检测不稳定的无细胞DNA的方法及使用其的装置
文献发布时间:2023-06-19 09:33:52
技术领域
本发明涉及一种无需基因扩增过程即可检测具有不稳定的双螺旋结构的无细胞DNA的方法和装置。
背景技术
近来,癌症疾病的早期诊断的重要性在世界范围内日益凸显。因此,对于癌症的早期诊断方法的研究越来越多。然而,到目前为止,癌症的诊断方法是通过例如收集组织样品和内窥镜检查等侵入性方法来进行。特别地,进行组织学检查,以提取一部分可疑的疾病部位并在显微镜下观察。因此,由于为了使用针、冲头、内窥镜或腹腔镜收集组织样品而需要切开人体,不仅另患者感到相当不舒服,而且还会留下疤痕且恢复时间很长。
作为侵入性诊断和检查方法的替代方法,使用液体活检的分子诊断方法引起了人们的注意。由于液体活检使用非侵入性方法,因此可以快速识别其结果。此外,与仅能对疾病的一部分进行分析的组织样本不同,液体活检可对疾病进行多边分析。特别地,液体活检有望在癌症诊断中发挥出色的功效。特别是,可以预见到,仅检查例如血液和尿液等体液,就可以分析血液中存在的各个身体部位的癌细胞来源的DNA,从而可以详细地观察癌症的发生和转移。
分子诊断方法是体外诊断的代表性技术,其通过数值或图像检测和诊断来自包含遗传信息(例如血液和尿液)的样本中DNA或RNA的变化。由于其准确性高且不需要组织学检查的优点,随着基因组分析技术的快速发展,基于其节省成本的优点,现已经尝试将分子诊断方法应用于癌症诊断技术。
另一方面,无细胞DNA(以下称为cfDNA)是指存在于血浆中且源自细胞的DNA。cfDNA通常具有双螺旋结构。另外,在许多情况下,cfDNA具有卷曲螺旋结构。cfDNA可以源自肿瘤细胞。另外,可以在从癌症患者获得的血液、血浆或尿液等生物样品中发现源自肿瘤细胞的cfDNA。
在癌症患者中发现的cfDNA可能源自坏死细胞、正常细胞和/或癌细胞。此类cfDNA通过各种过程释放到尿液、血液等中。因此,用于分离和检测例如血液、血浆或尿液等生物样品中的cfDNA的技术的发展使液体活检成为监测癌症风险患者的更有效和更可靠的工具。特别地,由于尿液、血浆、血液或体液是易于获得的样品,因此可以以非侵入性的方式收集大量样品。
但是,鉴于当前的技术水平,早期诊断癌症的方法,包括分析例如血液和尿液等液体样品中的cfDNA以及发现基因中存在的突变中存在许多困难。因此,需要开发一种易于检测cfDNA的方法以及用于提高检测灵敏度和精确地早期诊断癌症的技术。
同时,韩国专利第10-1751962号公开了一种能够通过使用引物进行聚合酶链式反应(PCR)来定量cfDNA以检测cfDNA的技术。但是,仍然存在需要单独的聚合酶和实验设备来进行聚合酶链式反应的问题,并且存在需要生成精确的引物以进行扩增且不容易进行现场诊断的问题。
此外,韩国专利第10-1701618号公开了一种纳米结构,其表面性质可以通过电场的变化而改变,以便有效地分离cfDNA。该纳米结构可以通过电场的变化结合或解离cfDNA,因此可以容易地从样品中分离cfDNA。但是,仍然存在一个限制,即,应该使用聚合酶链式反应来鉴定存在哪些cfDNA。
为了通过聚合酶链式反应扩增cfDNA,需要各种类型的引物对,并且要花费大量时间进行复杂的步骤。因此,人们一直在研究以克服必须进行PCR的局限性,并开发出用于高精度分析cfDNA的方法。
发明内容
技术问题
常规上,为了检测cfDNA,必须对cfDNA进行变性并使用与cfDNA互补的引物扩增的过程。然而,本发明人已经鉴定出血液中存在不稳定的cfDNA。另外,本发明人已经发现,与稳定的cfDNA不同,不稳定的cfDNA显示出与单链探针的异常反应。特别地,本发明人已经鉴定出这种不稳定的cfDNA衍生自患有癌症或传染性疾病的个体的细胞。基于这些发现,本发明人完成了本发明。
因此,本发明的目的是提供了一种鉴定不稳定的cfDNA存在与否的方法,并提供了一种通过鉴定这种不稳定的cfDNA来提供用于诊断癌症或各种疾病的信息的方法。
解决问题的方案
为了实现上述目的,本发明提供了一种从未经扩增的样品中检测不稳定的cfDNA的方法。另外,本发明提供了一种通过从未经扩增的样品中检测不稳定的cfDNA来提供用于诊断或预测癌症和传染性疾病的信息的方法。另外,本发明提供了一种从未经扩增的样品中检测不稳定的cfDNA的装置。
发明的有益效果
当使用一个实施方案的用于检测不稳定的cfDNA的方法时,不仅不需要扩增不稳定的cfDNA的过程,而且可以缩短用于分析cfDNA的时间。特别地,可以有效地检测含有基因突变部分的不稳定的双链cfDNA,从而有效地诊断或预测与基因突变有关的癌症或疾病。另外,当使用本发明的实施方案的方法时,可以从少量的例如尿液、脑脊液、胸膜液、腹水、血浆、血液或体液等生物样品中快速且准确地识别具有不稳定的双螺旋结构的cfDNA的存在与否。另外,由于已经确认由此检测到的不稳定的cfDNA与各种癌症或疾病有关,因此根据本发明的实施方案的方法可有效地用于癌症的诊断或治疗后的预后的鉴定。
附图说明
图1显示了带正电的纳米线(PEI/Ppy NW)的扫描电子显微镜(SEM)图像。
图2显示了带正电的纳米线(PEI/Ppy NWs)的透射电子显微镜(TEM)图像。
图3显示了通过扫描电子显微镜图像表示的纳米结构(PLL/Ppy NW)的表面,作为阳离子聚合物的聚赖氨酸(PLL)结合在其上。
图4显示了HRP/链菌亲和素结合的纳米颗粒的扫描电子显微镜图像。
图5显示了直径为50nm的PEI结合的纳米颗粒(PEI/Ppy NP)的扫描电子显微镜(SEM)图像。
图6显示了一种方法的示意图,其中使用表面结合有聚乙烯亚胺(PEI)的纳米线(PEI/Ppy NW)从患者的体液中获得cfDNA,然后通过与探针和HRP/链霉亲和素标记的纳米颗粒(HRP/st标记的NP)反应,在60分钟内分析基因突变。
图7示意性地显示了使用纳米线、探针和HRP/链菌亲和素标记的纳米颗粒检测不稳定的cfDNA的方法。
图8以时间顺序的流程图示出了用于检测诸如血液、脑脊液或胸膜液的样品中的不稳定cfDNA的方法。
图9以时间顺序的流程图示出了一种用于检测诸如尿液的样品中不稳定的cfDNA的方法。
图10示意性地显示了取决于从血液获得的cfDNA的状态的变性条件的差异。
图11示意性地显示了取决于从尿液、唾液和痰液中获取的cfDNA的状态的变性条件的差异。
图12显示了通过从宫颈癌患者的尿液样品和正常受试者的尿液样品中获得cfDNA,然后使用HPV 18或HPV 16探针检测cfDNA中基因突变的存在与否而获得的结果。
图13显示了通过吸光度鉴定HPV阳性宫颈癌患者(HPV16(+)和HPV18(+))以及HPV阴性健康对照患者(HPV-)的尿液中存在的cfDNA与特异于HPV 18或HPV 16的探针之间的结合所获得的结果。
图14显示了通过使从宫颈癌患者的尿液中分离出的cfDNA与对HPV 16、EGFR19缺失、HPV 18和EGFR 21 L858R具有特异性的探针进行顺序反应,然后鉴定cfDNA与每种探针之间的结合而获得的结果。
图15显示了通过向正常受试者的血浆中添加低范围(10bp-100bp),中范围(100bp-2kb)和高范围(3.5kb-21kb)的DNA梯状条带获得的结果,然后在使用PEI/Ppy NW捕获DNA梯状条带的情况下评估捕获效率。
图16显示了通过鉴定在没有超声处理的情况下降解A549细胞系(泳道1和2),HCC2279细胞系(泳道3和4)和H1975细胞系(泳道5和6)获得的细胞系基因组DNA(以下称为gDNA)的大小而获得的结果,以及鉴定通过超声处理每个细胞系的gDNA而获得的片段DNA(以下称为fDNA)的大小而获得的结果。
图17显示了通过使用PEI/Ppy NW捕获在未经超声处理的情况下从A549细胞系、HCC2279细胞系和H1975细胞系获得的gDNA,以及通过对各个细胞系进行超声处理而获得的fDNA,然后评估捕获效率所获得的结果。
图18和图19显示了通过使用PEI/Ppy NW捕获从未经超声处理的A549细胞系、HCC2279细胞系和H1975细胞系获得的gDNA以及通过对各自细胞系进行超声处理而获得的fDNA,然后通过添加对EGFR 19(19Del)、EGFR 20(T790M)和EGFR 21(L858R)具有特异性的探针以鉴定颜色变化和UV-Vis的可见光谱变化而获得的结果。已经鉴定出只有fDNA选择性结合靶探针。
图20显示了通过使用PEI/Ppy NW获得H1975细胞系、HCC2279细胞系和A549细胞系的fDNA,然后使用结合有荧光染料的DNA探针分析基因突变而获得的结果。
图21显示了通过利用HCC2279(EGFR外显子19缺失基因突变)细胞的fDNA和对EGFR外显子19缺失具有特异性的探针,并利用紫外线吸收率分析fDNA与探针的结合而获得的结果。通过这种方法,已经确定了可检测浓度的cfDNA。
图22显示了通过使用H1975(EGFR外显子20 T790M和外显子21 L858R基因突变)的fDNA和对EGFR外显子20 T790M具有特异性的探针,并利用UV吸收率分析fDNA与探针的结合(取决于fDNA的浓度)而获得的结果。通过这种方法,已经鉴定出可检测浓度的cfDNA。
图23显示了通过Bioanalyzer使用PEIA/Ppy NW分析从肺癌患者的200μL血浆中获得的cfDNA而获得的结果。根据文献,已知与癌症相关的cfDNA具有166bp的平均大小。如Bioanalyzer数据所示,已确定使用PEI/Ppy NW从肺癌患者血浆中获得的cfDNA在169bp处出现峰。
图24显示了通过鉴定从患有EGFR基因突变的肺癌患者血浆中分离的cfDNA与特异性结合EGFR外显子19缺失(Del)的探针或EGFR外显子21 L858R的探针反应的情况下获得的结果,图中显示了组织基因型相同的颜色变化和UV吸收率。
图25显示了通过鉴定从患有EGFR外显子19缺失(Del)、EGFR外显子20 T790M和EGFR外显子21 L858R基因突变的肺癌患者的血浆中分离出的cfDNA与特异性结合EGFR外显子19Del、EGFR外显子20 T790M或EGFR外显子21 L858R的探针反应而获得的结果,图中显示了组织基因型相同的颜色变化和UV吸收率。
图26和27显示了通过鉴定在使用PEI/Ppy NW从具有KRAS外显子2基因突变的肺癌患者血浆中分离的cfDNA与特异性结合KRAS外显子2的探针反应的情况下获得的结果,图中显示了组织基因型相同的颜色变化和UV吸收率。
图28显示了通过鉴定从患有ALK-EML4融合基因突变的肺癌患者血浆中分离的cfDNA与特异性结合ALK-EML4的探针反应的情况下获得的结果。图中显示了组织基因型相同的颜色变化和UV吸收率。
