用于治疗、缓解或预防与Tau聚集体相关的病症的新型化合物
文献发布时间:2023-06-19 09:44:49
发明领域
本发明涉及可用于治疗、缓解或预防与Tau(微管蛋白相关单位)蛋白聚集体相关的一组病症和异常,包括但不限于神经原纤维缠结(NFTs),如阿尔茨海默氏病(AD)的新型化合物。
发明背景
许多衰老疾病基于或与对该疾病的发病机理及进展作出贡献的淀粉样或类淀粉样(amyloid or amyloid-like)蛋白的细胞外或细胞内沉积物相关。形成细胞外聚集体的最充分表征的淀粉样蛋白是淀粉样蛋白β(Aβ)。形成细胞外聚集体的淀粉样蛋白的另一些实例是朊病毒、ATTR(运甲状腺素蛋白)或ADan(ADanPP)。主要形成细胞内聚集体的类淀粉样蛋白包括但不限于Tau、α-突触核蛋白、TAR DNA-结合蛋白43(TDP-43)和亨廷顿蛋白(htt)。涉及Tau聚集体的疾病通常被列为tau蛋白病,如AD。
淀粉样或类淀粉样沉积物起因于蛋白的错误折叠,接着聚集以产生β-折叠组装(β-sheet assemblies),其中多个肽或蛋白通过分子间氢键结合在一起。尽管淀粉样或类淀粉样蛋白具有不同的一级氨基酸序列,但它们的沉积物通常含有许多共有的分子成分,特别是β-折叠四级结构的存在。淀粉样沉积物与疾病之间的关联仍然在很大程度上不清楚。多种多样的蛋白聚集体,包括与疾病病理学相关和不相关的那些,已被发现有毒性,表明淀粉样蛋白的常见分子特征牵涉到疾病发作或对疾病发作负责(Bucciantini等人,Nature, 2002, 416, 507-11)。β-折叠聚集肽或蛋白的各种多聚体也已经与不同肽或蛋白的毒性相关联,范围从二聚体到可溶性低分子量低聚物、原纤维或不溶性纤维沉积物(insoluble fibrillar deposits)。
阿尔茨海默氏病(AD)是主要认为由淀粉样蛋白斑引起的神经病症,淀粉样蛋白斑是(淀粉样蛋白-β)Aβ聚集体在大脑中的异常沉积物的细胞外积聚。AD的其它主要的神经病理学标志是细胞内神经原纤维缠结(NFT),其源于过度磷酸化的Tau蛋白、错误折叠的Tau或病理性Tau及其构象异构体的聚集。AD与许多神经退行性tau蛋白病,特别是与指定类型的额颞叶痴呆(FTD)拥有共同的疾病发生学。Tau蛋白是易溶的“自然未折叠”蛋白,其贪婪地(avidly)结合到微管(MT)上以促进它们的组装和稳定性。MT对神经元的细胞骨架完整性和从而对神经元回路的适当形成和运作非常重要,因此对学习和记忆非常重要。如主要在体外和非神经元细胞中所证实的,通过动态磷酸化和脱磷酸化控制Tau与MT的结合。在AD大脑中,Tau病理(tau蛋白病)晚于淀粉样蛋白病理发生,但仍有争议地讨论Aβ蛋白是否是AD的致病因子,这构成所谓淀粉样蛋白级联假说的精髓(Hardy等人, Science 1992, 256,184-185;Musiek等人, Nature Neurosciences 2015, 18(6), 800-806)。将淀粉样蛋白与Tau病理联系起来的确切机制仍在很大程度上未知,但有提出涉及作用于GSK3和cdk5(作为主要“Tau-激酶”)或通过GSK3和cdk5作用的神经元信号通路的活化(Muyllaert等人, Rev.Neurol. (Paris), 2006, 162, 903-7;Muyllaert等人, Genes Brain and Behav. 2008,Suppl 1, 57-66)。即使tau蛋白病晚于淀粉样蛋白发生,但其不只是无辜的副作用,而是AD的主要病理学执行者(pathological executer)。在实验小鼠模型中,Tau蛋白的缺失几乎完全缓解由淀粉样蛋白病理造成的认知缺陷(Roberson等人, Science, 2007, 316(5825), 750-4)并且认知功能障碍和痴呆的严重度与tau蛋白病而非与淀粉样蛋白病理相关联。
涉及Tau聚集体的疾病通常被列为tau蛋白病,它们包括但不限于阿尔茨海默氏病(AD)、家族性AD、PART(原发性年龄相关性tau蛋白病)、克雅氏病、拳击员痴呆、唐氏综合征、Gerstmann-Sträussler-Scheinker病(GSS)、包涵体肌炎、朊病毒蛋白大脑淀粉样血管病、创伤性脑损伤(TBI)、肌萎缩性脊髓侧索硬化症(ALS)、关岛型帕金森-痴呆综合征、伴有神经原纤维缠结的非关岛型运动神经元病(non-Guamanian motor neuron disease withneurofibrillary tangles)、嗜银颗粒病、皮质基底节变性(CBD)、弥漫性神经原纤维缠结伴钙化症、17号染色体相关的额颞叶痴呆合并帕金森综合征(FTDP-17)、苍白球黑质红核色素变性(Hallervorden-Spatz disease)、多系统萎缩症(MSA)、C型尼曼匹克病、苍白球-脑桥-黑质变性(pallido-ponto-nigral degeneration)、皮克氏病(PiD)、进行性皮质下胶质增生、进行性核上性麻痹(PSP)、亚急性硬化性全脑炎、缠结优势型痴呆(tanglepredominant dementia)、脑炎后帕金森综合征、肌强直性营养不良、亚急性硬化型全脑病(subacute sclerosis panencephalopathy)、LRRK2中的突变、慢性创伤性脑病(CTE)、家族性英国型痴呆(familial British dementia)、家族性丹麦型痴呆、其它额颞叶变性、Guadeloupean帕金森综合征、脑组织铁沉积神经变性病、SLC9A6相关的智力迟钝、伴有球状胶质包涵体的白质tau蛋白病(white matter tauopathy with globular glialinclusions)、癫痫、路易体痴呆(LBD)、轻度认知损害(MCI)、多发性硬化症、帕金森氏病、HIV相关痴呆、成年型糖尿病(adult onset diabetes)、老年性心脏淀粉样变性、青光眼、缺血性卒中、AD中的精神病和亨廷顿舞蹈病。(Williams等人, Intern. Med. J., 2006, 36,652-60;Kovacs等人, J Neuropathol Exp Neurol. 2008;67(10): 963–975;Higuchi等人, Neuropsychopharmacology - 5th Generation of Progress, 2002, 第9部分, 94章: 1339-1354;Hilton等人, Acta Neuropathol. 1995;90(1):101-6;Iqbal等人,Biochimica et Biophysica Acta 1739 (2005) 198– 210;McQuaid等人, NeuropatholAppl Neurobiol. 1994 Apr;20(2):103-10;Vossel等人, Lancet Neurol 2017;16: 311–22;Stephan等人, Molecular Psychiatry (2012) 17, 1056–1076;Anderson等人, Brain(2008), 131, 1736-1748;Savica等人, JAMA Neurol. 2013;70(7):859-866;Brown等人Molecular Neurodegeneration 2014, 9:40;El Khoury等人, Front. Cell. Neurosci.,2014, Volume 8, Article22: 1-18;Tanskanen等人, Ann. Med. 2008;40(3):232-9;Gupta等人, CAN J OPHTHALMOL—VOL. 43, NO. 1, 2008: 53-60;Dickson等人, Int JClin Exp Pathol 2010;3(1):1-23;Fernández-Nogales等人, Nature Medicine, 20,881–885 (2014);Bi等人, Nature Communications volume 8, 文章编号: 473 (2017);Murray等人, Biol Psychiatry. 2014年4月1日;75(7): 542–552)。
在2017年,在临床试验中用于治疗阿尔茨海默氏病的所有药剂中,靶向Tau的非常少并仅构成II期临床试验的8%(Cummings等人, Alzheimer’s & Dementia:Translational Research & Clinical Interventions 3 (2017) 367-384)。靶向Tau蛋白的现有治疗方法主要包含基于抗体的方法,主要限制是仅靶向细胞外Tau。在使用小分子的方法中,已经开发出几种Tau激酶抑制剂,尽管在毒性和特异性方面极具挑战性。尽管如此,目前只有一种激酶抑制剂尼洛替尼在临床试验中测试。最后,在Tau聚集抑制剂中,只有一种LMTX目前在临床试验中(Cummings等人, 2017)。尽管近年来基于Tau的疗法已成为越来越受关注的点,但仍然非常需要另外的靶向已知或推定造成tau蛋白病的病理性Tau构象异构体的治疗剂。
WO2011/128455涉及适用于治疗与淀粉样蛋白或类淀粉样蛋白相关的病症的特定化合物。
