酒石酸匹莫范色林晶型A的制备方法
文献发布时间:2023-06-19 11:19:16
技术领域
本申请涉及药物和化学领域,具体涉及酒石酸匹莫范色林晶型A的制备方法。
背景技术
酒石酸匹莫范色林由阿卡迪亚(ACADIA)公司开发并于2016年4月获美国食品与药品监督管理局(FDA)批准上市,商品名为
目前匹莫范色林最优的制备路线如下:
匹莫范色林在溶剂中与酒石酸成盐制备得到酒石酸匹莫范色林。酒石酸匹莫范色林存在多晶型现象,CN101035759A中公开了6种晶型,即晶型A、B、C、D、E和F。其中晶型C为稳态晶型,其余晶型均为亚稳态晶型。药用晶型C主要由晶型A转晶制备得到。因此晶型A的制备成为了酒石酸匹莫范色林制备的关键步骤,但是现有技术制备得到的晶型A批间差异较大,其晶型纯度较低,含有其他未知晶型,使得制备得到的晶型C收率和晶型纯度较低。
基于上述现状,急需提出一种稳定的酒石酸匹莫范色林晶型A的制备方法。
发明内容
本发明提供一种酒石酸匹莫范色林晶型A的制备方法,该方法得到的晶型A纯度高,且批间稳定。
一种酒石酸匹莫范色林晶型A的制备方法,包括:
将式(I)化合物溶于第一溶剂中;
将所得溶液降温;以及
分离得到沉淀固体;
其中,所述第一溶剂为浓度为90.0%-98.5%的乙醇水溶液、90.0%-98.5%的异丙醇水溶液,或90.0%-98.5%的乙醇和异丙醇的混合水溶液。
发明人研究发现,采用上述第一溶剂进行重结晶,相对于现有方法的溶剂尤其是无水乙醇,可以更稳定地获得酒石酸匹莫范色林晶型A,并且提高了纯度。其可能的原因在于酒石酸匹莫范色林晶型A理论为半水合物,但是其中的水只是微弱的结合,温度在室温以上时就开始失去其中的水。本发明人研究发现,以含有一定水的溶液为结晶溶剂,有利于其在结晶过程中形成晶型A,避免了批间差异。进一步地,因为后期还需对结晶得到的产品进行干燥,如果含水量较大则增加干燥的困难,如果含水量较小则影响晶型A的纯度,所以采用以上浓度的结晶溶剂。
在一些实施例中,所述第一溶剂(即乙醇水溶液,异丙醇水溶液,乙醇和异丙醇的混合水溶液)的浓度为90.0%、95.0%、97.5%、98.0%或98.5%,优选为98%±0.5%。
在一些实施例中,所述乙醇和异丙醇的混合水溶液中,乙醇和异丙醇的重量比为1:1。
在一些实施例中,所述第一溶剂更优选为98%±0.5%的乙醇水溶液。
在一些实施例中,所述第一溶剂的用量为式(I)化合物投料量的3-8倍量(重量比),优选5±0.5倍量。
在一些实施例中,式(I)化合物可为其晶型例如A、B、C、D、E、F中任一种或不同晶型的混合物。其中,优选的是式(I)化合物的晶型A。实验证明,采用式(I)化合物的晶型A进行上述重结晶(精制)可以显著地提高其纯度,还可以提高批间的稳定性。
本文中,酒石酸匹莫范色林晶型A、B、C、D、E、F的定义参照CN101035759A,其全文引做本文作为参考。
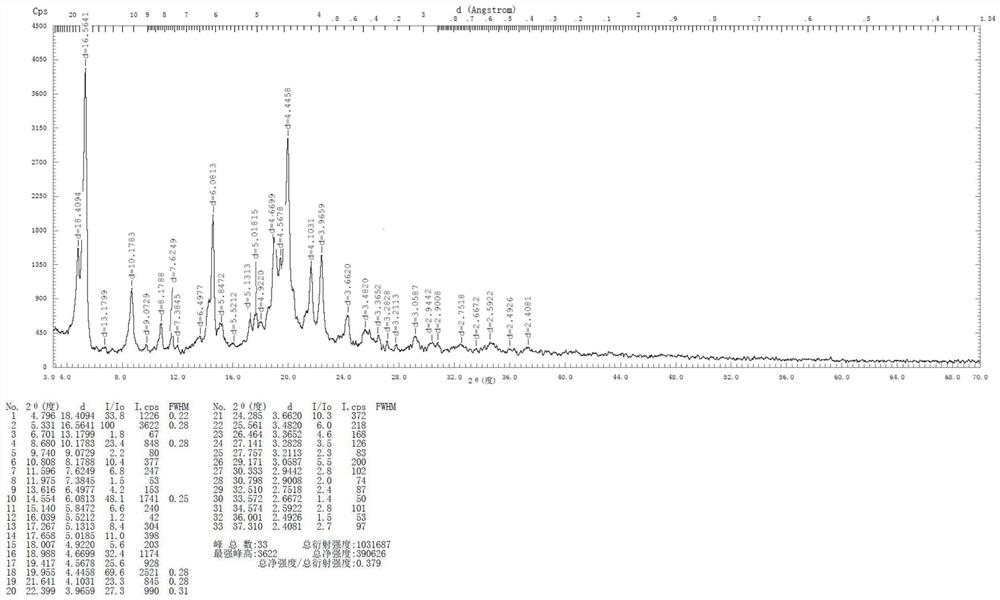
本文具体实施例中,酒石酸匹莫范色林晶型A的XRD图谱参见图1;该晶型A的DSC图谱参见图2。
在一些实施例中,将式(I)化合物与所述第一溶剂混合,搅拌升温至回流(75-78℃),待所得溶液溶清后再降温。这样可以将酒石酸匹莫范色林充分溶解,避免不溶物对精制工艺的影响。
在一些实施例中,在制备酒石酸匹莫范色林晶型A的过程中,将所得溶液降温至15-25℃(例如20℃)析晶。
在一些实施例中,将所得溶液降温的具体过程包括:将所得溶液先降温至45-50℃,保持一定时间(例如1-2h),再自然冷却至35℃,最后再降温并控温15-25℃析晶。