图29显示了在从患有EGFR外显子19缺失或EGFR外显子20 T790M基因突变的肺癌患者血浆中分离的cfDNA与特异性结合EGFR外显子19缺失或EGFR外显子20 T790M的各种探针反应的情况下,颜色变化和紫外线吸收率均显示出与组织匹配。然而,根据结果可以确认,与所使用的三种类型的探针无关,未观察到EGFR 21 L858R突变的颜色变化。结果可以看出,基因与探针之间的反应不限于特异性的探针,并且任何探针都可以结合具有突变的基因,只要该探针与该基因特异性结合即可。
图30显示了通过对无EGFR基因突变的肺癌患者血浆(野生型,WT)分离的cfDNA分别在95℃变性1分钟和10分钟,然后与特异性结合EGFR外显子19缺失、EGFR外显子20 T790M或EGFR外显子21 L858R的探针反应而获得的结果。已经发现,从EGFR WT患者血浆中提取的cfDNA在95℃变性1分钟的情况下与任何探针均无反应;而另一方面,cfDNA在95℃变性10分钟后与所有探针发生非特异性反应。
图31、图32和图33显示了通过纳米线从具有EGFR外显子19缺失和EGFR外显子20T790M基因突变的肺癌患者以及正常受试者的血浆中捕获cfDNA,使得该cfDNA在95℃下分别变性0分钟(图31)、1分钟(图32)和10分钟(图33),然后鉴定其与特异性结合EGFR 19缺失、20 T790M或21 L858R的探针反应后的结果。
图34显示了通过利用从151名肺癌患者的血浆中获得的cfDNA并分析肺癌患者中的基因突变而获得的表。
图35显示了通过从无EGFR突变的肺癌患者(野生型)、具有EGFR外显子19缺失的肺癌患者和具有EGFR外显子21 L858R的肺癌患者的血浆中获得cfDNA,将这些cfDNA与对EGFR外显子19 Del具有特异性的探针混合,然后通过分析UV光谱吸光度(ΔOD,500nm-650nm)值确定肺癌患者的基因突变而获得的结果。
图36显示了通过从患有EGFR外显子19缺失的肺癌患者的血浆中获得cfDNA,将cfDNA与对EGFR外显子19 Del具有特异性的探针混合,然后分析基因突变的特异性和敏感性而获得的结果。
图37显示了通过从无EGFR突变的肺癌患者(野生型)、具有EGFR外显子19缺失的肺癌患者和具有EGFR外显子21 L858R的肺癌患者的血浆中获得cfDNA,并添加对EGFR外显子21 L858R具有特异性的探针,然后通过分析UV光谱吸光度(ΔOD,500nm-650nm)值来确定患者的基因突变而获得的结果。
图38显示了通过从患有EGFR外显子21 L858R的肺癌患者的血浆中获得cfDNA,向其中添加对EGFR外显子21 L858R具有特异性的探针,然后分析患者中基因突变的特异性和敏感性而获得的结果。
图39显示了通过使用PEI/Ppy NP从具有EGFR外显子19缺失基因突变的肺癌患者血浆中分离的cfDNA与对EGFR外显子19 Del(E19)、EGFR外显子20 T790M(E20)或EGFR外显子21 L858R(E21)具有特异性的探针进行反应所获得的结果,结果发现观察到与组织相匹配的UV吸收率。
图40显示了通过使用经聚赖氨酸修饰的PLL/Ppy NW获得具有EGFR外显子20T790M和EGFR外显子21 L858R基因突变的H1975细胞系的fDNA,使fDNA与对EGFR外显子19 Del、EGFR外显子20 T790M或EGFR外显子21 L858R具有特异性的探针进行反应,然后分析基因突变而获得的结果。
图41显示了用于EGFR外显子19缺失的CP和DP的序列。在这项研究中,使用CP_1和DP分析了肺癌患者的cfDNA基因突变。在此,CP是指与包含突变部分或与其相邻的序列特异性结合的探针,DP是特异性结合与突变序列间隔的部分的探针。
图42显示了EGFR外显子20 T790M的CP和DP的序列。在这项研究中,使用CP2和DP分析了肺癌患者的cfDNA基因突变。
图43显示了EGFR外显子21 L858R的CP和DP的序列。在这项研究中,使用CP2和DP分析了肺癌患者的cfDNA基因突变。
图44显示了通过从患有EGFR外显子19缺失基因突变的肺癌患者的血浆中获得cfDNA,然后使用图41-42中的EGFR外显子19 CP_1、外显子20 CP2和外显子21 CP2来分析基因突变所获得的结果,未使用DP。
图45显示了通过对具有EGFR外显子19缺失的肺癌患者的血浆进行RNase和DNase处理,并通过PEI/Ppy纳米线获得cfDNA,然后使用对EGFR外显子19缺失具有特异性的探针检测不稳定的cfDNA而获得的结果。
图46显示了通过对具有EGFR外显子20 T790M的肺癌患者的血浆进行RNase和DNase处理,并通过PEI/Ppy纳米线获得cfDNA,然后使用对EGFR外显子20 T790M具有特异性的探针检测不稳定的cfDNA而获得的结果。
图47显示了通过将从患有EGFR外显子19缺失和EGFR外显子20 T790M基因突变的肺癌患者血浆中获得的cfDNA与对EGFR外显子19缺失(Del19)、EGFR外显子20 T790M和EGFR外显子21 L858R具有特异性的探针进行反应,然后向其中添加HRP/链霉亲和素纳米颗粒(包含大量HRP),以通过颜色变化和UV吸收检测cfDNA而获得的结果。
图48显示了通过使从图47中患有的具有EGFR外显子19缺失和EGFR外显子20T790M基因突变的相同肺癌患者的血浆中获得的cfDNA与对EGFR外显子19缺失(Del19)、EGFR外显子20 T790M和EGFR外显子21 L858R具有特异性的探针进行反应,然后向其中添加HRP/链霉亲和素复合物(通过在HRP和链霉亲和素之间以1:1结合获得的复合物)来通过颜色变化和UV吸收检测cfDNA而获得的结果。根据结果显示,与HRP/链霉亲和素纳米颗粒相比,在HRP/链霉亲和素复合物中产生了噪音。
图49显示了通过对从5名具有EGFR外显子19缺失和外显子20 T790M基因突变的肺癌患者血浆中提取cfDNA,然后将cfDNA与对EGFR外显子19 Del、EGFR外显子20 T790M和EGFR外显子21 L858R具有特异性的探针,以及HRP/链霉亲和素标记的纳米颗粒(HRP/st标记的NP)进行反应,并与对EGFR外显子19 Del、EGFR外显子20 T790M和EGFR外显子21 L858R具有特异性的探针,以及HRP/链霉亲和素复合物(其中HRP和链霉亲和素以1:1相互结合)进行反应而获得的两种结果进行分析,从而鉴定并比较与癌组织基因型的匹配而获得的图。
图50显示了通过将从患有EGFR外显子20 T790M和21 L858R基因突变的肺癌患者的胸膜液中获得的cfDNA与对EGFR外显子19缺失(19 Del)、EGFR外显子20 T790M和EGFR外显子21 L858R的探针(已结合有HRP/st标记的NP)反应而获得的结果,然后通过UV吸收检测基因突变。
图51显示了通过将从患有EGFR外显子20 T790M和EGFR外显子21 L861Q基因突变的肺癌患者的血浆中获得的cfDNA与对EGFR外显子19缺失(19 Del)、EGFR外显子20 T790M、EGFR外显子21 L858R和EGFR外显子L861Q具有特异性的探针,以及HRP/st标记的NP一次全部混合而获得的结果,用于检测cfDNA中的基因突变,结果通过UV吸收鉴定出,基因突变仅在EGFR外显子20 T790M和EGFR外显子21 L861Q中观察到,如同在癌症组织中一样。
图52显示了通过将从患有ALK-EML4融合和ALK点突变(I1171N/T)的基因突变的肺癌患者血浆中获得的cfDNA,所有同时与对ALK-EML4融合和ALK点突变(T1151,L1152P,L1152R,C1156Y,I1171N/T)具有特异性的探针和HRP/st标记的NP进行混合,从而检测cfDNA中的基因突变而获得的结果,结果显示,确定在癌症组织中检测到ALK-EML4融合和ALK点突变(I1171N/T)基因型。
图53显示了通过将从BRAF V600E基因突变的甲状腺癌患者血浆中获得的cfDNA与对BRAF V600E具有特异性的探针以及HRP/st标记的NP一次性混合来检测cfDNA中的基因突变所获得的结果,结果鉴定出,检测到与患者基因型相同的BRAF V600E基因突变。
图54显示了通过将从正常受试者的血液中收集的样品在各种温度条件下进行变性,然后针对各个治疗条件检测不稳定的cfDNA而获得的结果。
图55显示了通过将从患者血液中收集的样品在各种温度条件下进行变性,然后针对各个治疗条件检测不稳定的cfDNA而获得的结果。
图56显示了通过使从突变细胞系获得的fDNA在各种温度条件下变性,然后针对各个处理条件检测不稳定的cfDNA而获得的结果。
图57显示了通过将从突变细胞系获得的fDNA在37℃下用DNA酶处理30分钟,然后检测不稳定的cfDNA用于治疗条件所获得的结果。
图58显示了通过将从突变细胞系获得的fDNA在37℃下用DNase处理60分钟,然后检测不稳定的cfDNA用于治疗条件而获得的结果。
图59显示了通过将从突变细胞系获得的fDNA在37℃下用DNase处理120分钟,然后检测不稳定的cfDNA用于治疗条件所获得的结果。
图60显示了通过在24℃下用1μl或2μl的DNA酶对不稳定的cfDNA和稳定的cfDNA进行120分钟的处理而获得的结果,以便根据DNase的活性鉴定不稳定的cfDNA和稳定的cfDNA之间的差异。
图61显示了通过在3℃下用1μl或2μl的DNA酶对不稳定的cfDNA和稳定的cfDNA进行120分钟的处理而获得的结果,以便根据DNase的活性鉴定不稳定的cfDNA和稳定的cfDNA之间的差异。
<术语的定义>
如本发明所用,术语“无细胞DNA(cell-free DNA)”又称为cfDNA。此外,cfDNA也可以是循环肿瘤DNA(ctDNA),它是癌细胞衍生的DNA,可以在例如癌症患者的尿液、脑脊液、血浆、血液或体液等生物样品中被发现。另外,cfDNA还可能存在于例如尿液、脑脊液、胸膜液、腹水、血浆、血液、唾液、痰液或体液等生物样品中。此处,cfDNA的大小可以为80bp-10kbp,100bp-1kbp或120bp-500bp。另外,cfDNA的大小可以为150bp-200bp,并且通常可以具有165bp-170bp的大小。