WO2010/080253涉及可用于治疗受蛋白激酶信号转导抑制、调节和/或调制影响的疾病的二吡啶基-吡咯衍生化合物。
发明概述
本发明的一个目的是提供可用于治疗、缓解或预防与Tau蛋白聚集体相关的一组病症和异常,包括但不限于NFTs,如阿尔茨海默氏病(AD)的化合物。此外,在本领域中需要可用作治疗剂的化合物,所述治疗剂用于(a) 通过识别聚集的Tau和将Tau解聚,例如通过改变Tau聚集体分子构象来减少Tau聚集体/NFTs,和/或(b) 防止形成Tau聚集体,和/或(c) 细胞内干扰Tau聚集体,和/或(d) 减少体内Tau错误折叠和过度磷酸化和/或(f) 减少神经炎性标志物。本发明人已经意外地发现,可通过如下所述的式(I)的化合物实现这些目的。
式(I)的化合物(a)表现出通过识别聚集的Tau和将Tau解聚,例如通过改变Tau聚集体分子构象来减少Tau聚集体的高能力,和/或(b) 防止形成Tau聚集体,和/或(c) 细胞内干扰Tau聚集体,和/或(d) 减少体内Tau错误折叠和过度磷酸化和/或(f) 减少神经炎性标志物。尽管不希望受制于理论,但推测式(I)的化合物抑制Tau聚集或将预先形成的Tau聚集体解聚,包括存在于细胞内时。由于它们的独特设计特征,这些化合物表现出如适当的亲脂性和分子量、脑摄取和药代动力学、细胞渗透性、溶解度和代谢稳定性之类的性质,以成为用于治疗、缓解或预防tau蛋白病的成功药物。
通过组织病理学分析以及通过体内Tau PET成像,Tau NFT病变的积聚已表明与AD中的认知缺陷充分相关。本发明的化合物可防止形成Tau聚集体,或将已有的Tau聚集体解聚,并因此可预期防止或减轻AD中的相关认知缺陷。
超微结构分析已显示Tau包涵体由成对螺旋丝(PHF)或直丝(straightfilaments)(SF)组成。高分辨率结构分析已显示这些丝由包含Tau的氨基酸306-378的核心区组成,其适应(adapt)交叉β / β-螺旋结构。本发明的化合物可识别聚集的Tau和将Tau解聚,例如通过改变Tau聚集体分子构象,并因此可预期促进Tau清除。
此外,已经表明,Tau能够在细胞间传播并且某些形式的Tau(充当种子)能够诱发健康细胞内的天然Tau蛋白的结构变化以发生错误折叠和聚集。聚集的Tau被认为对该播种(seeding)和因此tau病理蔓延负责。本发明的化合物可细胞内干扰聚集的Tau并因此可预期减轻Tau病理蔓延并最终防止或减轻AD中的相关认知缺陷。
Tau聚集级联始于Tau错误折叠和过度磷酸化。相信这些事件先于细胞内Tau神经元包涵体的形成和因此Tau病理的建立和蔓延。本发明的化合物可减少体内Tau错误折叠和过度磷酸化并因此可预期有益于治疗、缓解或预防与Tau病理相关的疾病。
最后,Tau病理与神经炎症之间的联系现已充分确立。神经炎症是AD早期已存在的关键事件并相信是触发PHF中的Tau聚集的成因之一。此外,若干tau蛋白病小鼠模型显示一旦在脑中充分确立Tau病理,就会发生显著的神经炎症,表明Tau病理也可诱发神经炎性过程。这两个发现表明Tau病理和神经炎症在正反馈回路中联系在一起。本发明的化合物减少Tau病理背景下的神经炎性标志物。
本发明公开了式(I)的新型化合物,其有能力(a) 减少Tau聚集体、识别聚集的Tau和将Tau解聚,例如通过改变Tau聚集体分子构象
本发明概括为以下项目:
1. 式(I)的化合物:
及其所有立体异构体、外消旋混合物、互变异构体、可药用盐、前药、水合物、溶剂化物和多晶型物;
其中
B选自O和NR
E是N且V是S,E是S且V是N,E是N且V是O或E是O且V是N;
X选自
R独立地选自
R
R
2. 根据项目1的化合物,其是式(Ia)的化合物:
其中B、R和X如项目1中定义。
3. 根据项目1的化合物,其是式(Ib)的化合物:
其中B、R和R
4. 根据项目1的化合物,其是式(Ic)的化合物:
其中B和R如项目1中定义。
5. 一种药物组合物,其包含如项目1至4任一项中定义的化合物和任选可药用载体或赋形剂。
6. 如项目1至4任一项中定义的化合物,其用作药物。
7. 如项目1至4任一项中定义的化合物,其用于治疗、缓解或预防与Tau蛋白聚集体相关的病症或异常。
8. 如项目1至4任一项中定义的化合物,其用于减少tau聚集。
9. 如项目1至4任一项中定义的化合物,其用于防止形成Tau聚集体和/或用于抑制Tau聚集。
10. 如项目1至4任一项中定义的化合物,其用于细胞内干扰Tau聚集体。
11. 如项目1至4任一项中定义的化合物,其用于减少体内Tau错误折叠和过度磷酸化。
12. 如项目1至4任一项中定义的化合物,其用于减少神经炎性标志物。
13. 一种治疗、预防或缓解与Tau蛋白聚集体相关的病症的方法,所述方法包括向需要其的对象给药有效量的如项目1至4任一项中定义的化合物。
14. 一种减少tau聚集的方法,所述方法包括向需要其的对象给药有效量的如项目1至4任一项中定义的化合物。
15. 一种防止形成Tau聚集体和/或抑制Tau聚集的方法,所述方法包括向需要其的对象给药有效量的如项目1至4任一项中定义的化合物。
16. 一种细胞内干扰Tau聚集体的方法,所述方法包括向需要其的对象给药有效量的如项目1至4任一项中定义的化合物。
17. 一种减少体内Tau错误折叠和过度磷酸化的方法,所述方法包括向需要其的对象给药有效量的如项目1至4任一项中定义的化合物。
18. 一种减少神经炎性标志物的方法,所述方法包括向需要其的对象给药有效量的如项目1至4任一项中定义的化合物。
19. 如项目1至4任一项中定义的化合物在制备用于治疗与Tau蛋白聚集体相关的病症或异常的药物中的用途。
20. 如项目1至4任一项中定义的化合物在制备用于减少tau聚集的药物中的用途。
21. 如项目1至4任一项中定义的化合物在制备用于防止形成Tau聚集体和/或用于抑制Tau聚集的药物中的用途。
22. 如项目1至4任一项中定义的化合物在制备用于细胞内干扰Tau聚集体的药物中的用途。
23. 如项目1至4任一项中定义的化合物在制备用于减少体内Tau错误折叠和过度磷酸化的药物中的用途。
24. 如项目1至4任一项中定义的化合物在制备用于减少神经炎性标志物的药物中的用途。
25. 一种混合物,其包含如项目1至4任一项中定义的化合物和至少一种另外的生物活性化合物、可药用载体、稀释剂和赋形剂,所述至少一种另外的生物活性化合物选自不同于如项目1至4任一项中定义的化合物的治疗剂。
26. 根据项目25的混合物,其中所述另外的生物活性化合物是用于治疗淀粉样变性的化合物。
27. 根据项目25或26的混合物,其中所述另外的生物活性化合物选自抗氧化应激的化合物、抗凋亡化合物、金属螯合剂、DNA修复抑制剂如哌仑西平和代谢物、3-氨基-1-丙磺酸(3APS)、1,3-丙二磺酸盐(1,3PDS)、α-分泌酶激活剂、β-和γ-分泌酶抑制剂、Tau蛋白、神经递质、β-折叠阻断剂、淀粉样蛋白β 清除/消耗性(clearing/depleting)细胞组分的引诱剂(attractants)、N-端截短淀粉样蛋白β包括焦谷氨酸化淀粉样蛋白β 3-42的抑制剂、抗炎分子或胆碱酯酶抑制剂(ChEIs)如他克林、卡巴拉汀、多奈哌齐和/或加兰他敏、M1激动剂、其它药物,包括任何淀粉样蛋白或Tau修饰药物和营养补充剂,抗体,包括任何功能等效抗体或其功能部分或疫苗。
28. 根据项目27的混合物,其中所述另外的生物活性化合物是胆碱酯酶抑制剂(ChEI)。
29. 根据项目27的混合物,其中所述另外的生物活性化合物选自他克林、卡巴拉汀、多奈哌齐、加兰他敏、烟酸和美金刚胺。
30. 根据项目27的混合物,其中所述另外的生物活性化合物是抗体,特别是单克隆抗体,包括任何功能等效抗体或其功能部分。
31. 根据项目25至30任一项的混合物,其中所述化合物和/或所述另外的生物活性化合物以治疗有效量存在。
32. 用于根据项目7所述用途的化合物、根据项目13的方法或根据项目19的用途,其中所述病症选自阿尔茨海默氏病(AD)、家族性AD、原发性年龄相关性tau蛋白病(PART)、克雅氏病、拳击员痴呆、唐氏综合征、Gerstmann-Sträussler-Scheinker病(GSS)、包涵体肌炎、朊病毒蛋白大脑淀粉样血管病、创伤性脑损伤(TBI)、肌萎缩性脊髓侧索硬化症(ALS)、关岛型帕金森-痴呆综合征、伴有神经原纤维缠结的非关岛型运动神经元病(non-Guamanian motor neuron disease with neurofibrillary tangles)、嗜银颗粒病、皮质基底节变性(CBD)、弥漫性神经原纤维缠结伴钙化症、17号染色体相关的额颞叶痴呆合并帕金森综合征(FTDP-17)、苍白球黑质红核色素变性(Hallervorden-Spatz disease)、多系统萎缩症(MSA)、C型尼曼匹克病、苍白球-脑桥-黑质变性(pallido-ponto-nigraldegeneration)、皮克氏病(PiD)、进行性皮质下胶质增生、进行性核上性麻痹(PSP)、亚急性硬化性全脑炎、缠结优势型痴呆(tangle predominant dementia)、脑炎后帕金森综合征、肌强直性营养不良、亚急性硬化性全脑病(subacute sclerosis panencephalopathy)、LRRK2中的突变、慢性创伤性脑病(CTE)、家族性英国型痴呆(familial Britishdementia)、家族性丹麦型痴呆、其它额颞叶变性、Guadeloupean帕金森综合征、脑组织铁沉积神经变性病、SLC9A6相关的智力迟钝、伴有球状胶质包涵体的白质tau蛋白病(whitematter tauopathy with globular glial inclusions)、癫痫、路易体痴呆(LBD)、轻度认知损害(MCI)、多发性硬化症、帕金森氏病、HIV相关痴呆、成年型糖尿病(adult onsetdiabetes)、老年性心脏淀粉样变性、青光眼、缺血性卒中、AD中的精神病和亨廷顿舞蹈病,优选阿尔茨海默氏病(AD)、皮质基底节变性(CBD)、皮克氏病(PiD)和进行性核上性麻痹(PSP)。
