乐伐替尼杂质及其制备方法
文献发布时间:2023-06-19 10:02:03
技术领域
本发明涉及一种乐伐替尼杂质及其制备方法,尤其涉及可以精准控制特定杂质含量、提升中间体、原料药及相关制剂产品质量的乐伐替尼杂质及其制备方法。
背景技术
乐伐替尼(Lenvatinib)是由日本卫材公司开发的一种口服多酪氨酸激酶抑制剂。2012年8月,在日本被授予孤儿药(ODD)治疗甲状腺癌;2013年2月,获得美国FDA孤儿药认定,用于治疗滤泡状、髓样、未分化的转移性或局部晚期甲状腺乳头状癌;2015年,美国FDA和欧洲药品管理局EMA批准乐伐替尼用于治疗侵袭性、局部晚期或转移性分化型甲状腺癌;2016年,美国FDA和欧洲药品管理局EMA批准乐伐替尼联合依维莫司用于治疗晚期肾细胞癌;2018年3月,乐伐替尼在日本获批用于不可切除的肝细胞癌的一线治疗。
目前,乐伐替尼常用的合成路线如下所示:
研究发现,通过上述制备方法得到乐伐替尼原料药中常常存在若干未知结构的杂质化合物,这些杂质化合物含量低,难以分离,增加了乐伐替尼中间体及原料药的质量控制难度,进而影响其制剂相关产品的质量。
发明内容
发明目的:本发明的第一目的是提供一种乐伐替尼杂质,第二目的是提供所述乐伐替尼杂质的制备方法。
技术方案:本发明的乐伐替尼杂质,具有如下式VI的结构:
VI。
本发明制备得到的乐伐替尼杂质纯度为95%以上,甚至为97%以上。
在乐伐替尼的常规制备方法中,经研究发现,第一步反应得到的产物中会残留少量的起始原料3-氯-4-氨基苯酚,其与第一步产物继续反应,从而产生杂质中间体,杂质中间体会继续参与到乐伐替尼合成的后续反应中,最后生产杂质VI。杂质VI在乐伐替尼中间体及原料药中含量低、不易分离提纯,难以获得大量的、高纯度的上述杂质,无法对该杂质进行定性定量检测,从而难以对中间体、原料药及制剂相关产品进行精准质量控制。
上述乐伐替尼杂质的制备方法如下:
2-氟-4-羟基苯胺为起始物料,经两步烷基化反应制备得到式V的乐伐替尼杂质中间体,然后经缩合反应制备得到式VI的乐伐替尼杂质。
具体包括以下步骤:
第一步,将化合物I、化合物II溶于有机溶剂中,再加入碱,加热至反应温度,反应至终点,得到化合物III;
第二步,将化合物III、化合物IV溶于有机溶剂中,再加入碱,加热至反应温度,反应得到化合物V;
第三步,将化合物V、异氰酰基环丙烷溶于有机溶剂中,加热至反应温度,保温反应得到化合物VI。
其中,步骤(1)和(2)烷基化反应的反应溶剂为DMF、DMSO中的一种或多种,催化剂为氢氧化钠、氢氧化钾或碳酸铯,反应温度为80℃~120℃;
步骤(3)缩合反应的反应溶剂为DMF、DMSO、THF中的一种或多种,催化剂为无机碱或有机碱,反应温度优选为20℃~30℃;无机碱优选为碳酸氢钠、碳酸氢钾、碳酸钠或碳酸钾,有机碱优选为三乙胺、吡啶或DMAP,更优选为三乙胺或吡啶。
有益效果:与现有技术相比,本发明具有如下显著优点:
(1)经过对乐伐替尼制备过程产生的特定杂质进行结构确证,并制备出纯度(>95%)及批量(达到十克级)符合检测要求的杂质,可以对乐伐替尼中间体、原料药及相关制剂产品中相应杂质进行准确的定性与定量,有利于提升产品质量;
(2)杂质制备方法简便,所得杂质中间体、杂质均具有稳定的纯度和收率。
附图说明
图1为本发明实施例1制备化合物V的气相色谱图;
图2为本发明实施例1制备化合物V的质谱图;
图3为本发明实施例1制备化合物VI的气相色谱图;
图4为本发明实施例1制备化合物VI的质谱图。
具体实施方式
下面结合实施例对本发明的技术方案作进一步说明。
实施例1
(1)化合物III的制备方法
向1L反应釜中加入化合物I 12.71g、化合物II 23.66g,DMSO 500mL,氢氧化钠6.0g,升温至100℃反应2h,将反应液分散于水相与乙酸乙酯相中,分去水层,有机相减压浓缩至干,得到紫色固体31g,纯度96%。
(2)化合物V的制备方法
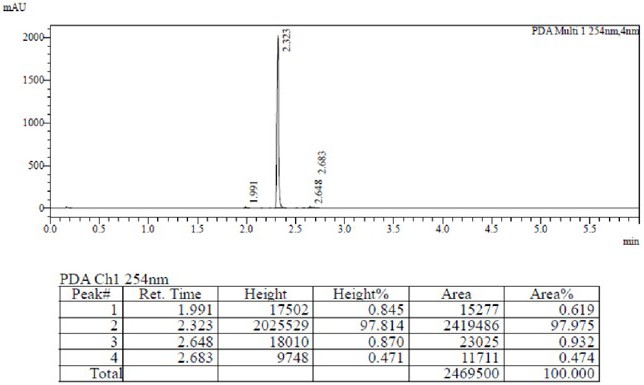
向1L反应釜中加入式III化合物 28.4g、化合物IV 14.57g,DMSO 800mL,氢氧化钠6.0g,升温至100℃反应2h,将反应液柱层析纯化,得到黄色固体41.4g,纯度100%,HPLC检测结果见图1。
(3)化合物VI的制备方法
向500mL反应釜中加入化合物V10g(0.022mol),DMSO 100mL,异氰酰基环丙烷10.96g(0.132mol),三乙胺13.36g(0.132mol),控温20~30℃反应3h。向反应液中滴加水淬灭,抽滤,得类白色固体12.7g,纯度98%,HPLC检测结果见图3。
实施例2
(1)化合物III的制备方法