如本发明所用,术语“不稳定的cfDNA”是指与“稳定的cfDNA”相比在热力学上不稳定的cfDNA。换句话说,与稳定的cfDNA发生变性的条件相比,不稳定的cfDNA可能在较不严重的条件下就发生变性。之所以会产生不稳定的cfDNA,是因为不稳定的cfDNA具有不稳定的双螺旋结构。
如本发明所用,术语“具有不稳定的双螺旋结构的cfDNA”的特征在于,相对于具有稳定的双螺旋结构的cfDNA而言其具有更低的Tm值,或者在具有稳定的双螺旋结构的cfDNA不变性的条件下其会发生变性。Tm是指将50%的双链DNA转化为单链DNA的解链温度。Tm值与DNA的长度成正比,并且可能随核苷酸序列的变化而变化。由于基因组DNA中大量核苷酸相互通过氢键结合,因此基因组DNA必须在92℃-95℃加热5分钟或更长时间,或者在98℃加热2分钟或更长时间。此外,基因组DNA在低于90℃的温度下不易变性。由于具有稳定的双螺旋结构的cfDNA平均具有170bp的核苷酸,因此可以具有与基因组DNA相似的Tm值。
但是,由于相对于具有稳定的双螺旋结构的cfDNA而言,“具有不稳定的双螺旋结构的cfDNA”具有更低的Tm值。因此,当具有稳定的双螺旋结构的cfDNA在选自以下的任意一种条件下时:i)在室温下放置1-120分钟的条件;ii)在90℃-95℃下加热1秒至3分钟的条件;iii)在75℃-90℃下加热1秒至5分钟的条件;iv)在60℃-75℃下加热30秒至60分钟的条件;v)在25℃-40℃下加热10-120分钟的条件;vi)用蛋白酶处理10秒至30分钟的条件;以及vii)用DNase处理10秒-30分钟的条件会发生变性,然后与15聚体(15-mer)至30聚体(30-mer)的探针进行结合反应,具有稳定的双螺旋结构的cfDNA不与该探针结合。在此,“室温”是指环境温度,可以为18℃-25℃。此外,除上述条件外,还可以包括在40℃-65℃下加热5-80分钟的条件。
然而,已经证实,当在上述i)-vii)的任何一种条件下对具有不稳定的双螺旋结构的cfDNA进行处理后,再与15聚体至30聚体的探针进行结合反应时,具有不稳定的双螺旋结构的cfDNA与该探针结合。在此,探针可以是15聚体至30聚体,或20聚体(20-mer)至25聚体(25-mer),并且可以是21聚体(21-mer),22聚体(22-mer),23聚体(23-mer)或24聚体(24-mer)的探针。
在此,具有不稳定的双螺旋结构的cfDNA可以是循环肿瘤DNA(以下称为ctDNA)。另外,具有不稳定的双螺旋结构的cfDNA可以是源自正常细胞中不存在的受损核酸序列的cfDNA。在此,正常细胞中不存在的受损核酸序列可能包含由于一部分基因的缺失、重复、倒位或易位而引起的结构异常。另外,受损核酸序列可以包含由于一部分核酸的错配而引起的结构异常,并且可以是由于核酸的部分序列的突变而引起的单核苷酸位点变异(SNV)。受损核酸序列可以具有至少一种基因的突变序列,所述基因选自由EGFR、KRAS、BRAF、TP53、PIK3CA、ROS1、RET、c-Met、PTEN、RB1、AR、BRCA、KIT、FGFR、IDH、ESR1、HER2、ALK-EML4和TMPRSS2-ERG组成的组。
具体而言,正常细胞中不存在的受损核酸序列可能是由以下原因之一引起的:1)单链裂解,2)双链裂解,3)复制叉停滞,4)核酸错配,5)染色体畸变,6)链内交联,7)链间交联,8)外源基因插入,9)部分基因缺失,10)部分核酸发生取代,11)核酸倒位,12)胸苷二聚体的形成,13)脱氨基,14)基因重复,15)染色体易位,或16)碱基缺乏(AP位点)。特别地,在肿瘤细胞中,双链DNA(dsDNA)经常被损坏,并且受损的dsDNA可能包含在肿瘤细胞中发现的特定结构内。特别地,由于核酸的错配,受损核酸序列可以具有摇摆碱基对。
如本发明所用,术语“探针”是指用于检测靶cfDNA的DNA或RNA。探针可以设计为具有能够与不稳定的cfDNA互补结合的序列。如本发明所用,术语“具有与cfDNA互补的序列的探针”是指具有能够特异性结合待检测的cfDNA的核酸序列的探针,其具有靶标双螺旋结构并且存在于血浆中。
在此,可以通过两种方式生产探针。一种是设计成能够与已经发生损伤的基因的一部分结合的第一探针(以下称为CP),另一种是设计成能够与基因的一部分受损部位的周边结合的第二探针(以下称为DP)。DP可以被设计成能够与距离靶DNA序列或已经发生损伤的区域有10bp-100bp,或20bp-50bp远的序列发生特异性结合。
在本说明书中,已经确认不仅当同时使用第一探针和第二探针时可以有效地检测到受损的cfDNA,而且当单独使用第一探针或第二探针时也能够有效地检测到受损的cfDNA。此外,探针可以是与生物素等物质结合的形式,以便结合标志物。或者,探针可以直接与标志物结合或者通过接头与标志物结合。在此,标志物可以是纳米颗粒、荧光染料、荧光蛋白或酶。另外,探针和标志物可以同时添加,也可以依次添加。
在本发明的一个实施方案中,能够与靶cfDNA互补结合的探针可以特异性地结合到包含对以下各种癌细胞类型具有特异性的序列的区域。例如,对于卵巢癌或乳腺癌具有特异性的序列可以是BRCA1外显子7、BRCA1外显子10、BRCA1外显子11或BRCA1外显子15中存在的SNP。另外,对于胃癌具有特异性的序列可以是TP53中存在的SNP,以及对结肠直肠癌具有特异性的序列可能是MSH2中存在的SNP。对肺癌具有特异性的序列可以是EGFR中存在的SNP。另外,对肝癌具有特异性的序列可以选自FGFR3中存在的SNP。
[表1]
如本发明所用,术语“分离的生物样品”是指已经从人体分离的尿液、唾液、脑脊液、胸膜液、腹水、血浆、血液、痰液或体液的样品。分离的生物样品可以是从人体分离的液体样品。在此,血浆可以从血液中获得。
如本发明所用,术语“带正电的物质”是指可以以纳米颗粒、纳米线、网状结构或过滤器的形式使用的物质。然而,带正电的物质的形状不限于此。“带正电的物质”的一个实施方案可以是带正电的纳米线或带正电的膜。可以使用导电聚合物来生产纳米线。导电聚合物可以是选自由聚(乙炔)、聚(吡咯)、聚(噻吩)、聚(对苯基)、聚(3,4-乙撑二氧噻吩)、聚(苯硫醚)、聚(对亚苯基亚乙烯基)和聚苯胺组成的组中的任何一种。根据不同的生产方法,可以适当地调节纳米线的长度和直径。在一个实施方案中,纳米线可以具有200nm的直径和18μm的长度。另外,在生产过程中可以使纳米线包含生物素。
纳米线的表面可以用阳离子聚合物改性。阳离子聚合物的类型不受限制。阳离子聚合物的一个实施方案可以是聚乙烯亚胺(PEI)或聚赖氨酸(PLL)。另外,阳离子聚合物可以是阳离子支链聚合物聚乙烯亚胺。用这种阳离子聚合物改性的纳米线可以具有带正电的表面。
在一个实施方案中,带正电的纳米线即使在低浓度下也可以成功且有效地捕获cfDNA。特别地,带正电的纳米线由于其具有一些特征例如与用于结合到包括DNA的靶分子的大表面积和用于促进与DNA相互作用的提高的迁移性,从而可以有效地捕获cfDNA。
如本发明所用,术语“标志物”是指用于有效检测具有不稳定的双螺旋结构的cfDNA的物质,并且可以具体包括量子点、降解特定底物并引起显色反应的物质以及当用特定波长的光照射时会导致发光的物质。具体地,标志物是荧光蛋白,并且可以是绿色荧光蛋白(GFP)、黄色荧光蛋白(YFP)、红色荧光蛋白(RFP)或青色荧光蛋白(CFP)。或者,标志物可以是能够将选自由ABTS、OPD、AmplexRed、DAB、AEC、TMB、高香草酸和鲁米诺组成的组中的任何一种底物转化为有色物质的物质,例如辣根过氧化物酶(HRP)。
标志物可以进一步包含能够与探针结合的物质。具体地,当生物素与探针结合时,标志物可以进一步包含选自由亲和素、链霉亲和素或其组合组成的组中的任意一种。在一个实施方案中,这种标志物可以以结合有链霉亲和素和HRP的纳米颗粒的形式使用,该纳米颗粒由导电聚合物和透明质酸组成。此处,导电聚合物如上所述,并且可以优选为聚吡咯。在另一个实施方案中,标志物可以以结合有链霉亲和素和荧光蛋白的纳米颗粒的形式使用,该纳米颗粒由导电聚合物和透明质酸组成。
<不稳定的cfDNA的检测方法>
在本发明的一个方面,提供了一种从未经扩增的样品中检测不稳定的无细胞DNA的方法,该方法包括将不稳定的cfDNA与结合了标志物的探针混合的步骤。
这种不稳定的cfDNA可以是源自肿瘤的循环肿瘤DNA(ctDNA)。在此,ctDNA可能如上所述具有受损的基因序列。另外,可以从表达高度活跃的基因中分离出ctDNA。在此,样品可以是生物样品,并且可以是从人体分离的样品。具体而言,样品可以是尿液、脑脊液、血浆、血液、胸膜液、腹水、唾液、痰液或体液。
因此,可以通过在以下任意条件下对样品进行处理的步骤,在与探针的反应性方面将不稳定的cfDNA与稳定的cfDNA区分开。具体而言,可以在选自以下的任意一种条件下进行处理:i)在室温下放置1-120分钟的条件;ii)在90℃-95℃下加热1秒至3分钟的条件;iii)在75℃-90℃下加热1秒至5分钟的条件;iv)在60℃-75℃下加热30秒至60分钟的条件;v)在25℃-40℃下加热10-120分钟的条件;vi)用蛋白酶处理10秒至30分钟的条件;以及vii)用DNase处理10秒-30分钟的条件,然后与15聚体(15-mer)至30聚体(30-mer)的探针进行结合反应,具有稳定的双螺旋结构的cfDNA不与该探针结合。在此,“室温”是指环境温度,可以为18℃-25℃。探针可以包含15聚体(15-mer)到30聚体(30-mer)的核苷酸,或20聚体(20-mer)到25聚体(25-mer)的核苷酸。在此,探针可以被设计为能够与不稳定的cfDNA的基因序列互补地结合。
当在以上任意条件下对稳定的cfDNA进行处理时,由于稳定的cfDNA在其中形成了牢固的双链,因此不会发生变性并且不会与探针互补结合。然而,在上述任意条件下进行处理的情况下,不稳定的cfDNA可能会与探针结合。这种不稳定性是由于以下事实:cfDNA中的某些核苷酸无法形成互补结合,因此cfDNA具有改变的双螺旋结构。
在本发明的另一方面,提供了一种从未经扩增的样品中检测具有不稳定的双螺旋结构的cfDNA的方法。在此,该方法包括:a)将含有cfDNA的样品与带正电的物质混合的步骤;b)分离与cfDNA结合的带正电的物质的步骤;c)将混合物与探针和标志物混合的步骤;d)去除不与cfDNA结合的探针和标志物;e)检测标志物。
具体地,该方法可以包括将含有cfDNA的样品与带正电的物质混合的步骤。
样品可以是如上所述的生物学样品。其一个实施方案可以是血浆或尿液。在血浆或尿液中,可以同时存在具有正常的双螺旋结构的cfDNA和具有不稳定的双螺旋结构的cfDNA。另外,带正电的物质可以具体地是带正电的纳米线。带正电的纳米线如上所述。