33. 如项目1至4任一项中定义的化合物作为分析参考物或体外筛选工具的用途。
定义
在本申请的含义内,适用下列定义:
“烷基”是指由碳和氢原子组成的直链或支链有机基团。合适的烷基的实例具有1至6个碳原子,优选1至4个碳原子,并包括甲基、乙基、丙基、异丙基、正丁基、叔丁基和异丁基。
具有一个或多个光学活性碳的本发明的化合物可作为外消旋物和外消旋混合物(包括所有比率的混合物)、立体异构体(包括非对映体混合物和独立的非对映体、对映体混合物和单一对映体、构象异构体混合物和单一构象异构体)、互变异构体、阻转异构体和旋转异构体存在。所有异构形式都包含在本发明中。本发明中描述的含有烯属双键的化合物包括E和Z几何异构体。在本发明中还包括所有可药用盐、前药、多晶型物、水合物和溶剂化物。
术语“多晶型物”是指本发明的化合物的各种结晶结构。这可包括,但不限于,晶体形态(和非晶材料)和所有晶格形式。本发明的盐可以是结晶的并可作为多于一种的多晶型物存在。
本发明也包含盐的溶剂化物、水合物以及无水形式。溶剂化物中包含的溶剂不受特别限制并可以是任何可药用溶剂。实例包括水和C
“可药用盐”被定义为所公开的化合物的衍生物,其中通过制备其酸式或碱式盐来改变母体化合物。可药用盐的实例包括,但不限于,碱性残基如胺的无机或有机酸盐;酸性残基如羧酸的碱金属盐或有机盐;等。可药用盐包括例如由无毒无机或有机酸形成的母体化合物的常规无毒盐或季铵盐。例如,这样的常规无毒盐包括衍生自无机酸,例如但不限于盐酸、氢溴酸、硫酸、氨基磺酸、磷酸和硝酸等的那些;和由有机酸,例如但不限于乙酸、丙酸、琥珀酸、乙醇酸、硬脂酸、乳酸、苹果酸、酒石酸、柠檬酸、抗坏血酸、扑酸、马来酸、羟基马来酸、苯基乙酸、谷氨酸、苯甲酸、水杨酸、磺胺酸、2-乙酰氧基苯甲酸、富马酸、甲苯磺酸、甲磺酸、乙二磺酸、草酸、羟乙磺酸等制成的盐。本发明的可药用盐可由含有碱性或酸性部分的母体化合物通过常规化学方法合成。通常,可通过使这些化合物的游离酸或碱形式与化学计算量的适当的碱或酸在水中或在有机溶剂中或在两者的混合物中反应来制备这样的盐。有机溶剂包括,但不限于,非水介质,如醚、乙酸乙酯、乙醇、异丙醇或乙腈。合适的盐的名单可见于Remington’s Pharmaceutical Sciences, 第18版, Mack PublishingCompany, Easton, PA, 1990, 第1445页,其公开内容通过引用特此并入。
本发明的化合物也可以前药形式提供,前药即在体内代谢成活性代谢物的化合物。如下面在本发明的说明书和权利要求书中所用,术语“前药”是指由于体内生物转化而释放活性母体药物的任何共价键合化合物。Goodman和Gilman所著的描述前药的参考文献(The Pharmacological Basis of Therapeutics, 8 ed, McGraw-Hill, Int. Ed. 1992,"Biotransformation of Drugs", 第13-15页)通过引用特此并入本文。
“可药用”被定义为在合理的医学判断范围内适合与人类和动物的组织接触使用而没有过度毒性、刺激、过敏反应或其它问题或并发症、与合理的效益/风险比相称的那些化合物、材料、组合物和/或剂型。
本发明中的患者或对象通常是动物,特别是哺乳动物,更特别是人类。
本文所用的“Tau”是指主要在神经元中发现的高度可溶的微管结合蛋白,并包括主要6种同种型(isoforms)、裂解或截短形式和其它修饰形式,如源于磷酸化、糖基化、糖化、脯氨酰异构化、硝化、乙酰化、多胺化、泛素化、类泛素化(sumoylation)和氧化。
“聚集Tau”是指折叠成低聚或聚合结构的Tau肽或蛋白的聚集单体。
本文所用的“神经原纤维缠结”(NFTs)是指含有成对螺旋丝(PHF)和直丝的过度磷酸化Tau蛋白的不溶性聚集体。它们的存在是AD和已知为tau蛋白病的其它疾病的标志。
除非另行规定,否则“定义”部分中给出的定义和优选定义适用于下述所有实施方案。
附图描述
图1:用实施例1和实施例12治疗的rTg4510小鼠的Morris水迷宫行为试验
图2:用实施例1和实施例12治疗的rTg4510小鼠的皮质萎缩(A)和Tau错误折叠(B)量化
图3:用实施例1和实施例12治疗的rTg4510小鼠的CD68量化
图4:用实施例2治疗的rTg4510小鼠的Morris水迷宫行为试验
图5:用实施例2治疗的rTg4510小鼠的sarkosyl不溶性Tau的量化
图6:用实施例2治疗的rTg4510小鼠的皮质萎缩(A)和Iba1阳性小胶质(B)量化。
发明详述
下面将描述本发明的化合物。要理解的是,也设想了下列定义的所有可能的组合。
在一个实施方案中,本发明涉及式(I)的化合物:
及其所有立体异构体、外消旋混合物、互变异构体、可药用盐、前药、水合物、溶剂化物和多晶型物。
式(I)的化合物的一个优选实施方案是
式(I)的化合物的另一优选实施方案是
式(I)的化合物的另一优选实施方案是
下列定义酌情适用于式(I)和它们的优选实施方案。
B选自O和NR
E是N且V是S,E是S且V是N,E是N且V是O或E是O且V是N。这意味着在式(I)的化合物中可存在下列基团:
更优选地,E是N且V是S或E是N且V是O。再更优选地,E是N且V是S。
X选自
R独立地选自
R
R
优选化合物也例示在实施例中。
在本发明中也设想了本文中公开的实施方案、优选实施方案和更优选实施方案的任何组合。
药物组合物
尽管本发明的化合物可以独自给药,但优选根据标准制药实践将它们配制成药物组合物。因此,本发明也提供了包含任选与可药用载体、稀释剂、辅助剂或赋形剂混合的治疗有效量的式(I)的化合物的药物组合物。
可药用赋形剂是制药领域众所周知的,并描述在例如Remington'sPharmaceutical Sciences, 15
可用于配制本发明的药物组合物的可药用赋形剂可包含例如载体、媒介物、稀释剂、溶剂如一元醇,如乙醇、异丙醇,和多元醇,如二醇,和食用油如大豆油、椰子油、橄榄油、红花油、棉籽油、油酯如油酸乙酯、肉豆蔻酸异丙酯,粘合剂、辅助剂、增溶剂、增稠剂、稳定剂、崩解剂、助流剂、润滑剂、缓冲剂、乳化剂、润湿剂、悬浮剂、甜味剂、着色剂、香料、包衣剂、防腐剂、抗氧化剂、加工剂、药物递送调节剂(modifiers)和增强剂,如磷酸钙、硬脂酸镁、滑石、单糖、二糖、淀粉、明胶、纤维素、甲基纤维素、羧甲基纤维素钠、右旋糖、羟丙基-ß-环糊精、聚乙烯基吡咯烷酮、低熔点蜡和离子交换树脂。
本发明的化合物的给药(递送)途径包括,但不限于,以下一种或多种:口服(例如作为片剂、胶囊或作为可食用溶液)、局部、粘膜(例如作为鼻喷雾剂或吸入用气溶胶)、鼻、肠胃外(例如通过可注射形式)、胃肠、脊柱内、腹膜内、肌肉内、静脉、子宫内、眼内、皮内、颅内、气管内、阴道内、脑室内、大脑内、皮下、眼(包括玻璃体内或前房内)、透皮、直肠、颊、硬膜外和舌下。
例如,该化合物可以片剂、胶囊、ovules、酏剂、溶液剂或混悬剂的形式口服给药,其可含有调味或着色剂,用于速释、迟释、调释、缓释、脉冲释放或控释应用。
片剂可含有赋形剂,如微晶纤维素、乳糖、柠檬酸钠、碳酸钙、磷酸氢钙和甘氨酸,崩解剂,如淀粉(优选玉米、马铃薯或木薯淀粉)、羟乙酸淀粉钠、交联羧甲基纤维素钠和某些复合硅酸盐,和造粒粘合剂,如聚乙烯基吡咯烷酮、羟丙基甲基纤维素(HPMC)、羟丙基纤维素(HPC)、蔗糖、明胶和阿拉伯树胶。另外,可包括润滑剂,如硬脂酸镁、硬脂酸、山嵛酸甘油酯和滑石。也可使用相似类型的固体组合物作为明胶胶囊中的填料。在这方面优选的赋形剂包括乳糖(lactose)、淀粉、纤维素、乳糖(milk sugar)或高分子量聚乙二醇。对于水性混悬剂和/或酏剂,可将药剂与各种甜味剂或调味剂、着色物质或染料、与乳化剂和/或悬浮剂和与稀释剂如水、乙醇、丙二醇和甘油及其组合合并。
如果本发明的化合物肠胃外给药,那么这样的给药的实例包括以下一种或多种:静脉内、动脉内、腹膜内、鞘内、脑室内、尿道内、胸骨内、颅内、肌肉内或皮下给药该化合物;和/或通过使用输液技术。对于肠胃外给药,该化合物最好以无菌水溶液的形式使用,其可含有其它物质,例如足够的盐或葡萄糖以使该溶液与血液等渗。如果必要,该水溶液应该合适地缓冲(优选到3至9的pH)。通过本领域技术人员公知的标准制药技术容易实现在无菌条件下制备合适的肠胃外制剂。