在一些实施例中,将所得溶液降温的具体过程包括:将所得溶液降温至45-50℃析晶0.5h,自然冷却降温至35℃,随后降温并控温15-25℃析晶2h。然后分离,将所得沉淀固体用所述第一溶剂淋洗(例如淋洗2次)。然后干燥。
在一些实施例中,还包括将分离得到的固体沉淀进行干燥的步骤。
在一些实施例中,采用真空干燥,优选控制干燥至干燥失重≤0.5%。研究发现,这样可以保证制备得到的晶型A能够顺利的转晶成药用晶型C。
在一些实施例中,真空干燥时优选在真空度≤-0.095MPa条件下进行,这样可以快速地使酒石酸匹莫范色林晶型A干燥完成。
在一些实施例中,真空干燥时优选在温度30-40℃条件下进行,这样可以避免干燥过程中酒石酸匹莫范色林受热分解。
在一些实施例中,上述酒石酸匹莫范色林晶型A的制备方法,还包括制备式(I)化合物的步骤,具体包括:
将式(II)化合物溶于第二溶剂中;
向所得溶液中加入L-酒石酸溶液,充分反应;
将所得溶液降温;以及
分离得到沉淀固体;
其中,所述第二溶剂为浓度为90.0%-98.5%的乙醇水溶液、90.0%-98.5%的异丙醇水溶液,或90.0%-98.5%的乙醇和异丙醇的混合水溶液;所述L-酒石酸溶液的溶剂为所述第二溶剂。
发明人研究发现,采用上述第二溶剂制备式(I)化合物可以更稳定地获得酒石酸匹莫范色林晶型A,并且提高了纯度。其可能的原因在于酒石酸匹莫范色林晶型A理论为半水合物,但是该水只是微弱的结合,仅在室温以上就开始失去。采用含有一定水的溶液有利于其在结晶过程中形成晶型A,避免了批间差异,但因为后期还需对结晶得到的产品进行精制,如果含水量较大则会降低产品收率,如果含水量较小则影响晶型A的纯度,所以采用以上浓度的结晶溶剂。
在一些实施例中,所述第二溶剂(即乙醇水溶液,异丙醇水溶液,乙醇和异丙醇的混合水溶液)的浓度为90.0%、95.0%、97.5%、98.0%或98.5%,优选为98%±0.5%。
在一些实施例中,所述第二溶剂优选为98%±0.5%的乙醇水溶液。
在一些实施例中,L-酒石酸溶液中,其溶剂的用量为L-酒石酸投料量的3-8倍量(重量比),优选5±0.5倍量。
在一些实施例中,制备式(I)化合物的过程中,L-酒石酸的用量为式(II)化合物的0.5-0.6当量,优选0.5-0.55当量。
在一些实施例中,制备式(I)化合物的过程中,将所得溶液降温至-5至5℃(例如0℃)析晶。
在一些实施例中,制备式(I)化合物的过程中,将所得溶液降温的具体过程包括:将所得溶液先降温至35-40℃,保持一定时间(例如0.5-2h),再降温并控温-5至5℃析晶。
在一些实施例中,制备式(I)化合物的过程中,将所得溶液降温的具体过程包括:将所得溶液降温至35-40℃析晶0.5h,降温并控温-5至5℃析晶2h。然后分离,将所得沉淀固体用所述溶剂淋洗(例如淋洗2次)。然后干燥。
在一些实施例中,制备式(I)化合物的方法具体包括:将式(II)化合物溶于加入乙醇水溶液中,搅拌升温至40-50℃,料液溶清后向体系中滴入L-酒石酸-乙醇水溶液,加毕,降温至35-40℃搅拌0.5h,待大量固体析出后,降温,并控温-5至5℃搅拌析晶2h,离心,滤饼用乙醇水溶液淋洗两次,抽干,获得式(II)化合物。其中,乙醇水溶液优选为98%±0.5%的乙醇水溶液。
在一些实施例中,酒石酸匹莫范色林晶型A的制备方法包括:
1)成盐
将式(II)化合物和乙醇水溶液加入反应釜中,所述乙醇水溶液的用量为式(II)化合物(匹莫范色林)投料量的3-8倍量(重量比),搅拌升温至40-50℃,料液溶清后向体系中滴入L-酒石酸-乙醇水溶液,L-酒石酸的用量为式(II)化合物(匹莫范色林)的0.5-0.6当量,加毕,降温至35-40℃搅拌(例如0.5h),待大量固体析出后,降温并控温-5至5℃搅拌析晶(例如2h),离心甩干;其中,所述乙醇水溶液的浓度为90.0%-98.5%,优选为98%±0.5%;