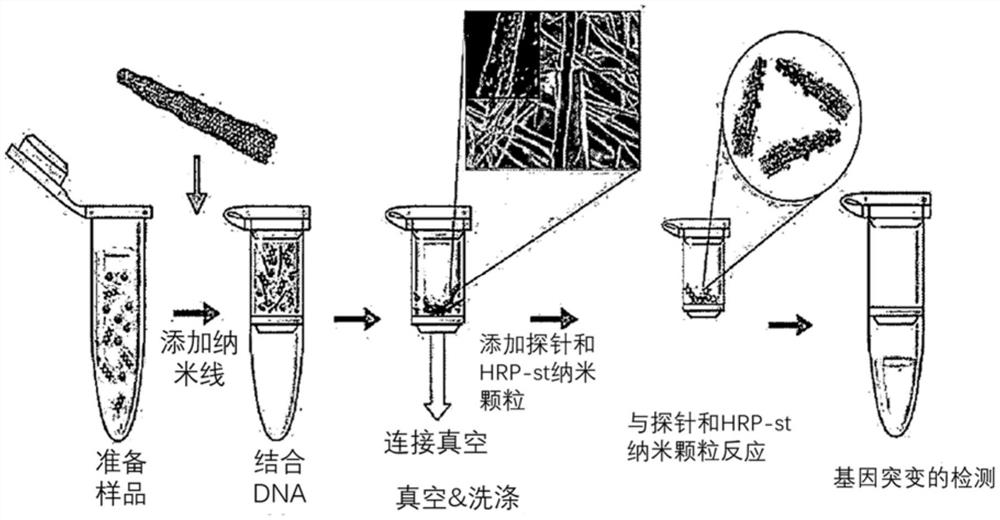
具体地,可以按照以下顺序并且在以下条件下执行以上步骤。首先,在收到患者的血浆、尿液、唾液、痰液等后,立即在4℃下以3,000×g离心10分钟。之后,将患者的血浆、尿液、唾液、痰液等以一定比例稀释在DPBS中。然后,在样品为血浆的情况下,将30μl血浆与120μl蒸馏水(DW)混合并置于旋转柱(G型或Q型)中。向其中添加表面用聚乙烯亚胺改性的聚吡咯(PEI/Ppy)纳米线(20μl),并使用热混合器在室温下以1200rpm的速度混合20分钟。
接着,该方法可以包括分离与cfDNA结合的带正电的物质的步骤。
分离带正电的物质的方法可以通过离心或施加负压例如真空来进行。cfDNA与带正电的物质结合。因此,当将与带正电的物质混合的样品置于旋转柱或真空柱中并进行离心分离或施加负压时,诸如纳米线之类的带正电的物质不会通过柱中的过滤器,而生物样品中的其他成分会通过过滤器。因此,可以通过诸如离心或真空的方法分离与cfDNA结合的带正电的物质。另外,为了从此类分离的纳米线中去除杂质,可以额外地执行清洗步骤一至三次。可以适当地使用常规方法进行洗涤。
具体地,可以按照以下顺序并且在以下条件下进行上述步骤。将旋转柱安装在用于施加真空的装置上,然后在550mBar的压力下进行抽吸。洗涤时,向其中加入400μl的1l0中加入,并再次进行抽吸。可以将同一过程再重复一次。
在一个实施方案中,当进行温度处理时,已经完成抽吸的旋转柱可以放置在预热至95℃的加热块中,在95℃下孵育1分钟,然后立即从中取出。在不需要温度变性步骤的条件下,样品可以不经过此过程。
接着,该方法可以包括将混合物与探针和标志物混合的步骤。
如上所述,探针可以由15聚体到30聚体的核苷酸,或者由20聚体到25聚体的核苷酸组成。探针的序列可以被设计成能够与待检测的具有不稳定的双螺旋结构的cfDNA互补结合。特别地,具有不稳定的双螺旋结构的cfDNA可以是肿瘤来源的ctDNA。另外,可以将具有不稳定的双螺旋结构的cfDNA设计成能够与称为癌症生物标志物的基因互补结合。
此外,标志物可以是在特定条件下被检测到的物质,例如荧光蛋白或HRP。在此,标志物可以包含能够与探针结合的物质。具体地,当生物素与标志物结合时,亲和素或链菌亲和素可以与标志物结合。在一个实施方案中,为了增加检测的灵敏度,标志物可以为纳米颗粒的形式,其中几个HRP分子和几个链霉亲和素分子被聚集。
具体地,可以按照以下顺序并且在以下条件下进行上述步骤。将适用于各个实验的探针(200μl)和HRP/STR纳米颗粒溶液(200μl)分别置于旋转柱中。在室温下以850rpm至1,000rpm的速度混合30分钟。
接下来,该方法可以包括去除未与cfDNA结合的探针和标志物的步骤。
这是去除未与cfDNA结合的探针的步骤。15聚体到30聚体的探针也带负电荷,可以与带正电的纳米线结合。因此,混合反应完成后,应从反应溶液中除去残留的探针和标志物。在此,可以通过离心或负压去除探针和标志物。此处,与短链探针相比,cfDNA与带正电的物质(特别是带正电的纳米线)能够强烈结合。在除去探针和标志物的步骤中,cfDNA与带正电的物质结合,因此不会被除去。
可以具体按照以下顺序进行上述步骤的实施方案。在执行上述步骤之后,向旋转柱施加负压,并进行抽吸。然后,向其中添加400μl的1×DPBS,并再次进行抽吸。可以将同一过程再重复一次。
最后,该方法可以包括检测标志物的步骤。
根据所使用的标志物,可以按不同方式执行检测标志物的方法。标志物的检测可以通过颜色的变化、UV吸收率的变化、荧光响应的变化或电化学的变化来进行测量。例如,当HRP用作标志物时,可以通过观察显色反应来检测标志物。另外,当标志物是诸如GFP的荧光蛋白时,可以通过用特定波长的光照射标志物后观察检测到的光来检测标志物的存在或不存在。
可以具体按照以下顺序进行上述步骤的实施方案。向离心柱中依次添加200μl乙酸钠缓冲液(0.2M,pH 7.0),50μl TMB(10mM)和50μl H2O2(0.1M)。然后,孵育3分钟。之后,以3,500rpm至5,000rpm的速度离心30秒。将收集在收集管中的溶液以每孔200μl转移到96孔中,然后使用UV/VIS分光光度计测量500nm至850nm波长范围内的吸光度。
此外,该方法可以另外包括额外的处理过程,以增加具有正常的双螺旋结构的cfDNA和具有不稳定的双螺旋结构的cfDNA与探针的反应性差异。可以在获得样品后或分离cfDNA之后进行额外的处理过程。
另外,在步骤c)之前,所述方法可以进一步包括以下步骤:使结合到带正电的物质上的样品或cfDNA在选自以下的任一条件下变性的步骤:i)在室温下放置1-10分钟的条件;ii)在90℃-95℃下加热1秒至1分钟的条件;iii)在75℃-90℃下加热10秒至3分钟的条件;iv)在60℃-75℃下加热1-30分钟的条件;v)在25℃-40℃下加热5-60分钟的条件;vi)用蛋白酶处理1-10分钟的条件;vii)用DNase处理1-10分钟的条件。此类变性过程不会使稳定的cfDNA发生变性,并且会使不稳定的cfDNA更不稳定地变性,从而不稳定的cfDNA可以更容易地结合到探针上。在获得样品后,可以在上述条件下进行变性。另外,可以在获得与带正电的物质结合的cfDNA之后进行变性。另外,可以适当地调节温度、蛋白酶和用DNase处理的时间,只要不使稳定的cfDNA变性即可。
在本发明的一个实施方案中,已经确定待检测的具有不稳定的双螺旋结构的cfDNA可以使用靶探针来检测而无需单独的变性步骤(图31)。此外,已经确定即使对cfDNA进行了温度处理过程,具有稳定的双螺旋结构的cfDNA和具有不稳定的双螺旋结构的cfDNA也仍显示出与相同探针的不同结合反应(图32和33)。
<通过检测不稳定的cfDNA来提供诊断信息的方法>
在本发明的又一方面提供了一种方法,该方法通过从未经扩增的样品中检测具有不稳定的双螺旋结构的cfDNA来提供用于诊断或预测癌症和传染性疾病的信息。在此,该方法包括:a)将含有cfDNA的样品与带正电的物质混合;b)分离与cfDNA结合的带正电的物质;c)将混合物与探针和标志物混合;d)去除不与cfDNA结合的探针和标志物;e)检测标志物;f)当检测到标志物时,确定存在与具有不稳定的双螺旋结构的cfDNA对应的基因相关的癌症或传染性疾病的步骤。具体地,用于检测不稳定的cfDNA的方法如上所述。
本说明书中使用的癌症的一个实施方案可以是选自由膀胱癌、骨癌、血液癌、乳腺癌、黑色素瘤、甲状腺癌、甲状旁腺癌、骨髓癌、直肠癌、喉癌、喉肿瘤、肺癌、食道癌、胰腺癌、结肠直肠癌、胃癌、舌癌、皮肤癌、脑瘤、子宫癌、头颈癌、胆囊癌、口腔癌、结肠癌、肛周癌、中枢神经系统肿瘤、肝癌和结肠直肠癌组成的组中的任意一种。特别地,癌症可以是胃癌、结肠直肠癌、肝癌、肺癌或乳腺癌。
在一个实施方案中,上述癌细胞中特异性存在的序列可以是癌细胞中存在的SNP。对于胃癌,癌症中特异性存在的序列的一个实施方案可以是肿瘤抑制基因p53和PTEN中的突变。另外,对于结肠直肠癌,其一个实施方案可以是APC和MSH2基因中的突变。另外,对于肝癌,由于其主要原因是感染了乙型肝炎病毒(HBV)或丙型肝炎病毒(HCV),因此HBV或HCV核酸可能成为靶标。另外,对于肺癌,表皮生长因子受体(EGFR)基因的突变可能成为靶点;对于乳腺癌,BRCA1/2基因的突变可能是主要的靶标。另外,对于宫颈癌,来源于人乳头瘤病毒DNA(HPV DNA)的cfDNA可能成为靶标。
不稳定的dsDNA的另一个实施方案可以是选自由EGFR、KRAS、BRAF、TP53、PIK3CA、ROS1、RET、c-Met、PTEN、RB1、AR、BRCA、KIT、FGFR、IDH、ESR1、HER2、ALK-EML4和TMPRSS2-ERG组成的组中的至少一种基因突变。
<用于检测不稳定的cfDNA的装置>
在本发明的又一个方面,提供了一种用于检测在室温下具有不稳定的双螺旋结构的cfDNA的装置,其包括:a)混合部分,用于将包含cfDNA的样品与带正电的纳米线混合;b)获取部分,用于除去与cfDNA结合的纳米线以外的样品;c)反应部分,用于向结合有cfDNA的纳米线中添加能够互补结合至所述cfDNA的结合有生物素的探针以及包含有链霉亲和素和标志物的纳米颗粒;d)检测部分,用于检测标志物;e)信息处理部分,用于确定样品中含有cfDNA,所述cfDNA具有与检测探针互补的序列,并且具有根据标志物的检测在室温下不稳定的双螺旋结构。
<使用不稳定的cfDNA检测的诊断装置>
在本发明的又一个方面,提供了一种装置,该装置通过从未经扩增的样品中检测具有不稳定的双螺旋结构的无细胞DNA来提供用于诊断或预测癌症或传染性疾病的信息。在此,该装置包括:a)混合部分,用于将包含cfDNA的样品与带正电的纳米线混合;b)获取部分,用于除去与cfDNA结合的纳米线以外的样品;c)反应部分,用于向结合有cfDNA的纳米线中添加能够互补结合至所述cfDNA的结合有生物素的探针以及包含有链霉亲和素和标志物的纳米颗粒;d)检测部分,用于检测标志物;e)信息处理部分,用于确定样品中含有cfDNA,所述cfDNA具有与检测探针互补的序列,并且具有根据标志物的检测在室温下不稳定的双螺旋结构。
本发明基于稳定的cfDNA和不稳定的cfDNA由于其热力学稳定性的差异而与探针的反应性的差异。在此,带正电的纳米线可以与基因组DNA和cfDNA结合。然而,由于其结合力和大小的差异,基因组DNA在洗涤时会从带正电的纳米线上分离出来。另外,在一个实施方案中,当纳米线被修饰时,可以使其包含生物素。然而,在用阳离子聚合物聚乙烯亚胺修饰纳米线的表面的过程中,包含在纳米线中并暴露在纳米线的表面上的生物素与阳离子聚合物结合。另外,纳米线被阳离子聚合物涂覆,因此含有链霉亲和素的标志物不与纳米线结合。另外,由于探针也带负电,因此它可以与带正电的纳米线结合。但是,已发现由于探针的结合力比cfDNA弱,因此在洗涤过程中会被除去。
另外,用能够特异性结合包含受损DNA序列的区域的探针(CP)和能够特异性结合不含受损DNA序列的周边区域的探针(DP)处理稳定的cfDNA和不稳定的cfDNA。结果,有可能在具有不稳定的双螺旋结构的cfDNA与具有稳定的双螺旋结构的cfDNA之间鉴定出与探针的不同结合反应。从这些结果中发现,具有不稳定的双螺旋结构的cfDNA不仅可以结合对受损DNA序列具有特异性的探针,而且可以结合能够与周边DNA序列特异性结合的探针。另外,已经证实仅使用一种探针就可以检测到不稳定的cfDNA。
