV位点的目的片段,所用引物序列如表6所示,方框标注的碱基为SNV位点。
表6 扩增FGFRL1目的片段的引物
PCR反应体系为:Ex Taq,25μL;FGFRL1-TA-WT-F或FGFRL1-TA-MUT-F,1μL;FGFRL1-TA-WT-R或FGFRL1-TA-MUT-R,1μL;基因组DNA 200ng以下;ddH
PCR反应程序为:预变性98℃,2min;变性98℃,10s;退火55℃,30s;延伸72℃,20s;29个循环。
2.PCR反应结束后进行1.5%核酸凝胶电泳,在目的大小位置观察到单一的条带证明扩增成功,进一步利用凝胶DNA回收试剂盒(DP209-03,TIANGEN)进行纯化回收,即可得到大量的目的片段。
3.将纯化好的目的基因片段连接到pMD19T载体(6013,TAKARA),方法参照相关产品说明书,即获得连接产物。
4.将获得连接产物转化至感受态细胞Ecoli.DH5α(KTSM101L,KT HEALTH)中,方法参照相关产品说明书。将转化后的Ecoli.DH5α接种于LB培养基,培养过夜。
5.挑取单克隆菌落于300μL含氨苄抗生素的LB培养基中,37℃,220rpm,培养5h。培养结束后进行菌落PCR鉴定阳性克隆,PCR所用引物、反应体系、反应程序同本实施例步骤(3)的第1步。
6.将所得阳性克隆送到华大基因公司测序,若序列正确无突变,对菌液进行扩大培养,用试剂盒(DP120-01,TIANGEN)提取质粒,详细步骤参考产品说明书,即获得pMD19T-FGFRL1-WT及pMD19T-FGFRL1-MUT质粒。
(4)pMD19T-FGFRL1-WT及pMD19T-FGFRL1-MUT质粒的线性化处理
为模拟人体所含SNV的基因组,对pMD19T-FGFRL1-WT质粒及pMD19T-FGFRL1-MUT质粒进行线性化处理。具体步骤如下:
1.使用BamHI-HF内切酶(R3136S,NEB)对pMD19T-FGFRL1-WT质粒及pMD19T-FGFRL1-MUT质粒进行单酶切,详细步骤以及反应体系可参照产品说明书,获得酶切产物。
2.将所得酶切产物分别用普通DNA产物纯化试剂盒(DP204-03,TIANGEN)进行回收,详细步骤参照产品说明书,即获得plasmid-FGFRL1-WT及plasmid-FGFRL1-MUT。
(5)PCR反应检测基因FGFRL1 3’UTR区的SNV
1.使用表1所示的引物配制PCR体系如表7~8所示,其中基因组DNA为本实施例所得人基因组DNA。
表7 检测基因FGFRL1 3’UTR区SNV的PCR体系
表8 检测基因FGFRL1 3’UTR区SNV的PCR体系
2.以表9所示反应程序进行PCR扩增。
表9 检测基因FGFRL1 3’UTR区SNV的PCR反应程序
3.琼脂糖凝胶电泳
配制3.5%的琼脂糖凝胶,上样量为5μL,120V电泳150min,拍照观察。
二、实验结果
如图3A所示,plasmid-FGFRL1-WT组只有一条目的条带,而plasmid-FGFRL1-MUT组有两条大小不一的目的条带,表明已成功鉴定出NTAFC1的3’UTR区存在SNV。
如图3B所示,a、b两组引物的最适退火温度为72.7~73.7℃,b组引物扩增出的目的条带更规整。用b组引物在第一步循环的退火温度为73℃时,PCR的扩增效果最好,在该条件下FGFRL1 3’UTR区的SNV的检测结果如图2C所示。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,对于本领域的普通技术人员来说,在上述说明及思路的基础上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
序列表
<110> 中山大学附属第一医院
<120> 一种检测基因SNV的方法
<160> 3
<170> SIPOSequenceListing 1.0
<210> 1
<211> 5
<212> DNA
<213> Homo sapiens
<400> 1
gcgcg 5
<210> 2
<211> 23
<212> DNA
<213> Homo sapiens
<400> 2
gatacgctgc tttaatgcct tta 23
<210> 3
<211> 5
<212> DNA
<213> Homo sapiens
<400> 3
cggcc 5