向1L反应釜中加入化合物I 12.71g、化合物II 23.66g,DMF 500mL,氢氧化钾12.6g,升温至120℃反应2h,将反应液分散于水相与乙酸乙酯相中,分去水层,有机相减压浓缩至干,得到紫色固体30.3g,纯度95.8%。
(2)化合物V的制备方法
向1L反应釜中加入化合物III 28.4g、化合物 IV14.57g,DMF 800mL,氢氧化钾12.6g,升温至80℃反应3h,将反应液柱层析纯化,得到黄色固体40.8g,纯度99.5%。
(3)化合物VI的制备方法
向500mL反应釜中加入化合物V10g,DMF 100mL,异氰酰基环丙烷10.96g,吡啶10g,控温20~30℃反应3h。向反应液中滴加水淬灭,抽滤,得类白色固体12g,纯度97%。
实施例3
(1)化合物III的制备方法
向1L反应釜中加入化合物I 12.71g、化合物II 23.66g,DMF 500mL,碳酸铯73.3g,升温至80℃反应2h,将反应液分散于水相与乙酸乙酯相中,分去水层,有机相减压浓缩至干,得到紫色固体30.2g,纯度96.5%。
(2)化合物V的制备方法
向1L反应釜中加入化合物III 28.4g、化合物IV 14.57g,DMF 800mL,碳酸铯73.3g,升温至120℃反应2h,将反应液柱层析纯化,得到黄色固体40g,纯度99.9%。
(3)化合物VI的制备方法
向500mL反应釜中加入化合物V 10g,THF 100mL,异氰酰基环丙烷10.96g,吡啶10g,控温20~30℃反应3h。向反应液中滴加水淬灭,抽滤,得类白色固体12g,纯度97%。
实施例4
(1)化合物III的制备方法
向1L反应釜中加入化合物I 12.71g、化合物II 23.66g,DMF 500mL,氢氧化钾12.6g,升温至90℃反应2h,将反应液分散于水相与乙酸乙酯相中,分去水层,有机相减压浓缩至干,得到紫色固体30.8g,纯度96.3%。
(2)化合物V的制备方法
向1L反应釜中加入化合物III 28.4g、化合物IV 14.57g,DMF 800mL,氢氧化钾12g,升温至110℃反应2h,将反应液柱层析纯化,得到黄色固体40.2g,纯度99.8%。
(3)化合物VI的制备方法
向500mL反应釜中加入化合物V 10g,THF 100mL,异氰酰基环丙烷10.96g,吡啶10g,控温20~30℃反应3h。向反应液中滴加水淬灭,抽滤,得类白色固体12.1g,纯度97.2%。
实施例5
(1)化合物III的制备方法
向1L反应釜中加入化合物I 12.7g、化合物II 23.6g,DMF 500mL,氢氧化钾12.6g,升温至110℃反应2h,将反应液分散于水相与乙酸乙酯相中,分去水层,有机相减压浓缩至干,得到紫色固体30.4g,纯度95.8%。
(2)化合物V的制备方法
向1L反应釜中加入化合物III 28.4g、化合物IV 14.57g,DMF 800mL,氢氧化钾12.5g,升温至120℃反应2h,将反应液柱层析纯化,得到黄色固体40.5g,纯度99.8%。
(3)化合物VI的制备方法
向500mL反应釜中加入化合物V 10g,THF 100mL,异氰酰基环丙烷11g,吡啶10.5g,控温20~30℃反应3h。向反应液中滴加水淬灭,抽滤,得类白色固体12.2g,纯度98.1%。
实施例6:化合物VI的制备
向500mL反应釜中加入化合物V 10g,DMF 200mL,异氰酰基环丙烷11g,碳酸钾19g,控温20~30℃反应5h。向反应液中滴加水淬灭,抽滤,得类白色固体9g,纯度97%。
实施例7:化合物VI的制备
向500mL反应釜中加入化合物V 10g,DMF 200mL,异氰酰基环丙烷11g,碳酸钠15g,控温20~30℃反应5h。向反应液中滴加水淬灭,抽滤,得类白色固体8.5g,纯度97.2%。
对比例1
(1)化合物III的制备方法
向1L反应釜中加入化合物I 12.71g、化合物II 23.66g,DMF 500mL,碳酸铯73.3g,升温至50℃反应8h,将反应液分散于水相与乙酸乙酯相中,分去水层,有机相减压浓缩至干,得到紫色固体12g。
(2)化合物V的制备方法
向1L反应釜中加入化合物III 28.4g、化合物IV 14.57g,DMF 800mL,碳酸铯73.3g,升温至160℃反应2h,将反应液柱层析纯化,得到黄色固体18g,纯度80.8%。
(3)化合物VI的制备方法
向500mL反应釜中加入化合物V 10g,THF 100mL,异氰酰基环丙烷10.96g,吡啶10g,控温0~5℃反应12h。向反应液中滴加水淬灭,抽滤,得类白色固体3g。
- 乐伐替尼杂质及其制备方法
- 一种乐伐替尼杂质的制备方法