如所示,本发明的化合物可鼻内或通过吸入给药,并方便地以干粉吸入器或气溶胶喷雾呈现的形式由加压容器、泵、喷雾器或雾化器借助使用合适的抛射剂,例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、氢氟烷烃如1,1,1,2-四氟乙烷(HFA134AT)或1,1,1,2,3,3,3-七氟丙烷(HFA 227EA)、二氧化碳或其它合适的气体递送。在加压气溶胶的情况下,可通过提供阀以递送计量的量来确定剂量单位。加压容器、泵、喷雾器或雾化器可含有活性化合物的溶液剂或混悬剂,例如使用乙醇和抛射剂的混合物作为溶剂,其可另外含有润滑剂,例如山梨糖醇酐三油酸酯。可配制用于吸入器或吹入器的胶囊和药筒(例如由明胶制成)以含有该化合物和合适的粉末基质如乳糖或淀粉的粉末混合物。
或者,本发明的化合物可以栓剂或子宫托的形式给药,或其可以凝胶、水凝胶、洗剂、溶液剂、乳膏剂、软膏剂或撒粉剂的形式局部施加。本发明的化合物也可真皮或透皮给药,例如通过使用皮肤贴剂。
它们也可通过肺或直肠途径给药。它们也可通过经眼途径给药。对于眼科应用,该化合物可配制为在等渗的、pH调节的无菌盐水中的微粉化混悬剂,或优选为在等渗的、pH调节的无菌盐水中的溶液剂,任选与防腐剂如苯扎氯铵组合。或者,它们可配制在软膏剂如矿脂中。
为了局部施加于皮肤,本发明的化合物可配制为合适的软膏剂,其含有悬浮或溶解在例如以下一种或多种的混合物中的活性化合物:矿物油、液体矿脂、白矿脂、丙二醇、乳化蜡和水。或者,它们可配制为合适的洗剂或乳膏剂,悬浮或溶解在例如以下一种或多种的混合物中:矿物油、山梨糖醇酐单硬脂酸酯、聚乙二醇、液体石蜡、聚山梨酯60、鲸蜡酯蜡、鲸蜡硬脂醇、2-辛基十二烷醇、苄醇和水。
通常,医师将确定最适合个体对象的实际剂量。用于任何特定个体的具体剂量水平和给药频率可变并取决于各种因素,包括所用具体化合物的活性、该化合物的代谢稳定性和作用时长、年龄、体重、一般健康状况、性别、饮食、给药模式和时间、排泄率、药物组合、特定病况的严重度和接受治疗的个体。
给药于人类(大约70 kg体重)的根据本发明的化合物的建议剂量是0.1 mg至1 g,优选1 mg至500 mg活性成分/单位剂量。该单位剂量可以每天给药例如1至4次。剂量取决于给药途径。要认识到,可能必须根据患者的年龄和体重以及要治疗的病况的严重度对剂量作出常规改变。精确剂量和给药途径最终由主治医师或兽医决定。
本发明的化合物也可与其它治疗剂组合使用。当本发明的化合物与对相同疾病有活性的第二治疗剂组合使用时,各化合物的剂量可能不同于该化合物独自使用时的剂量。
上文提到的组合可方便地以药物制剂的形式呈现以供使用。这样的组合的各个组分可在分开或组合的药物制剂中通过任何方便的途径依序或同时给药。当依序给药时,本发明的化合物或第二治疗剂可首先给药。当同时给药时,该组合可在相同或不同的药物组合物中给药。当组合在相同制剂中时,要认识到,这两种化合物必须稳定和与彼此和与该制剂的其它组分相容。当分开配制时,它们可在任何方便的制剂中提供,方便地以本领域中已知用于此类化合物的方式。
本发明的药物组合物可以如例如Remington's Pharmaceutical Sciences, 15
可用本发明的化合物治疗、缓解或预防的疾病或病况是与Tau蛋白聚集体相关的病症或异常,如神经退行性病症。可治疗、缓解或预防的疾病和病况的实例由神经原纤维病变的形成造成或与其相关。这是tau蛋白病中的主要脑病理学。所述疾病和病况包含一组多样化的神经退行性疾病或病况,包括表现出Tau和淀粉样蛋白病理的共存的疾病或病况。
可治疗、缓解或预防的疾病和病况的实例包括但不限于阿尔茨海默氏病(AD)、家族性AD、PART(原发性年龄相关性tau蛋白病)、克雅氏病、拳击员痴呆、唐氏综合征、Gerstmann-Sträussler-Scheinker病(GSS)、包涵体肌炎、朊病毒蛋白大脑淀粉样血管病、创伤性脑损伤(TBI)、肌萎缩性脊髓侧索硬化症(ALS)、关岛型帕金森-痴呆综合征、伴有神经原纤维缠结的非关岛型运动神经元病(non-Guamanian motor neuron disease withneurofibrillary tangles)、嗜银颗粒病、皮质基底节变性(CBD)、弥漫性神经原纤维缠结伴钙化症、17号染色体相关的额颞叶痴呆合并帕金森综合征(FTDP-17)、苍白球黑质红核色素变性(Hallervorden-Spatz disease)、多系统萎缩症(MSA)、C型尼曼匹克病、苍白球-脑桥-黑质变性(pallido-ponto-nigral degeneration)、皮克氏病(PiD)、进行性皮质下胶质增生、进行性核上性麻痹(PSP)、亚急性硬化性全脑炎、缠结优势型痴呆(tanglepredominant dementia)、脑炎后帕金森综合征、肌强直性营养不良、亚急性硬化性全脑病(subacute sclerosis panencephalopathy)、LRRK2中的突变、慢性创伤性脑病(CTE)、家族性英国型痴呆(familial British dementia)、家族性丹麦型痴呆、其它额颞叶变性、Guadeloupean帕金森综合征、脑组织铁沉积神经变性病、SLC9A6相关的智力迟钝、伴有球状胶质包涵体的白质tau蛋白病(white matter tauopathy with globular glialinclusions)、癫痫、路易体痴呆(LBD)、轻度认知损害(MCI)、多发性硬化症、帕金森氏病、HIV相关痴呆、成年型糖尿病、老年性心脏淀粉样变性、青光眼、缺血性卒中、AD中的精神病和亨廷顿舞蹈病。可治疗、缓解或预防的疾病和病况优选包括阿尔茨海默氏病(AD),以及其它神经退行性tau蛋白病,如克雅氏病、拳击员痴呆、肌萎缩性脊髓侧索硬化症(ALS)、嗜银颗粒病、皮质基底节变性(CBD)、17号染色体相关的额颞叶痴呆合并帕金森综合征(FTDP-17)、皮克氏病(PiD)、进行性核上性麻痹(PSP)、缠结优势型痴呆(tangle predominantdementia)、关岛型帕金森-痴呆综合征、苍白球黑质红核色素变性(Hallervorden-Spatzdisease)、慢性创伤性脑病(CTE)、创伤性脑损伤(TBI)和其它额颞叶变性。更优选阿尔茨海默氏病(AD)、皮质基底节变性(CBD)、皮克氏病(PiD)和进行性核上性麻痹(PSP)。
本发明的化合物也可用于减少蛋白聚集,特别是Tau聚集。可以例如使用ThT测定法(Hudson等人, FEBS J., 2009, 5960-72)确定化合物减少Tau聚集的能力。
本发明的化合物可用于治疗多种多样的病症,其中神经炎性过程与Tau蛋白的错误折叠和/或病理性聚集相关。
本发明的化合物可用作用于表征具有Tau病理的组织和用于测试靶向这样的组织上的Tau病理的化合物的分析参考物或体外筛选工具。
根据本发明的化合物也可以与至少一种另外的生物活性化合物和/或可药用载体和/或稀释剂和/或赋形剂的混合物的形式提供。该化合物和/或另外的生物活性化合物优选以治疗有效量存在。
另外的生物活性化合物的性质取决于该混合物的预期用途。另外的生物活性物质或化合物可通过与根据本发明的化合物相同或类似的机制或通过不相关的作用机制或通过多种相关和/或不相关的作用机制发挥其生物学效应。
通常,另外的生物活性化合物可包括神经元传递增强剂(neutron-transmissionenhancers)、精神病治疗药物、乙酰胆碱酯酶抑制剂、钙通道阻断剂、生物胺、苯二氮䓬安定剂、乙酰胆碱合成、存储或释放增强剂、乙酰胆碱突触后受体激动剂、单胺氧化酶-A或单胺氧化酶-B抑制剂、N-甲基-D-天冬氨酸谷氨酸受体拮抗剂、非甾体抗炎药、抗氧化剂和5-羟色胺受体拮抗剂。特别地,另外的生物活性化合物可选自用于治疗淀粉样变性的化合物、抗氧化应激的化合物、抗凋亡化合物、金属螯合剂、DNA修复抑制剂如哌仑西平和代谢物、3-氨基-1-丙磺酸(3APS)、1,3-丙二磺酸盐(1,3PDS)、α-分泌酶激活剂、β-和γ-分泌酶抑制剂、Tau蛋白、神经递质、β-折叠阻断剂、淀粉样蛋白β 清除/消耗性(clearing / depleting)细胞组分的引诱剂(attractants)、N-端截短淀粉样蛋白β包括焦谷氨酸化淀粉样蛋白β 3-42的抑制剂、抗炎分子或胆碱酯酶抑制剂(ChEIs)如他克林、卡巴拉汀、多奈哌齐和/或加兰他敏、M1激动剂、其它药物,包括任何淀粉样蛋白或Tau修饰药物和营养补充剂,抗体,包括任何功能等效抗体或其功能部分,或疫苗。
在进一步实施方案中,根据本发明的混合物可包含烟酸或美金刚胺以及根据本发明的化合物和任选地可药用载体和/或稀释剂和/或赋形剂。
在本发明的又一实施方案中,提供了混合物,其包含作为另外的生物活性化合物的“非典型抗精神病药物”例如氯氮平、齐拉西酮、利培酮、阿立哌唑或奥氮平以及根据本发明的化合物和任选地可药用载体和/或稀释剂和/或赋形剂,所述另外的生物活性化合物用于治疗阳性和阴性精神病症状,包括幻觉、妄想、思维障碍(表现为明显的不连贯、思维脱轨、言不及义)和离奇或紊乱行为,以及快感缺乏、情感平淡(flattened affect)、冷漠和社交退缩。