2)精制
将乙醇水溶液和步骤1)所得滤饼加至反应釜中,所述乙醇水溶液的用量为式(I)化合物(酒石酸匹莫范色林)投料量的3-8倍量(重量比)搅拌升温至回流(75-78℃);料液溶清后,将反应液降温至45-50℃析晶(1-2h,例如0.5h),自然冷却降温至35℃,随后降温并控温15-25℃析晶(例如2h),离心,淋洗,甩干,将滤饼于温度30-40℃、真空度≤-0.095MPa条件下干燥至干燥失重≤0.5%,得到酒石酸匹莫范色林晶型A;其中,所述乙醇水溶液的浓度为90.0%-98.5%,优选为98%±0.5%。
本发明采用乙醇水溶液为成盐和精制溶液,还优化了真空干燥时的温度和真空度,并且通过控制干燥失重控制≤0.5%,可稳定的制备出高纯度的酒石酸匹莫范色林晶型A。与现有技术相比,可以显著地提高酒石酸匹莫范色林晶型A的纯度,且批间稳定。
为了快速准确地检测晶型纯度,本发明人意外地发现无水四氢呋喃可用作酒石酸匹莫范色林晶型A的晶型纯度检测。
因此,本发明还提供一种酒石酸匹莫范色林晶型A的晶型纯度检测方法,包括:将酒石酸匹莫范色林晶型A溶于无水四氢呋喃中,加热;观察是否形成澄清溶液或出现不溶固体。
若形成澄清溶液,则所述酒石酸匹莫范色林晶型A为纯净的晶型A;若溶液浑浊或有不溶固体,则所述酒石酸匹莫范色林晶型A为不纯净的晶型A。
在一些情况下,所述不纯净的晶型A指存在混晶,例如酒石酸匹莫范色林晶型B、C、D、E、F中的一种或几种。
在一些实施例中,所述无水四氢呋喃中水分含量小于等于0.01wt%。因为酒石酸匹莫范色林溶于水,所以使用水分含量小于等于0.01wt%的无水四氢呋喃。
在一些实施例中,所述无水四氢呋喃的用量为酒石酸匹莫范色林晶型A重量的6-8倍。这样可以更好地保证检测结果的准确性。另外,如果无水四氢呋喃的用量过低,则不能使晶型A全部溶解;如果用量过高,则会增加其中混晶的溶解,影响检测结果的准确性。
在一些实施例中,所述加热的温度优选为65-70℃。如果温度过低,则不能保证晶型A全部溶解。
在一些实施例中,所述加热的时间优选为20-30min。如果加热的时间过短不能保证晶型A全部溶解。
在一些实施例中,所述酒石酸匹莫范色林晶型A的晶型纯度检测方法具体包括:将酒石酸匹莫范色林晶型A溶于6-8倍重量的无水四氢呋喃中,加热至65-70℃,保持20-30min;若形成澄清溶液,则所述酒石酸匹莫范色林晶型A为纯净的晶型A;若溶液浑浊或有不溶固体,则所述酒石酸匹莫范色林晶型A为不纯净的晶型A。
进一步地研究发现,无水四氢呋喃仅对酒石酸匹莫范色林晶型A有较好的溶解性,对其他晶型溶解性较差。
附图说明
图1为本发明实施例酒石酸匹莫范色林晶型A的XRD图谱。
图2为本发明实施例酒石酸匹莫范色林晶型A的DSC图谱。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
以下所用匹莫范色林均为相同工艺制备得到的。
以下所述纯度采用HPLC法检测,方法同文献CN105153016A。
以下XRD的检测方法:取本品适量,照粉末X射线衍射法(中国药典2020年版四部通则0451项下第二法)测定,以CuKa为光源,光管电压和光管电流分别为40kV和40mA,步长为0.02°,停留时间为0.15s,测角仪半径、发射狭缝、防散射狭缝分别为280mm、0.6mm、5.7mm,在衍射角(2θ)3°~70°的范围内扫描,记录衍射图谱。