在下文中,将通过以下实施例更详细地描述本发明。然而,以下实施例仅旨在解释说明本发明,但本发明的范围不局限于此。
I.实验方法以及纳米线、标志物和探针的生产
实验方法1.检测不稳定的cfDNA的方法
步骤1:样品制备和纳米线的添加
收到患者的血浆、尿液、唾液、痰液等后,立即在4℃下以3,000×g离心10分钟。将患者的血浆、尿液、唾液、痰液等以一定比例稀释在DPBS中。当为血浆时,将30μl血浆与120μl的DW混合并置于离心柱(G型或Q型)中。向其中添加PEI/Ppy纳米线(20微升),并使用热混合器在室温下以1200rpm的速度混合20分钟。
步骤2:真空/清洗/温度变性
将旋转柱安装在真空抽吸装置上,然后在550mBar下进行抽吸。向其中添加400μl的1×DPBS,并再次进行抽吸。再重复一次相同的过程。仅通过两步过程获得的纳米线-DNA复合物被捕获在离心柱中。如果需要温度变性,将已经完成吸力的旋转柱放在预热至95℃的加热块中,在95℃孵育1分钟,然后立即从中取出。不需要温度变性步骤的样品不经过此过程。
步骤3:添加探针和HRP/STR NP
将适合各个实验的HRP/STR NP的探针(200μl)和溶液(200μl)分别置于离心柱中。使用热混合器在室温下以850rpm-1,000rpm的速度混合30分钟。将旋转柱安装在真空装置上,然后进行抽吸。向其中加入400μl的1×DPBS,并再次进行抽吸。再重复一次相同的过程。
步骤4:TMB响应以检测基因突变
用新的收集管替换旧收集管后,使用注射泵将200μl乙酸钠缓冲液(0.2M,pH 7.0)和50μl H2O2(0.1M)依次添加到离心柱中。然后,孵育3分钟。孵育结束时,将离心柱在3500rpm-5,000rpm的速度下离心30秒。以每孔200μl将收集在收集管中的溶液转移至96孔中,然后使用UV/VIS分光光度计测量在500nm-850nm的波长范围内的吸光度。
制备实施例1.带正电的纳米线的制备
如图1中所示,产生表面缀合有作为阳离子聚合物的聚乙烯亚胺(PEI)的纳米线。使用Q150T模块化涂层系统(Quorum Technologies,UK)在5×10
为了制备表面用阳离子聚合物处理过的纳米线(PEI/Ppy NWs),通过在1.0V(vs.Ag/AgCl)下对AAO模板的孔施加7分钟计时电流法以及包含0.01M的聚(4-苯乙烯磺酸)和1mg/ml的生物素的0.01M的吡咯溶液,以进行电化学沉积。
将所得的AAO模板用蒸馏水洗涤数次,浸入2M氢氧化钠(NaOH)溶液中3小时,然后放入Bioruptor UCD-200(Diagenode)中进行超声处理,以获得掺杂有生物素分子的自立式Ppy NW。然后,向所得纳米线中添加30mM N-(3-二甲基氨基丙基)-N-乙基碳二亚胺盐酸盐(EDC)和6mM N-羟基琥珀酰亚胺(NHS),从而活化羧酸(-COOH)基团。随后,在添加PEI溶液之后,使反应在室温下进行1小时并且用水洗涤。将表面结合有聚乙烯亚胺的纳米结构(PEI/Ppy NW)分散在去离子水中,并保持在室温下直至使用。
使用这种制备方法,在选择性溶解AAO模板后,从AAO模板中释放出每个聚吡咯(Ppy)纳米线,并通过生物素-链霉亲和素相互作用将阳离子支化聚乙烯亚胺(PEI)(25kDa)偶联到纳米线上。
制备实施例2.表面用阳离子聚合物处理过的纳米颗粒(PEI/Ppy NPs)的制备
为了制备表面缀合了聚乙烯亚胺的纳米颗粒(PEI/Ppy NP),将0.2g聚乙烯吡咯烷酮(PVP)添加到12.5ml三重蒸馏水中,并搅拌30分钟。然后,向其中加入65μl吡咯,然后将所得物进一步搅拌10分钟。然后,向其中加入浓度为0.75g/ml的0.5ml FeCl
使用具有50,000MWCO孔径的膜对三重蒸馏水进行透析2天。以1200rpm离心3分钟以去除大尺寸的颗粒聚集体,然后将所得物冻干。将200μg的Ppy-HA-NP添加至1ml的三重蒸馏水中。然后,向其中加入100mM EDC/50mM NHS溶液,并反应45分钟,以使透明质酸的羧基活化。以15,000rpm离心10分钟以除去上清液,在此期间进行两次洗涤。
随后,向其中加入100mg/ml的PEI溶液(溶剂:0.2M碳酸氢钠),并在4℃下反应过夜。然后,以15,000rpm离心10分钟以除去上清液,接着将所得物保持在三重蒸馏水中。使用扫描电子显微镜观察其表面缀合有聚乙烯亚胺的纳米颗粒(PEI/Ppy NPs)的形状。如图6A所示,用扫描电子显微镜图像(比例尺为200μm)检查与PEI结合的平均为50nm的纳米颗粒(NP)的形状。
制备实施例3.用HRP和链霉亲和素标记的聚吡咯纳米颗粒的制备
为了制备包含HRP和链霉亲和素的纳米颗粒,将0.5g聚乙烯吡咯烷酮(PVP)添加到12.5ml三重蒸馏水中,并搅拌30分钟。然后,向其中加入65μl吡咯,并将其进一步搅拌10分钟。此后,向其中加入浓度为0.75g/ml的0.5ml FeCl
使用具有50,000MWCO孔径的膜对三重蒸馏水进行透析2天。以1200rpm离心3分钟以去除大尺寸的颗粒聚集体,然后将所得物冻干。将200μg含聚吡咯和透明质酸的Ppy-HA-NP加入1ml三重蒸馏水中。然后,向其中加入100mM EDC/50mM NHS溶液,并反应45分钟,以使透明质酸的羧基活化。以15,000rpm离心10分钟以去除上清液,在此期间进行两次洗涤。将1mg HRP和1mg链霉亲和素添加到Ppy-HA-NP中,并在4℃的温度下混合。随后,以15,000rpm离心10分钟以去除上清液,然后将所得物保持在三重蒸馏水中。使用扫描电子显微镜观察其表面上缀有HRP和链菌亲和素的纳米颗粒(HRP/st-标记的NP)的形状(图4)。
制备实施例4.探针的制备
制备探针以检测具有不稳定的双螺旋结构的cfDNA。探针的制备因待检测的cfDNA而异。在此,探针以生物素与之结合的形式进行制备。探针分为两种类型,即,与包含引起不稳定双螺旋结构的受损DNA的区域互补结合的第一探针(下文称为CP),与受损DNA的周边区域互补结合的第二探针(下文称为DP)。
II.使用两个探针检测cfDNA
实施例1.尿液中HPV衍生的cfDNA的分析
为了从尿液样本中分离出无细胞的HPV DNA,将10μg/ml的纳米线(PEI/Ppy NW)的表面用实施例1中生产的阳离子聚合物处理过后,加入从HPV阳性患者获得的200μl尿液中,在室温下混合30分钟。分离的cfDNA在95℃变性1分钟。然后,加入1pM在其末端结合有生物素的第一探针(CP)和第二探针(DP),并在37℃下反应1小时。然后,将以辣根过氧化物酶(HRP)和链霉亲和素标记的聚吡咯纳米颗粒(以下称为HRP/st-标记的NP)添加到样品中,并在37℃下孵育30分钟。
随后,向HPV衍生的cfDNA中添加25μl的10mM 3,3',5,5'-四甲基联苯胺(TMB)、25μl的0.1M H2O2和200μl的0.2M乙酸钠三水合物缓冲液(pH 5.0)。使反应与DNA样品在黑暗中于室温下进行3分钟。为了鉴定HPV DNA的浓度和吸光度之间的相关性,使用DU 730紫外可见分光光度计(Beckman Coulter,USA)在652nm波长下进行UV-Vis(紫外可见)的检测。
结果显示,可以识别通过放大比色信号而获得的结果,放大的程度使得可以视觉观察比色信号。下表2显示了用于检测HPV的探针序列。
表2
此外,进行无PCR比色测定,以评估HPV阳性宫颈癌患者(HPV16(+)和HPV18(+))和HPV阴性健康对照(HPV-)的尿液样本。使用PBS作为阴性对照。结果如图12所示,将使用纳米线分离的靶标HPV DNA与CP和DP混合,并向其中添加HRP/st标记的NP。然后观察到颜色变化。在95℃下变性1分钟以分析分离的cfDNA。
总共收集并检测了24份HPV阳性和HPV阴性的尿液样本。结果鉴定出从HPV阳性宫颈癌患者(HPV16(+)和HPV18(+))和HPV阴性健康对照(HPV-)的尿液中分离的cfDNA具有不同的吸光度值(图13)。此处,使用与HPV16或HPV18特异性结合的探针,在95℃下变性1分钟以分析分离的cfDNA。
此外,当使用非HPV探针例如EGFR 19和EGFR 21时,未观察到任何反应。并且发现由于靶标HPV及其互补探针之间的类型特异性结合而导致了颜色变化和紫外线吸收率变化(图14)。此处,在95℃下变性1分钟以分析分离的cfDNA。
实施例2.通过肺癌细胞系的DNA分析鉴定EGFR基因的突变
实施例2.1通过纳米线鉴定cfDNA的大小
将低范围(10bp-100bp)、中范围(100bp-2kb)和高范围(3.5kb-21kb)的DNA梯状条带添加到正常受试者的血浆中,然后使用纳米线(PEI/Ppy NW)检查捕获效率。已确定纳米结构可有效捕获小尺寸DNA(图15)。
使用纳米线,从H1975(具有EGFR外显子20 T790M、21 L858R基因突变的细胞系)、HCC2279(具有EGFR外显子19缺失基因突变的细胞系)和A549细胞系(无EGFR外显子基因突变的细胞系)获得基因。然后,将未经超声处理的基因组DNA(gDNA)和经过超声处理的片段DNA(fDNA)用于比较纳米结构捕获的效率。结果发现使用纳米结构时,观察到fDNA的捕获效率比gDNA高(图16和17)。
实施例2.2通过分析不稳定的cfDNA检测EGFR突变
从EGFR外显子20、21阳性的H1975细胞系、EGFR外显子19阳性的HCC2279细胞系和无EGFR基因突变的A549细胞系中,使用纳米结构进行了无PCR比色测定,以分离gDNA和fDNA。之后,向其中添加对EGFR外显子19、20和21具有特异性的探针,并进行混合。然后向其添加HRP/st标记的NP,并观察到颜色变化。结果可以确定纳米结构在捕获小尺寸DNA(即fDNA)方面更为有效,并且与gDNA相比具有明显的颜色变化和UV-Vis光谱变化(图18和图19)。
实施例2.3使用荧光染料检测cfFNA
为了鉴定是否可以使用靶标探针从肺癌患者的血浆样本中检测到基因突变,通过使用荧光染料检查了这种可能性。对H1975(具有EGFR外显子20 T790M、21 L858R基因突变的细胞系)、HCC2279(具有EGFR外显子19缺失基因突变的细胞系)和A549细胞系(无EGFR外显子基因突变的细胞系)进行超声处理,然后通过纳米线捕获获得的DNA(fDNA)。之后,将fDNA在95℃变性1分钟,然后与对EGFR19、20和21具有特异性的探针混合。使用与探针结合的荧光染料(Alexa488)检查细胞系的基因突变。
结果显示,在A549细胞系中,当与对EGFR 19和21具有特异性的探针反应时,未检测到荧光。但是,在H1975和HCC2279中,对EGFR 19和20具有特异性的探针可以检测到荧光(图20)。