可合适地与根据本发明的化合物组合在混合物中使用的其它化合物例如描述在WO 2004/058258中(尤其参见第16和17页),包括治疗药物靶标(第36至39页)、链烷磺酸和链烷醇硫酸(第39至51页)、胆碱酯酶抑制剂(第51至56页)、NMDA受体拮抗剂(第56至58页)、雌激素(第58至59页)、非甾体抗炎药(第60和61页)、抗氧化剂(第61和62页)、过氧化物酶体增殖物激活受体(PPAR)激动剂(第63至67页)、降胆固醇药(第68至75页)、淀粉样蛋白抑制剂(第75至77页)、淀粉样蛋白形成抑制剂(第77至78页)、金属螯合剂(第78和79页)、抗精神病药和抗抑郁药(第80至82页)、营养补充剂(第83至89页)和提高生物活性物质的脑利用率的化合物(参见第89至93页)和前药(第93和94页),该文献通过引用并入本文。
本发明还包括本发明的化合物的所有合适的同位素变体。本发明的化合物的同位素变体被定义为是其中至少一个原子被具有相同原子序数但原子质量不同于自然界中通常发现的原子质量的原子替代的化合物。可并入本发明的化合物中的同位素的实例包括氢、碳、氮、氧、硫、氟和氯的同位素,分别如
本发明的化合物可通过下列方案中显示的通用方法之一合成。这些方法仅为举例说明给出并且不应被解释为限制。
本发明的化合物可通过下列方案中显示的通用方法之一合成。这些方法仅为举例说明给出并且不构成限制。
用于制备其中X = NCH(CH
方案1
通过市售1,4-二氧杂螺[4.5]癸-8-酮用二甲胺的还原胺化制备
用于制备其中X = NCH(CH
方案2
市售2,6-二-溴吡啶或2-溴-6-氟吡啶与肼或甲肼在适当溶剂中的加热在提纯后提供相应的2-溴-6-肼基吡啶衍生物(R
用于制备苯并噻唑和苯并噁唑
方案3
其中E是N且V是S,或E是S且V是N,或E是N且V是O或E是O且V是N的氯
用于制备本发明的化合物的通用合成方案。
方案4
使其中Y = Br或NH
可使用本领域中已知的任何合适的测定法测量Tau K18和全长(fl) Tau的解聚。描述了用于测量解聚能力的标准体外测定法。
实施例
所有试剂和溶剂获自商业来源并且不经进一步提纯使用。在Bruker AV 300和400MHz光谱仪上在氘代溶剂中记录
向市售2,6-二溴吡啶(10 g, 42.2 mmol)在乙醇(50 mL)中的悬浮液中加入市售
将4-(甲基氨基)环己酮缩(2,2-二甲基-1,3-丙二醇)盐酸盐(4-(methylamino)cyclohexanone-2,2-dimethyltrimethylene ketal hydrochloride)(3.7 g, 14.8 mmol)和来自上述步骤A的标题化合物(3 g, 14.8 mmol)在二氧杂环己烷(30 mL)中的悬浮液置于冰浴中。向搅拌的悬浮液中缓慢加入浓硫酸(3 mL)。在添加完成后,反应混合物使用砂浴(~140℃)在回流温度下加热5小时。将反应混合物冷却到室温,弃置二氧杂环己烷层,并加入冰水(20 mL)。搅拌混合物直至胶质物质溶解。然后使用NaOH水溶液将反应混合物的pH调节到pH = 14。用二氯甲烷(200 mL)萃取水层,用水和盐水洗涤有机相。分离有机相,经Na
向来自上述步骤B的标题化合物(4.7 g)在THF(50 ml)中的溶液中加入三乙胺(5 mL)和二碳酸二叔丁酯(10 g, 45.8 mmol)。反应混合物在室温下搅拌整夜。然后,将反应混合物浓缩并将残留物溶解在二氯甲烷(200 mL)中。有机相用水、盐水洗涤,经Na
将来自上述步骤C的标题化合物(1 g, 2.54 mmol)溶解在二氯甲烷(32 mL)中并在室温下加入盐酸在二氧杂环己烷中的4M溶液(16 mL)。混合物在室温下搅拌整夜并在减压下除去溶剂。将残留物悬浮在二氯甲烷(25 mL)和水(20 mL)中并加入1 M氢氧化钠水溶液直至pH ~12。分离有机相且水相用二氯甲烷萃取(2 x 20 mL)。合并的有机相经Na
将来自上述步骤D的标题化合物(0.745 g, 2.53 mmol)溶解在水(2 mL)和乙酸(0.61g, 10.12 mmol)中。然后加入37%-甲醛水溶液(1.11 g, 13.77 mmol)并将混合物在室温下搅拌10分钟。在加入锌粉(1.19 g, 18.35 mmol)后,反应混合物在室温下搅拌4小时。然后加入浓氨水(10 mL)并将混合物声处理2分钟。浆料用二氯甲烷萃取(3 x 30 mL),分离有机相,经Na
将手性原料(2 g, 5.67 mmol, 化学纯度: 99.2%;手性纯度: 99.2% ee)(以与WO2011/128455制备实施例337中报道的类似方式,通过使用DMF而非MeOH作为溶剂制备)溶解在二氯甲烷(80 mL)中并加入40毫升氯化氢在1,4-二氧杂环己烷中的4 M溶液。反应混合物在室温下搅拌18小时并在减压下除去溶剂。将残留物溶解在二氯甲烷(250 mL)和水(120mL)中并通过加入氢氧化钠丸粒将水相的pH调节到pH~12。分离有机相且水相用二氯甲烷(120 mL)萃取。合并的有机相经Na
将上述步骤A的化合物(1.46 g, 4.96 mmol)悬浮在甲醇(30 mL)中并加入37%甲醛水溶液(3 mL, 35.7 mmol)。反应混合物在室温下搅拌10分钟并加入氰基硼氢化钠(1.25 g,20.26 mmol)。然后加入乙酸(4.5 mL)(放热!)并将反应混合物在室温下搅拌18小时。在减压下除去溶剂并将残留物溶解在二氯甲烷(80 mL)和水(40 mL)中。通过加入氢氧化钠丸粒将水相的pH调节到pH~12。分离有机相且水相用二氯甲烷(40 mL)萃取。合并的有机物用水和盐水的混合物(40 mL (1/1))洗涤,分离,经Na
市售1-异丙基哌啶-4-酮(500 g, 3.541 mol)、羟胺盐酸盐(369.1 g, 5.315 mol)和碳酸钾(1221 g, 8.855 mol)在乙醇(2.5 L)中在油浴中在80℃下加热3小时。在通过TLC确定反应完全后,将反应混合物冷却到25℃并过滤固体,用水(5 L)和乙醚(2.5 L)洗涤。化合物在管路真空(line vaccum)下干燥24小时以获得为白色固体的1-异丙基哌啶-4-酮肟(525 g, 95%)。
在0℃下向来自上述步骤A的标题化合物(500 g, 3.201 mol)在DMF(5 L)中的搅拌溶液中逐份加入在矿物油中的NaH 60%(192 g, 4.801 mol)。在添加后,反应混合物在25℃下搅拌1小时,然后再将反应混合物冷却到0℃。在0℃下向其中逐滴加入溶解在DMF(1 L)中的市售2-溴-6-氟吡啶(563 g, 3.201 mol),并将反应混合物在25℃下搅拌2小时。在通过TLC确定反应完全后,将反应混合物冷却到0℃并用冰水(1 L)淬灭,接着使用乙酸乙酯(10 L)萃取。乙酸乙酯层用水(3 x 5 L)和盐水溶液(1 x 5 L)洗涤。有机层经Na
在0℃下向来自上述步骤B的标题化合物(650 g, 2.08 mol)中加入10%浓H
将市售1-异丙基哌啶-4-酮(1.56 g, 7 mmol)和2-溴-6-(1-甲基肼基)吡啶(2.23g,11.0 mmol)在1,4-二氧杂环己烷(9 mL)中的悬浮液置于冰浴中。向搅拌的悬浮液中缓慢加入浓H
向2-溴-6-(1-甲基肼基)吡啶(20.00 g, 99 mmol)和4-氧代哌啶-1-甲酸叔丁酯(19.72 g, 99 mmol)在1,4-二氧杂环己烷(100 ml)中的溶液中缓慢加入H
向来自上述步骤B的标题化合物(1.1 g, 4.13 mmol)和异丙醇钛(IV)(1.453 ml,4.96 mmol)在丙酮(10 ml)中的溶液中加入三乙酰氧基硼氢化钠(1.314 g, 6.20 mmol)。然后,反应在室温下搅拌18小时。加入1N NaOH,粗产物用DCM萃取几次。合并的有机物经Na
向1,4-二氧杂螺[4.5]癸-8-酮(30 g, 192.3 mmol)在THF(30 mL)中的悬浮液中缓慢加入在THF中的二甲胺(195 mL)。在添加后将反应混合物冷却到0℃,逐滴加入丙醇钛(IV)(84 ml, 288.45 mmol)并在室温下搅拌4小时。然后将反应混合物冷却到0℃并以分批方式加入甲醇(350 ml)、硼氢化钠(10.91 g, 288.45 mmol),然后搅拌12小时。在反应混合物完成后,加入水(200 mL)并充分搅拌。过滤固体,滤液用二氯甲烷(200 mL)萃取,用水和盐水洗涤有机相。分离有机相,经Na
来自上述步骤A的标题化合物(20 g, 1 eq)在6N浓HCl(150 mL)中在回流温度下加热2小时(~100℃)。在反应完成后,反应混合物用碳酸钾碱化(pH = 14)并充分搅拌。过滤固体,滤液用二氯甲烷(200 mL)萃取并用盐水洗涤有机相。分离有机相,经Na
向来自上述步骤B的标题化合物(10.0 g, 70.82 mmol)在乙醇中的搅拌溶液中加入羟胺盐酸盐(7.38 g, 106.23 mmol)和碳酸钾(29.3 g, 212.46 mmol)并将反应混合物在油浴中在80℃下加热2小时。将反应混合物冷却到室温并在减压下除去溶剂。粗产物用乙酸乙酯(150 mL)和水(100 mL)稀释。分离有机相,经Na