以下DSC的检测方法:取本品适量,照热分析法(中国药典2020年版四部通则0661)测定,以10℃/min加热到300℃,记录熔融温度及熔化热。
实施例1以98%乙醇水溶液制备酒石酸匹莫范色林晶型A
成盐:将9.94kg匹莫范色林和98%乙醇水溶液(49.71kg)加入反应釜中,搅拌升温至50℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-98%乙醇水溶液(8.70kg),加毕,降温至40℃搅拌0.5h,待大量固体析出后,降温至-5℃,并控温-5℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和98%乙醇水溶液(49.71kg)加至于2反应釜中,搅拌升温至75℃,料液溶清后降温至45℃析晶0.5h,大量固体析出后降温至15℃,控温15℃析晶2h,离心,用98%乙醇水溶液淋洗,甩干,30℃真空干燥设备干燥至干失失重为0.3%,(真空度≤-0.095MPa),得9.84kg酒石酸匹莫范色林晶型A。
收率85.0%,纯度99.6%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱见图1,DSC图谱见图2。
实施例2以98.5%乙醇水溶液制备酒石酸匹莫范色林晶型A
成盐:将9.94kg匹莫范色林和98.5%乙醇水溶液(49.71kg)加入反应釜中,搅拌升温至40℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-98.5%乙醇水溶液(13.92kg),加毕,降温至40℃搅拌0.5h,待大量固体析出后,降温至0℃,并控温0℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和98.5%乙醇水溶液(49.71kg)加至于2反应釜中,搅拌升温至78℃,料液溶清后降温至45℃析晶0.5h,大量固体析出后降温至20℃,控温20℃析晶2h,离心,用98.5%乙醇水溶液淋洗,甩干,40℃真空干燥设备干燥至干失失重为0.2%,(真空度≤-0.098MPa),得9.85kg酒石酸匹莫范色林晶型A。
收率:85.1%,纯度99.7%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
实施例3以95%乙醇水溶液制备酒石酸匹莫范色林晶型A
成盐:将9.94kg匹莫范色林和95%乙醇水溶液(49.71kg)加入反应釜中,搅拌升温至50℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-95%乙醇水溶液(5.22kg),加毕,降温至45℃搅拌0.5h,待大量固体析出后,降温至5℃,并控温5℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和95%乙醇水溶液(49.71kg)加至于2反应釜中,搅拌升温至75℃,料液溶清后降温至45℃析晶0.5h,大量固体析出后降温至15℃,控温15℃析晶2h,离心,用95%乙醇水溶液淋洗,甩干,35℃真空干燥设备干燥至干失失重为0.3%,(真空度≤-0.096MPa),得9.26kg酒石酸匹莫范色林晶型A。
收率:80.0%,纯度99.7%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
实施例4以90%乙醇水溶液制备酒石酸匹莫范色林晶型A