实施例2.4通过体外cfDNA分析确定EGFR基因突变的检出限(LOD)
对H1975(具有EGFR外显子20 T790M、21 L858R基因突变的细胞系)和HCC2279(具有EGFR外显子19缺失基因突变的细胞系)进行超声处理,然后将获得的片段化DNA以不同浓度(fDNA;1ag ml-1至100ng ml-1)添加到健康人的血浆中(200μl)。然后,通过纳米线捕获了fDNA。此后,在95℃下变性1分钟,然后使用对EGFR外显子19 Del和EGFR外显子20 T790M具有特异性的探针和HRP/st标记的NP检查检测限(LOD)。结果鉴定出当施加3倍的信噪比时,HCC2279细胞系的fDNA可检测到高达10ag ml-1,而H1975细胞系的fDNA可检测到高达100ag ml-1(图21和22)。
实施例3.通过分析血浆或脑脊液样品(肺癌患者)中存在的cfDNA,鉴定EGFR、KRAS和ALK-EML4基因的突变
实施例3.1通过分析不稳定的cfDNA鉴定EGFR基因突变
在一个实施方案中,使用纳米结构从癌症患者的体液样品中分离cfDNA,并通过与探针杂交并随后与HRP/st标记的纳米颗粒结合来检测基因突变,总共需要60分钟(图6)。
为了从血浆或脑脊液样本中分离出cfDNA,在室温下将200μl EGFR阳性患者的血浆和5μg/ml PEI/Ppy NW混合30分钟,以从患者血浆中提取cfDNA。通过Bioanalyzer分析使用PEI/Ppy NW从200μl肺癌患者血浆中提取的cfDNA的大小(图23)。一般而言,与已知与癌症相关的cfDNA的平均大小为166bp的事实类似,在本生物分析仪实验中,已确定结果显示使用PEI/Ppy NW从肺癌患者血浆中提取的cfDNA在169bp处观察到一个峰。
随后,将在纳米结构中捕获的DNA在95℃下变性1分钟,向其中加入1pM的生物素结合的CP和生物素结合的DP,并在37℃下反应1小时。然后,将用HRP和链霉亲和素标记的聚吡咯纳米颗粒(HRP/st标记的NP)添加到样品中,并在37℃下反应30分钟。
此后,向反应溶液中加入25μl 10mM TMB,25μl 0.1M H2O2和2001 0.2M乙酸钠三水合物缓冲液(pH 5.0)。然后,遮光,使反应在室温下发生3分钟。之后,使用DU 730UV-Vis分光光度计(Beckman Coulter,USA)在652nm的波长下进行UV-Vis检测。
肉眼观察到由TMB的氧化反应引起的颜色差异。下表3中显示了用于捕获和检测具有EGFR外显子19缺失、20 T790M和21 L858R的突变cfDNA的探针序列。
表3
在本实验中,除非另有说明,否则将EGFR外显子19缺失-探针1(靶标特异性)、EGFR外显子20 T790M-探针2(靶标特异性)和EGFR外显子21 L858R-探针2(靶标特异性)用作CP和DP。结果确定了根据检测探针特异性检测了分离的EGFR突变的cfDNA(图24和25)。已确定,当从患有EGFR外显子19缺失、20 T790M或21 L858R基因突变的肺癌患者血浆中捕获的cfDNA分别与靶向EGFR外显子19缺失、20 T790M或21L858R的探针和与HRP/链霉亲和素结合的纳米颗粒(NP)反应时,当使用靶向与患者组织中相同的基因突变的探针时,会出现颜色变化和UV-Vis光谱变化(图24)。
另外,将从具有另一个EGFR突变的肺癌患者中捕获的cfDNA与靶向EGFR 19、20和21的相同探针反应。结果可以确定,当使用靶向与患者组织中相同基因突变的探针时,则会出现颜色变化和UV-Vis光谱变化(图25)。
实施例3.2通过cfDNA分析鉴定KRAS和ALK-EML4基因突变
此外,类似于EGFR,为了分析KRAS和ALK-EML4基因的突变,从血浆或脑脊液样本中分离出cfDNA,然后将捕获的DNA在95℃下变性1分钟。加入1pM的结合有生物素的CP和结合有生物素的DP,并使反应在37℃下进行1小时。然后,将标记有HRP和链霉亲和素的聚吡咯纳米颗粒添加到样品中,并在37℃下反应30分钟。
结果显示,可以鉴定出从患有KRAS外显子2基因突变和ALK-EML4融合基因突变的肺癌患者血浆中分离的cfDNA对KRAS外显子2的探针和ALK-EML4的探针有反应,并因此显示出与患者组织相匹配的颜色变化和紫外线吸收(图26-28)。下表4中显示了用于检测具有KRAS外显子2突变(图26和27)和ALK-EML4变体1和3(图28)的cfDNA的探针(CP和DP)的序列。
表4
实施例3.3通过温度变性鉴定非特异性反应
将表3中的三种探针(包括靶标特异性或靶标非特异性的探针)中的每一种添加至具有EGFR外显子19缺失和20 T790M基因突变的肺癌患者的血浆中,并进行混合。结果显示,在95℃下DNA变性1分钟后,所有使用的探针(即,无论探针是靶标特异性还是靶标非特异性)都仅针对EGFR外显子19缺失和20 T790M显示出了颜色和紫外线吸收率的变化,与组织类似,这使得识别特定的基因突变称为可能(图29)。但是,对于EGFR外显子21,无论探针类型如何,均未观察到颜色和紫外线吸收的变化。这表明不稳定的cfDNA对EGFR外显子19和EGFR外显子20突变的特异性探针有反应,从而可以分析基因突变。
另外,cfDNA是从无EGFR基因突变的肺癌患者血浆中获得的。分别在95℃变性1分钟和10分钟后,检查对EGFR 19、20和21突变具有特异性的探针与不稳定的cfDNA之间的结合。结果显示,在95℃变性1分钟的情况下,在无EGFR基因突变的肺癌患者中未观察到颜色变化和UV吸收率变化。但是,在95℃变性10分钟的情况下,在所有对EGFR外显子19、20和21特异性的探针中都可以观察到通过非特异性杂交引起的颜色变化和紫外线吸收率变化(图30)。
另外,通过纳米线从其他患有EGFR 19缺失和20 T790M基因突变的肺癌患者和正常受试者的血浆中捕获cfDNA。然后,通过在95℃下分别变性0、1和10分钟,检查其与对EGFR19、20和21具有特异性的探针的杂交反应性。结果如图31-33所示,在EGFR基因没有突变的正常受试者中,在95℃下变性0分钟(图31)和1分钟(图32)时,未观察到颜色变化和UV吸收率变化。然而,通过在95℃下变性10分钟(图33),在所有对EGFR外显子19、20和21具有特异性的探针中均观察到了通过非特异性杂交引起的颜色变化和紫外线吸收率变化。因此,可以确定cfDNA中的基因突变无需变性即可进行分析。
实施例4.通过分析肺癌患者的样品来鉴定检测不稳定的cfDNA的方法的准确性
在本发明的一个实施方案中,已经确定通过分析从151名肺癌患者血浆中获得的cfDNA中基因突变的特异性和敏感性获得的结果与患者癌症组织中的基因突变的结果相匹配(图34)。将对EGFR外显子19 Del具有特异性的探针添加到EGFR无突变的肺癌患者(EGFR野生型)、EGFR外显子19缺失的肺癌患者(Del)和EGFR外显子21 L858R的肺癌患者的cfDNA中,并且通过分析UV光谱的吸光度值(ΔOD,500nm-650nm)检测产生的基因突变。结果确定所获得的结果显示有98.4%与患者癌症组织中基因突变的结果相符(图35和36)。
此外,将对EGFR外显子21 L858R具有特异性的探针添加到EGFR无突变的肺癌患者、EGFR外显子19缺失(Del)的肺癌患者和EGFR外显子21 L858R的肺癌患者的cfDNA中,并通过分析UV光谱(ΔOD,500nm至650nm)的吸光度值来检测产生的基因突变。结果确定获得的结果显示出98.0%与患者癌症组织中基因突变的结果相符(图37和38)。
实施例5.使用带正电的纳米颗粒检测cfDNA
与上述EGFR实验类似,为了从具有EGFR 19缺失(Del)基因突变的肺癌患者的血浆样品中分离cfDNA,将在制备实施例中制得的PEI结合的纳米颗粒(PEI-Ppy NP,5μg/ml)加入到EGFR阳性患者的200μl血浆中,并在室温下混合30分钟。此后,捕获的DNA要么未变性,要么在95℃变性1分钟。然后,以1pM的量加入结合有生物素的CP和结合有生物素的DP,并在37℃下孵育30分钟。此后,将HRP/st标记的NP加入样品中,并在37℃下反应15分钟。通过TMB的氧化反应鉴定出仅在对EGFR 19具有特异性的探针中观察到UV-Vis光谱的吸光度变化(图39)。
实施例6.通过分析血浆中存在的cfDNA鉴定EGFR基因突变
使用H1975细胞系(具有EGFR外显子20 T790M和21 L858R基因突变),通过超声处理制备fDNA。此后,向其中添加通过制备实施例中的方法制备的表面结合有聚赖氨酸的纳米结构(PLL/Ppy NW),然后进行无PCR比色测定(图40)。使用纳米线分离了EGFR外显子20、21阳性H1975细胞系的fDNA。然后,向其中添加对EGFR外显子19、20和21具有特异性的探针,并使其发生反应。此后,向其中添加HRP/st-标记的NP,然后观察到颜色变化。如图40所示,仅在EGFR 20和EGFR 21中可以识别出清晰的颜色变化和UV-Vis光谱变化。
III.使用单个探针检测不稳定的cfDNA
实施例7.仅使用仅能结合受损部分的探针检测不稳定的cfDNA
从肺癌患者的血浆样品中分离出cfDNA。然后,为了检测基因突变,将两种类型的探针CP和DP混合使用。如图41-43所示,为了检测EGFR外显子19缺失、外显子20T790M和外显子21 L858R基因突变,将对含有上述区域的cfDNA具有特异性的探针CP_1(EGFR外显子19缺失)、CP2(EGFR外显子20 T790M)和CP2(EGFR外显子21 L858R)与DP混合使用。
但是,如图44所示,即使在不添加DP而仅单独添加CP的情况下,也可能在患者的癌组织中鉴定出相同的EGFR外显子19缺失基因突变结果(EGFR外显子19缺失)。
IV.根据样品处理方法鉴定不稳定的cfDNA的可检测性
实施例8.鉴定基于DNase或RNase处理的不稳定的cfDNA的可检测性
首先用DNase或RNase处理肺癌患者的血浆样品,然后使用纳米结构从中分离出cfDNA,以鉴定基因突变。如图45所示,在用RNase A对患有EGFR外显子19缺失基因突变的肺癌患者的血浆进行预处理后,用纳米线从中获得cfDNA,然后与对EGFR外显子19del具有特异性的探针反应。结果鉴定出与患者癌症组织相同的EGFR外显子19缺失基因突变的UV-vis峰,与对照类似(对照cfDNA)。但是,可以确定用DNase I预处理后,由于cfDNA可能降解,因此未观察到紫外线吸收(图45)。