在0℃下向来自上述步骤C的粗制标题化合物(8.8 g, 56.4 mmol)在DMF(80 mL)中的搅拌溶液中加入NaH (60%)(4.02 g, 112.8 mmol)并搅拌30分钟。然后加入2-溴-6-氟吡啶(9.9 g, 56.4 mmol)并将反应混合物在室温下搅拌4小时。粗产物用乙酸乙酯(150 mL)和水(100 mL)稀释。分离有机相,经Na
将来自上述步骤D的标题化合物(5 g, 24.2 mmol)在1,4-二氧杂环己烷(50 mL)中的悬浮液置于冰浴中。向搅拌的悬浮液中缓慢加入浓硫酸(5 mL)。在添加完成后,反应混合物在回流温度下加热12小时(~110℃)。将反应混合物冷却到室温,弃置二氧杂环己烷层,并加入冰水(20 mL)。搅拌混合物直至胶质物质溶解。然后使用NaHCO
对该化合物施以SFC分离以提供纯对映体。
SFC方法: 流速: 3 ml/min,柱名称: Chirapak OX-H
助溶剂: 35%。助溶剂名称: 0.5% DEA/IPA,注射体积: 15 μl
出口压力: 100巴,温度: 40℃。
通过SFC手性柱分离异构体(0.150 g)以提供为灰白色固体的第一洗脱异构体(Rt=3.79)(100%对映体纯度)(0.05 g)和为灰白色固体的第二洗脱异构体(Rt=4.63)(100%对映体纯度)(0.04 g)。
向市售2,6-二溴吡啶(10 g, 42.2 mmol)在乙醇(50 mL)中的悬浮液中加入市售N-甲基肼(11.11 mL, 211 mmol)。混合物在80℃(反应混合物温度)下加热6小时。将反应混合物浓缩至干,残留物用二氯甲烷(200 mL)萃取,用水和盐水洗涤有机相。分离有机相,经Na
向市售1,4-二氧杂螺[4.5]癸-8-酮(30 g, 192.3 mmol)在THF(30 mL)中的悬浮液中缓慢加入在THF中的二甲胺(195 mL)。在添加后将反应混合物冷却到0℃,逐滴加入异丙醇钛(IV)(84 ml, 288.45 mmol)并在室温下搅拌4小时。然后将反应混合物冷却到0℃并以分批方式加入甲醇(350 ml)、硼氢化钠(10.91 g, 288.45 mmol),然后搅拌12小时。在反应混合物完成后,加入水(200 mL)并充分搅拌。过滤固体,滤液用二氯甲烷(200 mL)萃取,用水和盐水洗涤有机相。分离有机相,经Na
将来自上述步骤B的标题化合物(3.78 g, 27.2 mmol)和来自上述步骤A的标题化合物(5 g, 24.2 mmol)在1,4-二氧杂环己烷(50 mL)中的悬浮液置于冰浴中。向搅拌的悬浮液中缓慢加入浓硫酸(5 mL)。在添加完成后,反应混合物在回流温度下加热12小时(~110℃)。将反应混合物冷却到室温,弃置二氧杂环己烷层,并加入冰水(20 mL)。搅拌混合物直至胶质物质溶解。然后使用NaHCO
对该棕色固体施以SFC手性分离以提供纯对映体。
SFC方法: 柱名称: YMC Cellulose C
助溶剂: 20mM氨/甲醇;助溶剂流速:1.6
出口压力: 100巴,温度: 35℃。
通过SFC手性柱分离异构体(2.2 g)以提供为浅棕色固体的第一洗脱异构体(Rt=3.13)(100%对映体纯度)(0.7 g)和作为浅棕色固体的第二洗脱异构体(Rt=4.32)(100%对映体纯度)(0.6 g)。
向市售2-溴-6-氟吡啶(20 g, 113 mmol)在乙醇(200 mL)中的悬浮液中加入市售N-甲基肼(36.6 mL, 568 mmol)。混合物在115℃(反应混合物温度)下加热6小时。将反应混合物浓缩至干以提供为灰白色固体的产物(18 g, 85%)。
将来自制备实施例6步骤B的标题化合物(2.0 g, 10.79 mmol)和来自上述步骤A的标题化合物(2.02 g, 10.79 mmol)在1,4-二氧杂环己烷(10 mL)中的悬浮液置于冰浴中。向搅拌的悬浮液中缓慢加入浓硫酸(2mL)。在添加完成后,反应混合物在微波中在150℃下加热1小时。将反应混合物冷却到室温,弃置二氧杂环己烷层,并加入冰水(20 mL)。搅拌混合物直至胶质物质溶解。然后使用NaHCO
向微波管中加入来自制备实施例6的三环溴化物(第二洗脱峰)(2 g, 6.4 mmol)和30%NH
在微波管中加入2-氯苯并[d]噻唑-6-胺(950 mg, 5.16 mmol)和吗啉(10 mL)。将管密封并在微波反应器(Biotage)中在180℃下搅拌10分钟。在减压下除去溶剂并将水添加到粗产物中,并在室温下研制整夜。过滤固体,用水(2x10 mL)洗涤并干燥以提供为灰白色固体的标题化合物(2.00 g, 82%)。
在微波管中加入2-氯苯并[d]噻唑-5-胺(400 mg, 2.17 mmol)和吗啉(4 mL)。将管密封并在微波反应器(Biotage)中在180℃下搅拌10分钟。在减压下除去溶剂并将水添加到粗产物中,并在室温下研制整夜。过滤固体,用水(2x10 mL)洗涤并干燥以提供为灰白色固体的标题化合物(0.38 g, 76%)。
在微波管中加入2-氯苯并[d]噻唑-5-胺(250 mg, 1.35 mmol)和4-氟哌啶盐酸盐(419mg, 3.00 mmol)并加入DMF(4 mL),接着DIPEA(0.402 ml, 2.3 mmol)。将管密封并在微波反应器(Biotage)中在150℃下搅拌30分钟。在减压下除去溶剂,粗产物用DCM(15 mL)和饱和氯化铵溶液(15 mL)稀释。分离有机相,且有机相用水再洗涤两次。有机相经Na
根据如制备实施例11中所述的胺化程序,制备下列化合物。
向2,5-二氯苯并[d]噁唑(0.35 g, 1.86 mmol)和(R)-3-氟哌啶(0.42 g 2.97mmol)的溶液中加入乙醇(12.5 mL),反应混合物使用微波在100℃下加热60分钟。将反应冷却到室温,溶解在乙酸乙酯(100 mL)中,用水、盐水洗涤并经Na
将2-氨基硝基酚(1 g, 6.48 mmol)置于吡啶中,加入乙基黄原酸钾(1.14 g, 7.13mmol)。混合物在120℃下加热8小时。在反应完成后,混合物用1.5N HCl酸化。通过过滤收集所得固体并在真空下干燥以提供为黄色固体的标题化合物(1 g, 78%)。
向来自上述步骤A的标题化合物(1 g, 5.09 mmol)在二甲苯(10 mL)中的溶液中加入三乙胺(2.14 mL, 15.29 mmol)和3-氟哌啶(0.52 g, 5.09 mmol)。所得混合物在100℃下加热16小时。在反应完成后,蒸发混合物,将残留物溶解在乙酸乙酯中并用水洗涤。有机层经无水Na
向来自上述步骤B的标题化合物(1.1 g, 4.15 mmol)在乙醇和水(2:1比率)中的溶液中加入铁粉(1.14 g, 20.75 mmol)和氯化铵(2.19 g, 41.5 mmol)。所得悬浮液在90℃下加热2小时。在反应完成后,混合物经硅藻土过滤,蒸发滤液且粗产物通过快速色谱法提纯以产生2-(3-氟哌啶-1-基)-2,3-二氢苯并[d]噁唑-6-胺(0.2 g, 20%)。
通过手性分离法分离两种异构体以获得各对映体。
方法: 流动相:0.1% DEA/正己烷:ETOH::60:40
柱: CHIRALCEL OJ-H(250X4.6)mm,5μm。
通过手性柱分离异构体(0.2 g)以提供为灰白色固体的第一洗脱异构体(Rt=14.2)(100%对映体纯度)(0.04 g)和为灰白色固体的第二洗脱异构体(Rt=21.7)(100%对映体纯度)(0.04 g)。
1,4-二氧杂环己烷(4 mL)用氩气脱气10分钟。加入乙酸钯(II)(0.0675 g, 0.301mmol)和二环己基(2',4',6'-三异丙基-[1,1'-联苯]-2-基)膦(XPhos, 0.43 g, 0.902mmol)。悬浮液然后在110℃下加热2分钟。然后加入来自制备实施例4的标题化合物(0.927mg, 3.01 mmol)、来自制备实施例9的标题化合物(0.849 g, 3.61 mmol)和叔丁醇钠(0.867 g, 9.02 mmol)并将反应混合物在110℃下搅拌2小时。将反应冷却到室温并浓缩至干。加入1 N NaOH,水相用二氯甲烷/甲醇萃取几次。合并的有机物经Na
抽空烘干的schlenk烧瓶并用氩气回填。然后加入2-二环己基膦基-2′,4′,6′-三异丙基联苯(XPhos, 0.046 g, 0.10 mmol)和乙酸钯(II)(0.007 g, 0.03 mmol),接着加入干燥1,4-二氧杂环己烷(8 mL)并在砂浴中在110℃下加热90秒以生成催化剂(清澈红色溶液)。加入来自制备实施例6第二洗脱峰的标题化合物(0.100 g, 0.32 mmol)、来自制备实施例10的标题化合物(0.091 g, 0.39 mmol)和叔丁醇钠(0.102 g, 1.07 mmol)。混合物在砂浴中在~110℃下加热4小时。混合物用二氯甲烷(200 mL)稀释并用水(100 mL)和盐水溶液(100 mL)洗涤。有机相经Na