成盐:将9.94kg匹莫范色林和90%乙醇水溶液(49.71kg)加入反应釜中,搅拌升温至50℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-90%乙醇水溶液(8.70kg),加毕,降温至40℃搅拌0.5h,待大量固体析出后,降温至-5℃,并控温-5℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和90%乙醇水溶液(49.71kg)加至于200L反应釜中,搅拌升温至75℃,料液溶清后降温至50℃析晶0.5h,大量固体析出后降温至25℃,控温25℃析晶2h,离心,用90%乙醇水溶液淋洗,甩干,40℃真空干燥设备干燥至干失失重为0.5%,(真空度≤-0.097MPa),得8.68kg酒石酸匹莫范色林晶型A。
收率:75.0%,纯度99.6%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
实施例5以95%异丙醇水溶液制备酒石酸匹莫范色林晶型A
成盐:将9.94kg匹莫范色林和95%异丙醇水溶液(49.71kg)加入反应釜中,搅拌升温至50℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-95%异丙醇水溶液(8.70kg),加毕,降温至40℃搅拌0.5h,待大量固体析出后,降温至-5℃,并控温-5℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和95%异丙醇水溶液(49.71kg)加至于200L反应釜中,搅拌升温至75℃,料液溶清后降温至50℃析晶0.5h,大量固体析出后降温至25℃,控温25℃析晶2h,离心,用95%异丙醇水溶液淋洗,甩干,40℃真空干燥设备干燥至干失失重为0.4%,(真空度≤-0.095MPa),得9.24kg酒石酸匹莫范色林晶型A。
收率:79.8%,纯度99.6%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
实施例6以95%异丙醇/无水乙醇(1:1)水溶液制备酒石酸匹莫范色林晶型A
成盐:将9.94kg匹莫范色林和95%异丙醇/无水乙醇(1:1)水溶液(49.71kg)加入反应釜中,搅拌升温至50℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-95%异丙醇/无水乙醇(1:1)水溶液(8.70kg),加毕,降温至40℃搅拌0.5h,待大量固体析出后,降温至-5℃,并控温-5℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和95%异丙醇/无水乙醇(1:1)水溶液(49.71kg)加至于200L反应釜中,搅拌升温至78℃,料液溶清后降温至50℃析晶0.5h,大量固体析出后降温至25℃,控温25℃析晶2h,离心,用95%异丙醇/无水乙醇(1:1)水溶液淋洗,甩干,35℃真空干燥设备干燥至干失失重为0.4%,(真空度≤-0.098MPa),得9.03kg酒石酸匹莫范色林晶型A。
收率:78.0%,纯度99.6%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
实施例7以98%乙醇水溶液制备酒石酸匹莫范色林晶型A
本实施例所用酒石酸匹莫范色林参照CN101035759A的方法制备。
精制:将9.94kg酒石酸匹莫范色林和98%乙醇水溶液(49.71kg)加至于反应釜中,搅拌升温至75℃,料液溶清后降温至45℃析晶0.5h,大量固体析出后降温至15℃,控温15℃析晶2h,离心,用98%乙醇水溶液淋洗,甩干,35℃真空干燥设备干燥至干失失重为0.3%,(真空度≤-0.095MPa),得6.75kg酒石酸匹莫范色林晶型A。