类似地,如图46所示,在用RNase A预处理具有EGFR外显子20 T790M基因突变的肺癌患者的血浆之后,用纳米线从中获得cfDNA,然后与对EGFR外显子20 T790M具有特异性的探针反应。结果鉴定出与患者癌症组织相同的EGFR外显子20 T790M基因突变的紫外线吸收。但是,可以确定在用DNase I处理后未观察到紫外线吸收(图46)。这是由于cfDNA可能因在患者血浆中添加DNase I而被降解了。
V.根据标志物鉴定cfDNA的可检测性
实施例9.使用HRP/链霉亲和素复合物鉴定cfDNA的可检测性
如图47所示,从具有EGFR外显子19缺失和外显子20 T790M基因突变的肺癌患者血浆中提取cfDNA,并与对EGFR外显子19 Del、20 T790M和21 L858R具有特异性的探针以及HRP/st-标记的NP反应,然后通过紫外线吸收和颜色变化鉴定出与患者癌症组织中的结果相同的EGFR外显子19缺失和外显子20 T790M基因突变的结果。
但是,如图48所示,当从同一患者的血浆中提取cfDNA,然后与对EGFR外显子19Del、20 T790M、21 L858R具有特异性的探针以及HRP-链霉亲和素复合物(其中HRP和链霉亲和素以1:1的比例彼此结合)以代替HRP/st标记的NP进行反应时,基因突变的结果与患者癌症组织的结果完全不同。发现当使用HRP/链霉亲和素复合物代替HRP/链霉亲和素标记的纳米颗粒(NP)时,由于增加的非特异性结合,观察到了不准确的基因突变结果。
如图49所示,对从5名具有EGFR外显子19缺失和外显子20 T790M基因突变的肺癌患者血浆中提取cfDNA,然后将该cfDNA与对EGFR外显子19 Del、20 T790M和21 L858R具有特异性的探针,以及带有HRP/st标记的NP进行反应;并且与对EGFR外显子19 Del、20 T790M和21 L858R具有特异性的探针,以及HRP/链霉亲和素复合物(其中HRP和链霉亲和素以1:1的比例彼此结合)以代替HRP/st标记的NP进行反应获得的结果进行分析。结果显示,由于通过探针和HRP/st标记的NP的结合而引起的反应特异性的提高,通过纳米线获得的cfDNAs显示出与癌组织匹配的UV吸收。此外,已确定HRP/st标记的NP在确定基因突变中甚至在5名患有EGFR外显子20 T790M和21 L858R基因突变的肺癌患者的血浆中也起着重要作用。
VI.使用探针和标志物相互结合的复合物检测cfDNA
实施例10.使用结合有HRP/st标记的NP的探针检测基因突变
如图50所示,不同于将cfDNA分别与探针和HRP/st标记的NP进行顺序反应,而是将HRP/st标记的NP首先与对EGFR外显子19 Del、20 T790M和21 L858R具有特异性的探针结合,以产生探针-HRP标志物形式的结合产物,并使cfDNA与这种结合产物反应;结果发现,即使在具有EGFR外显子20 T790M和21 L858R基因突变的肺癌患者的胸膜液中也检测到与癌组织相同的基因型。
VII.基于混合顺序的cfDNA检测
实施例11.通过同时混合探针和标志物鉴定不稳定的cfDNA的检测
如图51所示,不同于将cfDNA分别与CP和DP探针以及HRP/st标记的NP进行顺序反应,而是将cfDNA与探针和HRP/st标记的NP一次混合在一起,并进行反应;结果发现,即使在具有EGFR外显子20 T790M和21 L858R基因突变的肺癌患者的血浆中也检测到与癌组织相同的基因型。此外,如图52所示,将cfDNA与CP和DP探针以及HRP/st标记的NP一次性混合在一起,并使得反应发生。结果显示,即使在具有ALK-EML4融合和ALK点突变(I1171N/T)基因突变的肺癌患者的血浆中,也检测出与癌组织相同的基因型。
VIII.根据样品变性条件检测不稳定的cfDNA
实施例12.样品变性后根据温度条件检测cfDNA
进行实验以确定不稳定的cfDNA和稳定的cfDNA是否可以根据样品变性条件进行区分。具体地,使用能够检测EGFR 19缺失的探针,对从正常受试者和肺癌患者中收集的血浆(0208-343,20190311_LC#1,来自组织:E19del)进行各种变性条件,然后检查不稳定的cfDNA和稳定的cfDNA是否能够彼此区分。
对于探针,使用能够检测EGFR外显子19缺失的探针ggaattaaga gaagcaacat ctcc(SEQ ID NO:9)。在此,使用生物素结合的探针。使用PEI/Ppy纳米线,并使用聚集了HRP/链霉亲和素的纳米颗粒作为标志物。
具体地,在用纳米线分离cfDNA之前,将样品在不同条件下进行如下处理。将样品分别在30℃加热15分钟和0分钟。此外,将样品分别在60℃加热5分钟和0分钟。此外,将样品分别在95℃加热1分钟和0分钟。其他步骤以图8所示的方法进行。
结果显示,在没有EGFR 19缺失突变的正常受试者中,在任何变性条件下均未检测到不稳定的cfDNA(图54)。但是,在E19del患者中,已确定在所有不同的变性条件下均检测到不稳定的cfDNA(图55)。根据这些结果,通过不稳定的cfDNA的存在或不存在,有可能提供有关肺癌患者发生E19del突变的信息。
实施例13.根据温度条件检测细胞系来源的不稳定cfDNA
与从人体获得的样品相同,进行实验以鉴定细胞系中存在的突变位置。具体地,从HCC2279(外显子19Del)、HCC827(外显子19Del)、H1975(T790M,L858R)和A549(EGFR野生型)中的每一个获得具有与cfDNA相似大小的fDNA。具体而言,通过实施例2的方法获得fDNA。
结果发现在分别于95℃加热1分钟和0分钟的变性条件下,仅不稳定的cfDNA特异性地与探针结合并且以与标志物结合的状态被检测到(图56)。从这些结果中,证实了在从细胞系获得的样品中也可以鉴定不稳定的cfDNA的存在与否。
实施例14.依赖于用DNase处理检测不稳定的cfDNA
为了鉴定不稳定的cfDNA和稳定的cfDNA之间的不同是否不仅取决于温度条件而且还取决于DNA降解酶,将不稳定的cfDNA和稳定的cfDNA用DNase进行处理,然后检查其与探针的反应性。在此,样品没有使用高温进行变性。
具体而言,将使用PEI/Ppy纳米线从HCC2279(外显子19Del)、HCC827(外显子19Del)、H1975(T790M,L858R)和A549(EGFR野生型)分别获得的fDNA悬浮在PBS中,然后用1μl的DNase进行处理。在37℃下处理30分钟,结果发现不稳定的cfDNA和稳定的cfDNA与探针的反应性不同(图57)。另外,已经确认即使在37℃下使用DNase处理60分钟显示出相同的效果(图58)。根据这些结果,可以确定稳定的cfDNA不容易被DNase酶降解。
但是,在37℃下用1μl或2μl DNase处理120分钟,结果发现,当延长DNase的处理时间或增加DNase的量时,稳定的cfDNA也会对探针产生反应(图59)。从这些结果可以确定不稳定的cfDNA和稳定的cfDNA之间的稳定性差异。
另外,为了鉴定不稳定的cfDNA和稳定的cfDNA之间的差异是否取决于DNase的活性,图60显示了在24℃下用1μl或2μl的DNase处理120分钟所获得的结果。结果显示,尽管该酶的活性在24℃时有所降低,但是已确定不稳定的cfDNA和稳定的cfDNA在与探针的反应性方面有所不同(图60)。
此外,为了确定不稳定的cfDNA和稳定的cfDNA之间的差异是否取决于DNase的活性,图58显示了在3℃下用1μl或2μl DNase处理120分钟获得的结果。结果显示,尽管酶的活性在3℃时有所降低,但是已确定不稳定的cfDNA和稳定的cfDNA在与探针的反应性方面有所不同(图61)。
SEQUENCE LISTING
<110> 健诺心理股份有限公司
<120> 检测不稳定的无细胞DNA的方法及使用其的装置
<130> P20116737WP
<150> KR 10-2018-0063258
<151> 2018-06-01
<160> 48
<170> PatentIn version 3.5
<210> 1
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> HPV 16-CP
<400> 1
gaggaggagg atgaaataga tggt 24
<210> 2
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> HPV 16-DP
<400> 2
ttggaagacc tgttaatggg c 21
<210> 3
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> HPV 18-CP
<400> 3
cacattgtgg cacaatcttt ta 22
<210> 4
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> HPV 18-DP
<400> 4
gccatatcgc tttcatctgt 20
<210> 5
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR 19-CP
<400> 5
ggaattaaga gaagcaacat ctcc 24
<210> 6
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR 19-DP
<400> 6
aacctcaggc ccacctttt 19
<210> 7
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR 21-CP
<400> 7
ccaggaacgt actggtgaaa a 21
<210> 8
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR 21-DP
<400> 8
ggaagagaaa gaataccatg ca 22
<210> 9
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子19-探针1 CP1
<400> 9
ggaattaaga gaagcaacat ctcc 24
<210> 10
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子19-探针1 DP