将乙酸钯(II)(0.00583 g, 0.026 mmol)和4,5-双(二苯基膦基)-9,9-二甲基呫吨(XantPhos, 0.0451 g, 0.078 mmol)添加到反应小瓶中并加入脱气的1,4-二氧杂环己烷(4 ml)。用氩气填充小瓶并密封。悬浮液在110℃下加热1分钟并加入来自制备实施例6的标题化合物(0.08 g, 0.260 mmol)、来自制备实施例11的标题化合物(0.078 g, 0.311mmol)和碳酸铯(0.254 g, 0.779 mmol),并将溶液在110℃下加热4小时。反应混合物用乙酸乙酯(10 mL)和水(10 mL)稀释。分离有机相,水相用乙酸乙酯再萃取两次。合并的有机相经Na
将乙酸钯(II)二乙酰氧基钯(0.0038 g, 0.017 mmol)和二环己基(2',4',6'-三异丙基-[1,1'-联苯]-2-基)膦(XPhos, 0.024mg, 0.050 mmol)添加到反应小瓶中并加入脱气的1,4-二氧杂环己烷(4 ml)。用氩气填充小瓶并密封。悬浮液在110℃下加热1分钟并加入来自制备实施例4的标题化合物(0.050 g, 0.16 mmol)、来自制备实施例10的标题化合物(0.0485 g, 0.20 mmol)和叔丁醇钠(0.0540 g, 0.56 mmol),并将溶液在110℃下加热3小时。反应混合物用乙酸乙酯(5 mL)和水(5 mL)稀释。分离有机相,水相用乙酸乙酯再萃取两次。合并的有机相经Na
来自制备实施例5的标题化合物(0.10 g, 0.33 mmol)和来自制备实施例10的标题化合物(0.063 g, 0.27 mmol)在1,4-二氧杂环己烷(5 mL)中的搅拌溶液用氮气脱气15分钟。然后加入叔丁醇钠(0.086 g, 0.99 mmol)、2-二环己基膦基-2′,6′-二异丙氧基联苯(RuPhos, 0.016 g, 0.033 mmol)和三(二亚苄基丙酮)二钯(0)(Pd
根据如制备实施例1、2、3、4和5中所述的钯偶联程序,制备下列化合物。
生物测定法描述
通过硫磺素T (ThT)测定全长Tau (flTau)解聚
在细菌中表达并纯化人类Tau的最长同种型(2N4R;441个氨基酸)。为了通过ThT测定Tau解聚,使PBS中的35 µM重组全长(fl)Tau在37℃下在50 µM的肝素(Sigma-Aldrich)和10mM的DTT(Sigma-Aldrich)存在下在以750 RPM摇动下聚集24小时。将化合物溶解在无水二甲亚砜(DMSO, Sigma-Aldrich)中以达到10 mM的浓度。将flTau聚集体和化合物的连续稀释液在PBS(体积50 µL)中混合在一起至2 μM的flTau聚集体和160至0.04 µM的化合物的最终浓度。混合物在室温(RT)下孵育30分钟,然后将40 µL这种混合物转移到黑色384孔检测板(Perkin-Elmer)中并与10 µL的100 µM ThT/250 mM甘氨酸(两者都来自Sigma-Aldrich)在PBS中混合。在Tecan读取器(激发: 440 nm;发射: 485 nm)上一式一份(monoplicate)或一式两份测量荧光(相对荧光单位;RFU)。然后计算flTau解聚的百分比并使用GraphPadPrism版本5(GraphPad软件)假设单结合位点拟合模型测定半最大效应浓度(EC
通过ThT测定Tau K18解聚
在细菌中表达并纯化包含人类Tau441的最长同种型(2N4R)的氨基酸244至372的TauK18片段,或购自SignalChem。为了通过ThT测定K18解聚,使PBS中的35 µM重组K18在37℃下在50 µM的肝素(Sigma-Aldrich)和10 mM的DTT(Sigma-Aldrich)存在下在以750 RPM摇动下聚集24小时。将化合物溶解在无水二甲亚砜(DMSO, Sigma-Aldrich)中以达到10 mM的浓度。将K18聚集体和化合物的连续稀释液在PBS(体积50 µL)中混合在一起至2 μM的K18聚集体和160至0.04 µM的化合物的最终浓度。混合物在室温(RT)下孵育30分钟,然后将40 µL这种混合物转移到黑色384孔检测板(Perkin-Elmer)中并与10 µL的100 µM ThT/250 mM甘氨酸(两者都来自Sigma-Aldrich)在PBS中混合。在Tecan读取器(激发: 440 nm;发射: 485nm)上一式一份(monoplicate)或一式两份测量荧光(相对荧光单位;RFU)。然后计算K18解聚的百分比并使用GraphPad Prism版本5(GraphPad软件)假设单结合位点拟合模型测定半最大效应浓度(EC
测量下列实施例化合物:
细胞内Tau聚集的减少
过度表达携带P301L突变的全长形式的人类Tau的人神经母细胞瘤细胞系在完全培养基 [DMEM-F12 4.5 g/L Glutamax (Invitrogen), 15% FBS (Biochrom), 1% Peni/Strep(Invitrogen),补充了2.5 µg/ml的G418 (Sigma-Aldrich)选择抗生素]中培养。在实验前一天,在6孔板中在3毫升完全培养基中接种5x10
通过AlphaLisa使用下列抗体对量化磷酸化Tau、聚集Tau和总Tau:
·HT7-受体珠 + 生物素(BT)-Tau13-供体珠: 总Tau
·HT7-受体珠+ 生物素(BT)-HT7-供体珠: 聚集人Tau
Tau13 (Abcam)使用EZ-Link® NHS-PEO Solid Phase生物素化试剂盒(ThermoScientific)生物素化,而HT7-生物素来自商业来源(Thermo Scientific)。
对于每个抗体对,优化受体珠和生物素化抗体的浓度。所有样品首先在PBS中连续稀释测试,以确定用于各样品和测定的线性范围和最佳稀释。对于最终程序,在384孔白色OptiPlate (PerkinElmer)中加入下列试剂:
·5 µL的受试稀释样品
·20 µL的以下最终浓度的混合物生物素-mAb受体珠:
·1.25 nM的 HT7-BT以及10 µg/ml的HT7-Acc珠
·5 nM的Tau13-BT以及2.5 µg/ml的HT7-Acc珠
在室温下孵育这种混合物1小时后,在暗处加入25 µL的25 µg/mL的链霉亲和素供体珠(Streptavidin Donor beads)(Perkin Elmer)。在孵育30分钟后使用EnSpire Alpha仪器和EnSpire Workstation版本3.00分析板。将聚集Tau的数据相对于总Tau归一化,然后表示为DMSO处理细胞的百分比。
测量下列实施例化合物:
本发明的化合物的体内效力
双转基因rTg4510小鼠是在四环素诱导型CaMKII启动子的控制下表达携带P301L突变的全长人类Tau(Tau4R0N-P301L)的侵袭型(aggressive)Tau蛋白病模型(Ramsden等人, J.Neurosci., 2005 • 25(46):10637–10647)。使用野生型动物和仅表达四环素控制的反式激活因子(tTA)的单转基因小鼠作为基因型对照。该研究包括6个治疗组(n = 20只小鼠/组),组分布如下(参见表5)。在5.5周龄开始,每天一次通过灌胃给药化合物或媒介物对照共12.5周。在治疗12.5周后经过连续5天进行Morris水迷宫(MWM)行为试验。在MWM测试过程中,继续每天给药。仅对雌性小鼠(大约n=11只雌性/组)进行行为测试(MWM)。对每组7只动物(不包括野生型动物)进行皮质萎缩和错误折叠的Tau (MC1)和CD68的组织学的测量。
表5: 用于测试实施例1和实施例12的体内研究设计
(a) 媒介物: 0.5% w/v羟丙基甲基纤维素4000 cps;0.5% w/v Tween 80。
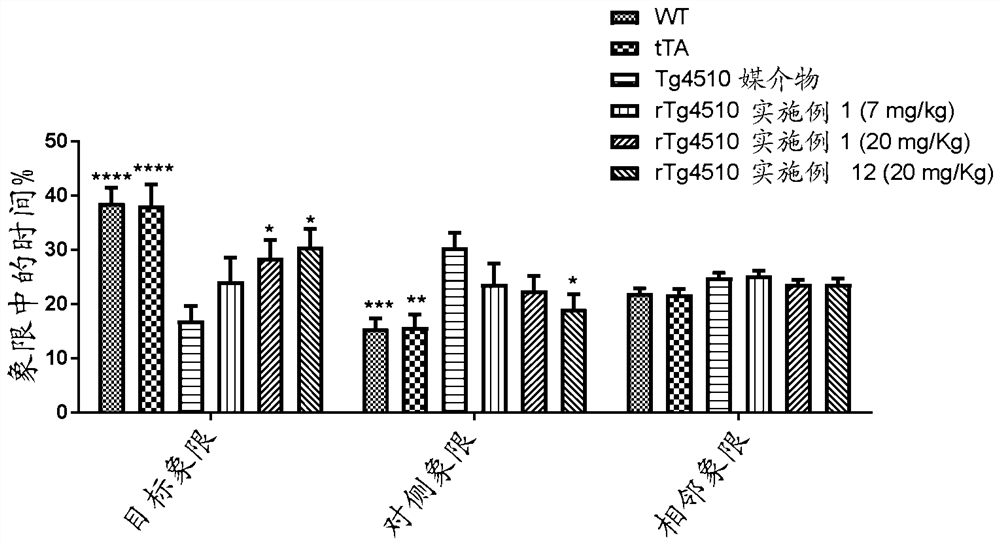
在测得为48”直径的圆形槽中进行MWM。水深大约24-25”,并用无毒漆染为白色。水保持在25℃ ±1℃。在水槽周围安装迷宫外暗示。视频记录试验并通过计算机程序(WaterMaze;Actimetrics, Wilmette, IL)分析。在第一周的连续5天,用浸没在水面下大约1.5 cm的平台进行训练周期(training sessions)。每天有4个60秒长的试验,试验之间间隔大约15分钟。在训练的第五天,最后第四个试验由无平台的60秒组成(探索试验)。在治疗组之间平衡平台位置和释放点。对探索试验计算在每个象限(目标象限、左和右相邻象限和对侧象限)中花费的时间%。将与相邻象限对应的数据平均化。通过双因素ANOVA、接着事后比较来分析数据,所示统计学参考与媒介物治疗的rTg4510小鼠相比的差异。
如图1中所示,本发明的化合物,实施例1,表现出在目标象限中花费的时间%的剂量依赖性增加。另一种本发明的化合物,实施例12,也表现出在目标象限中花费的时间%的显著增加。这些数据表明在rTg4510中用这两种受试化合物治疗都显著改善这种侵袭型(aggressive)Tau转基因模型的记忆表现。