收率67.9%,纯度99.6%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
实施例8以98%乙醇水溶液制备酒石酸匹莫范色林晶型A
与实施例1相比,区别仅在于将成盐及精制步骤的98%乙醇水溶液用量均替换为79.52kg。
收率81.5%,纯度99.6%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
实施例9以98%乙醇水溶液制备酒石酸匹莫范色林晶型A
与实施例1相比,区别仅在于将成盐及精制步骤的98%乙醇水溶液用量均替换为29.82kg。
收率84.9%,纯度99.4%(HPLC法),晶型纯度为纯净的晶型A(检测方法见下文实验例1)。
本实施例制备的酒石酸匹莫范色林晶型A的XRD图谱与图1近似,DSC图谱与图2近似。
对比例1
成盐:将9.94kg匹莫范色林和无水乙醇(49.71kg)加入反应瓶中,搅拌升温至45℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-无水乙醇(8.70kg),加毕,降温至35℃搅拌0.5h,待大量固体析出后,降温至-5℃,并控温-5℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和无水乙醇(49.71kg)加至于反应瓶中,搅拌升温至75℃,料液溶清后降温至50℃析晶0.5h,大量固体析出后降温至15℃,控温15℃析晶2h,离心,用无水乙醇淋洗,甩干,35℃真空干燥设备干燥至干失失重为0.3%,(真空度≤-0.096MPa),得7.39kg酒石酸匹莫范色林。
收率:63.3%,纯度99.5%(HPLC法),晶型纯度为含有晶型A的混合晶型(检测方法见下文实验例1)。
对比例2
成盐:将9.94kg匹莫范色林和无水乙醇(49.71kg)加入反应瓶中,搅拌升温至45℃,料液溶清后向体系中滴入L-酒石酸(1.74kg)-无水乙醇(8.70kg),加毕,降温至35℃搅拌0.5h,待大量固体析出后,降温至-5℃,并控温-5℃析晶2h,离心甩干,湿品直接进行精制。
精制:将上述酒石酸匹莫范色林滤饼和无水乙醇(49.71kg)加至于反应瓶中,搅拌升温至75℃,料液溶清后降温至50℃析晶0.5h,大量固体析出后降温至15℃,控温15℃析晶2h,离心,用无水乙醇淋洗,甩干,35℃真空干燥设备干燥至干失失重为0.3%,(真空度≤-0.096MPa),得8.25kg酒石酸匹莫范色林。
收率:70.6%,纯度99.5%(HPLC法),晶型纯度为含有晶型A的混合晶型(较对比例1的不溶物更多,检测方法见下文实验例1)。
实验例1
分别检测以上实施例1-9和对比例1-2制备的酒石酸匹莫范色林晶型A的晶型纯度。
晶型纯度的检测方法:称取1.0g酒石酸匹莫范色林晶型A,加入无水四氢呋喃(水分含量小于等于0.01wt%),加热至70℃,保持30min。如可形成澄清溶液即为纯净的晶型A;如溶液混浊或有不溶固体则存在混晶。
检测结果见下表:
通过上表结果可知,采用本发明方法制备得到的酒石酸匹漠范色林为纯净的晶型A,采用现有技术制备得到的酒石酸匹漠范色林为混有其他晶型的晶型A。
本发明方法制备得到的酒石酸匹莫范色林晶型A工艺稳定,重现性好,且晶型纯度较高。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 酒石酸匹莫范色林晶型A的制备方法
- 一种匹莫范色林的新晶型及其制备方法