<400> 10
aacctcaggc ccaccttt 18
<210> 11
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子19-探针2 CP2
<400> 11
aaaattcccg tcgctatcaa g 21
<210> 12
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子19-探针2 DP
<400> 12
aacctcaggc ccacctttt 19
<210> 13
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子19-探针3 CP3
<400> 13
ggactctgga tcccagaagg tgag 24
<210> 14
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子19-探针3 DP
<400> 14
aacctcaggc ccacctttt 19
<210> 15
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子20-探针1 CP1
<400> 15
ccatgagtac gtattttgaa actc 24
<210> 16
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子20-探针1 DP
<400> 16
gcaagagttt gccatgggga tatg 24
<210> 17
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子20-探针2 CP2
<400> 17
ccaccgtgca gctcatcacg cagctca 27
<210> 18
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子20-探针2 DP
<400> 18
gcaagagttt gccatgggga tatg 24
<210> 19
<211> 23
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子20-探针3 CP3
<400> 19
gaagcctacg tgatggccag cgt 23
<210> 20
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子20-探针3 DP
<400> 20
gcaagagttt gccatgggga tatg 24
<210> 21
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子21-探针1 CP1
<400> 21
ccaggaacgt actggtgaaa a 21
<210> 22
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子21-探针1 DP
<400> 22
ggaagagaaa gaataccatg ca 22
<210> 23
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子21-探针2 CP2
<400> 23
aagatcacag attttgggcg gg 22
<210> 24
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子21-探针2 DP
<400> 24
ggaagagaaa gaataccatg ca 22
<210> 25
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子21-探针3 CP3
<400> 25
ggcatgaact acttggagga ccgt 24
<210> 26
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFR外显子21-探针3 DP
<400> 26
ggaagagaaa gaataccatg ca 22
<210> 27
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> KRAS外显子2-探针 CP1
<400> 27
aaatgactga atataaactt g 21
<210> 28
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> KRAS外显子2-探针 DP
<400> 28
gagtgccttg acgatacagc t 21
<210> 29
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> ALK-EML4变体1-探针 CP2
<400> 29
tagagcccac acctgggaaa 20
<210> 30
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> ALK-EML4变体1-探针 DP
<400> 30
cggagcttgc tcagcttgta 20
<210> 31
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> ALK-EML4变体3-探针 CP3
<400> 31
gcataaagat gtcatcatca accaag 26
<210> 32
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> ALK-EML4变体3-探针 DP
<400> 32
cggagcttgc tcagcttgta 20
<210> 33
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的BRCA1外显子7
<400> 33
caaagtatgg gctacagaaa ccgtgccaaa ag 32
<210> 34
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的BRCA1外显子7
<400> 34
caaagtatgg gcttcagaaa ccgtgccaaa ag 32
<210> 35
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的BRCA1外显子10
<400> 35
tgggaaaacc tatcggaaga aggcaagcct cc 32
<210> 36
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的BRCA1外显子10
<400> 36
tgggaaaacc tatcggtaga aggcaagcct cc 32
<210> 37
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的BRCA1外显子11
<400> 37
ggggccaaga aattagagtc ctcagaagag 30
<210> 38
<211> 31
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的BRCA1外显子11
<400> 38
ggggccaaga aaattagagt cctcagaaga g 31
<210> 39
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的BRCA1外显子15
<400> 39
atatacagga tatgcgaatt aagaagaaac aaa 33
<210> 40
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的BRCA1外显子15
<400> 40
atatacagga tatgtgaatt aagaagaaac aaa 33
<210> 41
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的TP53
<400> 41
taggaggccg agctctgttg cttcgaactc ca 32
<210> 42
<211> 31
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的TP53
<400> 42
taggaggccg agctctttgc ttcgaactcc a 31
<210> 43
<211> 31
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的MSH2
<400> 43
tgaggaggtt tcgacatggc ggtgcagccg a 31
<210> 44
<211> 31
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的MSH2
<400> 44
tgaggaggtt tcgacctggc ggtgcagccg a 31
<210> 45
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的EGFR
<400> 45
aaaaagatca aagtgctggg ctccggtgcg tt 32
<210> 46
<211> 32
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的EGFR
<400> 46
aaaaagatca aagtgctgag ctccggtgcg tt 32
<210> 47
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> 正常细胞的FGFR3
<400> 47
atcctctctc tgaaatcact gagcaggaga aag 33
<210> 48
<211> 33
<212> DNA
<213> Artificial Sequence
<220>
<223> 癌症细胞的FGFR3
<400> 48
atcctctctc tgaaatcact gcgcaggaga aag 33
- 检测不稳定的无细胞DNA的方法及使用其的装置
- 用于检测及量化无细胞DNA片段的方法和装置