在行为测试后,通过标准注射麻醉将小鼠深度麻醉并用冷PBS经心脏灌注。然后取出大脑并半矢状切成对半。左半球在干冰中速冻并在用于生化分析前储存在-80℃。右半球在4%低聚甲醛/PBS中在室温下固定3小时,通过浸在15%蔗糖中在4℃下冷藏保存3天,并准备好冷冻切片。然后将固定的半球在干冰中冷冻在冷冻模具(cryo molds)中的OCT介质上并用Leica CM3050S低温切片机矢状冷冻切片(10 μm厚)。从中线外侧0.2 mm开始,从大约10个中外侧水平(mediolateral levels)收集来自每组7只小鼠(不包括野生型动物)的切片,安装在载片上,并用于量化皮质大小。来自水平 4的切片用于MC1组织学染色。简言之,切片用免疫组化笔(pap pen liquid blocker)划圈并在PBS中洗涤三次5分钟。用PBS中的10%纯山羊血清(NGS)和0.25% Triton X-100在室温下进行封闭和透化2小时。然后将切片用纸吸干并在湿室中在4℃下用1:1000稀释在含5% NGS和0.25% Triton-X 100的PBS中的小鼠单抗MC-1抗体孵育整夜。在PBS中洗涤三次5分钟后,切片用1:1000在PBS中的山羊抗小鼠IgGAlexaFluo 555 (Invitrogen)二抗在室温下孵育30分钟,随后在PBS中洗涤三次5分钟。为了减少组织的自体荧光,切片用0.1% Sudan Black (Sigma)在70%乙醇中的溶液在室温下孵育15分钟,然后在PBS中洗涤四次5分钟,并使用ProLong Gold Antifade试剂(Invitrogen)封固(mounted)。在Nikon Eclipse Ti显微镜上目视检查染色,用Nikon DS-Fi2摄像机成像并使用NIS-Element AR4.13.1软件量化。报道的值是每个样品三张单独照片的量化平均值。
每只动物五个矢状切片的一组系统随机切片(总共175个切片)用于CD68组织学染色。简言之,针对大鼠抗小鼠CD68克隆FA-11 (BD Biosciences)标记切片并用DAPI复染色。使用高度交叉吸收的(highly cross-absorbed)荧光标记二抗(Thermo Fisher)使抗体结合可视化。在抗体稀释剂(Dako)中稀释抗体,在初次孵育前用M.O.M.血清(Vector)阻断非特异性内源性IgG结合。封固(mounted)的切片整体在通过ZEN软件驱动的Axio.Scan Z1玻片扫描仪上在20x放大率(平场复消色差物镜)下使用LED (Colibri2)照明和灵敏的OrcaFlash 4.0单色摄像机成像。通过单因素ANOVA、接着事后比较来分析数据,所示统计学参考与媒介物治疗的rTg4510小鼠相比的差异。
如图2A中所示,本发明的化合物,实施例1,表现出皮质大小的剂量依赖性增加。另一种本发明的化合物,实施例12,也表现出皮质大小的显著增加。这些数据表明在rTg4510中用这两种受试化合物治疗都缓解了这种侵袭型(aggressive)Tau转基因模型的皮质萎缩。
对皮质萎缩的修复(rescue)还辅以由MC1组织学测得的Tau错误折叠的减少。如图2B中所示,本发明的化合物,实施例1,表现出错误折叠的Tau的剂量依赖性减少。另一种本发明的化合物,实施例12,也表现出错误折叠的Tau的显著减少。这些数据表明在rTg4510中用这两种受试化合物治疗都减少了这种侵袭型(aggressive)Tau转基因模型中的病理性Tau的含量。
为了评估受试化合物对Tau的上述效应是否也对神经炎症标志物有效,使用将活动过度的小胶质细胞染色的小胶质细胞吞噬标志物(microglia phagocytosis marker)CD68评估本发明的化合物对神经炎症的作用。如图3中所示,本发明的化合物,实施例1,表现出CD68阳性小胶质细胞的减少。另一种本发明的化合物,实施例12,也表现出CD68阳性小胶质细胞的显著减少。这些数据表明在rTg4510中用这两种受试化合物治疗都降低了这种侵袭型(aggressive)Tau转基因模型中的神经炎症水平。
双转基因rTg4510小鼠是在四环素诱导型CaMKII启动子的控制下表达携带P301L突变的全长人类Tau(Tau4R0N-P301L)的侵袭型(aggressive)Tau蛋白病模型(Ramsden等人,2005)。使用野生型动物和仅表达四环素控制的反式激活因子(tTA)的单转基因小鼠作为基因型对照。治疗并测试小鼠。该研究包括4个治疗组(n = 15只雌性小鼠/组),组分布如下(参见表6)。在5.5周龄开始,每天一次通过灌胃给药本发明的化合物或媒介物对照共12周。在治疗12.5周后经过连续5天进行Morris水迷宫(MWM)行为试验。在MWM测试过程中,继续每天给药。对所有小鼠(n=15只小鼠/组)进行行为测试(MWM)和sarkosyl不溶性Tau的生化量化。对10只小鼠/组进行皮质萎缩和Iba1的组织学的测量。
表6: 用于测试实施例2的体内研究设计
(a) 媒介物: 0.5% w/v羟丙基甲基纤维素4000 cps;0.5% w/v Tween 80。
在测得为48”直径的圆形槽中进行MWM。水深大约24至25”,并用无毒漆染为白色。水保持在25℃±1℃。在水槽周围安装迷宫外暗示。视频记录试验并通过计算机程序(WaterMaze;Actimetrics, Wilmette, IL)分析。在第一周的连续5天,用浸没在水面下大约1.5 cm的平台进行训练周期。每天有4个60秒长的试验,试验之间间隔大约15分钟。在训练的第五天,最后第四个试验由无平台的60秒组成(探索试验)。在治疗组之间平衡平台位置和释放点。对探索试验计算在每个象限(目标象限、左和右相邻象限和对侧象限)中的穿越%。通过双因素 ANOVA、接着事后比较来分析数据,所示统计学参考与媒介物治疗的rTg4510小鼠相比的差异。
如图4中所示,本发明的化合物,实施例2,表现出与对侧象限相比在目标象限中的平台穿越%的剂量依赖性增加,在20 mg/kg的剂量下达到统计显著性。这些数据表明在rTg4510中用受试化合物治疗显著改善这种侵袭型(aggressive)Tau转基因模型的记忆表现。
在行为测试后,通过标准注射麻醉将小鼠深度麻醉并用冷PBS经心脏灌注。然后取出大脑并半矢状切成对半。从左脑半球剖开皮质,称重,迅速浸在液氮中并储存在-80℃直至用于生化分析。为了制备皮质全脑匀浆(Cx-TBH),将冷冻的皮质在9体积(ml)/重量(g)的冰冷均质缓冲液[25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA,含磷酸酶抑制剂(30 mM NaF, 0.2 mM Na
如图5中所示,本发明的化合物,实施例2,表现出sarkosyl不溶性Tau的剂量依赖性减少,在20 mg/kg的较高剂量下达到统计显著性。这些数据表明在rTg4510中用受试化合物治疗减少这种侵袭型(aggressive)Tau转基因模型中的病理性Tau。
在行为测试后,通过标准注射麻醉将小鼠深度麻醉并用冷PBS经心脏灌注。然后取出大脑并半矢状切成对半。右半球在4%低聚甲醛/PBS中在室温下固定3小时,通过浸在15%蔗糖中在4℃下冷藏保存3天,并准备好冷冻切片。然后将固定的半球在干冰中冷冻在冷冻模具(cryo molds)中的OCT介质上并用Leica CM1950低温切片机矢状冷冻切片(10 μm厚)。从大约12个中外侧水平(mediolateral levels)收集来自每组10只小鼠的切片。来自每只动物七个矢状水平切片的一组系统随机切片用于量化皮质大小。如图6A中所示,本发明的化合物,实施例2,表现出皮质大小的剂量依赖性增加,在较高剂量下达到统计显著性。这些数据表明在rTg4510中用受试化合物治疗缓解这种侵袭型(aggressive)Tau转基因模型的皮质萎缩。
也使用来自层6的每只动物的一个切片量化Iba1免疫反应性。简言之,从-20℃取出冷冻切片并在室温下晾干25分钟。脑组织用免疫组化笔(pap pen liquid blocker)划圈并在PBS中在室温下洗涤5分钟一次和10分钟一次。用PBS中的10%正常山羊血清(NGS)和0.25% Triton X-100在室温下进行封闭和透化2小时。然后将切片用纸吸干并在湿室中在4℃下用1:450稀释在含5% NGS和0.25% Triton X-100的PBS中的兔多克隆Iba1抗体(Wako)孵育整夜。切片在室温下在PBS中洗涤三次10分钟,并用1:1000稀释在PBS中的Cy3-标记的山羊抗兔IgG (H+L)二抗在室温下避光孵育30分钟。在PBS中洗涤三次10分钟后,切片用0.1% Sudan Black (Sigma)在70%乙醇中的溶液在室温下孵育30秒以减少组织的自体荧光。切片在PBS中洗涤三次10分钟,使用含DAPI的ProLong Gold Antifade试剂 (MolecularProbes)封固(mount)并封片。切片使用数字玻片扫描仪(Pannoramic 250 BF Flash + FL,3D Histech Ltd.)成像并使用图像可视化软件CaseViewer和图像分析软件模块HistoQuant (3D Histech Ltd.)量化。
如图6B中所示,本发明的化合物,实施例2,在20 mg/kg下表现出Iba1免疫反应面积的显著减少。这些数据表明在rTg4510中用受试化合物治疗降低这种侵袭型(aggressive)Tau转基因模型中的小胶质细胞增生和因此神经炎症。
- 用于治疗、缓解或预防与Tau聚集体相关的病症的新型化合物
- 用于治疗、缓解或预防与Tau聚集物相关病症的四氢苯并呋喃并2.3-c吡啶和β-咔啉化合物