多接枝位点纳米碳材料和活性纳米碳材料及其制备方法和超低渗油藏用驱油体系
文献发布时间:2023-06-19 11:30:53
技术领域
本发明属于油田化学领域,具体涉及一种多接枝位点纳米碳材料和活性纳米碳材料及其制备方法和超低渗油藏用驱油体系。
背景技术
随着我国国民经济的迅速增长,对石油资源的需求不断提高。因此,加大油气资源的勘查力度,提高已有区块的开采效率是我国从源头节约石油资源的最有效途径之一。当前,我国新探明储量中超低渗透油藏原油占比明显增大,但该类油藏动用程度较低。
压裂技术是改善超低渗透油藏开发的重要技术。通过压裂改造,在超低渗透油藏储层内形成众多的微纳米级人工裂缝,提高储层导流能力。人工裂缝的产生弥补了基质渗透率低的缺陷,同时也会加剧储层的非均质性。另外,由于超低渗透油藏储层中天然微裂缝发育,进一步加剧储层的非均质性。注水开发过程中,由于裂缝的渗透率远远大于基质渗透率,注入水很容易沿微裂缝窜进,使参与渗流的主要裂缝全部或大部分被水充满,而次生裂缝和基质系统仍为高含油饱和度区,导致注水开发效果差,油藏水驱采收率低。因此,提高超低渗透油藏的高效开采是目前该类油藏开发面临重点。
超低渗油藏已用的驱油体系主要包括表面活性剂类、聚合物类、聚合物微球、纳米二氧化硅类等。
但超低渗油藏物性差,储层非均质严重,注入的表面活性剂易沿裂缝系统窜流,降低了表面活性剂的驱油效果;同时,超低渗油藏多存在高压欠注难题,聚合物类驱油体系黏度高难以有效注入。为此,发展了纳米级、微米级聚合物微球,并在超低渗油藏广泛应用,取得了显著增油降水效果。但聚合物微球以丙烯酰胺单体为制备原料,环保性差且在地层中膨胀后稳定性变差,难以保证长期有效的驱油效果。近年来,发展的纳米二氧化硅类驱油体系由于其小尺寸和界面活性效应在超低渗油藏中开始应用。但纳米二氧化硅类驱油体系为刚性颗粒,受地层理化性质、地层水长期侵蚀的影响,纳米颗粒表面性质发生改变导致稳定性较差。此外,纳米二氧化硅颗粒密度较大,会形成重力差异效应,导致颗粒驱油效果变差。
因此,超低渗油藏驱油用活性纳米碳材料具有重要意义。
发明内容
本发明的目的是为了克服现有技术存在的超低渗油藏采用的驱油体系效果差的缺陷问题,提供一种多接枝位点纳米碳材料和活性纳米碳材料及其制备方法和超低渗油藏用驱油体系。该超低渗油藏用驱油体系具有较高界面活性,油水界面张力降低达10
为了实现上述目的,本发明第一方面提供了一种多接枝位点纳米碳材料,其中,所述多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示的结构单元;
其中,R选自甲苯基、二苯基甲烷基、异佛尔酮基和二环己基甲烷基中的一种或多种。
本发明第二方面提供了一种前述所述的多接枝位点纳米碳材料的制备方法,其中,所述的制备方法包括:
(F
(F
(F
其中,所述氧化纳米碳材料具有式(3)所示的结构:
其中,所述接枝剂选自甲苯二异氰酸酯、二苯基甲烷二异氰酸酯、异佛尔酮二异氰酸酯和二环己基甲烷二异氰酸酯中的一种或多种。
本发明第三方面提供了一种活性纳米碳材料的制备方法,其中,所述的制备方法包括:
(F
(F
(F
其中,所述活性剂具有式(4)所示的结构:
其中,R”选自十二烷基、十四烷基、十六烷基和十八烷基中的一种或多种。
本发明第四方面提供了一种由前述所述的制备方法制备得到的活性纳米碳材料。
本发明第五方面提供了一种超低渗油藏用驱油体系,其中,所述驱油体系含有前述所述的活性纳米碳材料。
通过上述技术方案,本发明具有如下优点:
(1)本发明的超低渗透油藏用驱油体系以活性纳米碳材料为基元,平均粒径为70-200nm,具有软体、自润滑、油藏中物理化学性质稳定特征,能够实现超低渗透油藏“注得进、走得远、驱得动”。
(2)本发明的超低渗透油藏用驱油体系具有较高界面活性,油水界面张力降低达10
(3)本发明的超低渗透油藏用驱油体系具有水溶即分散特点,易于配制,具备现场操作简单、施工灵活特点。
(4)本发明的超低渗透油藏用驱油体系制备方法简单,原材料易得,利于产品推广。
附图说明
图1是本发明实施例4制备的多接枝位点纳米碳材料的宏观形貌图;
图2是本发明实施例7制备的超低渗透油藏用驱油体系的宏观形貌图;
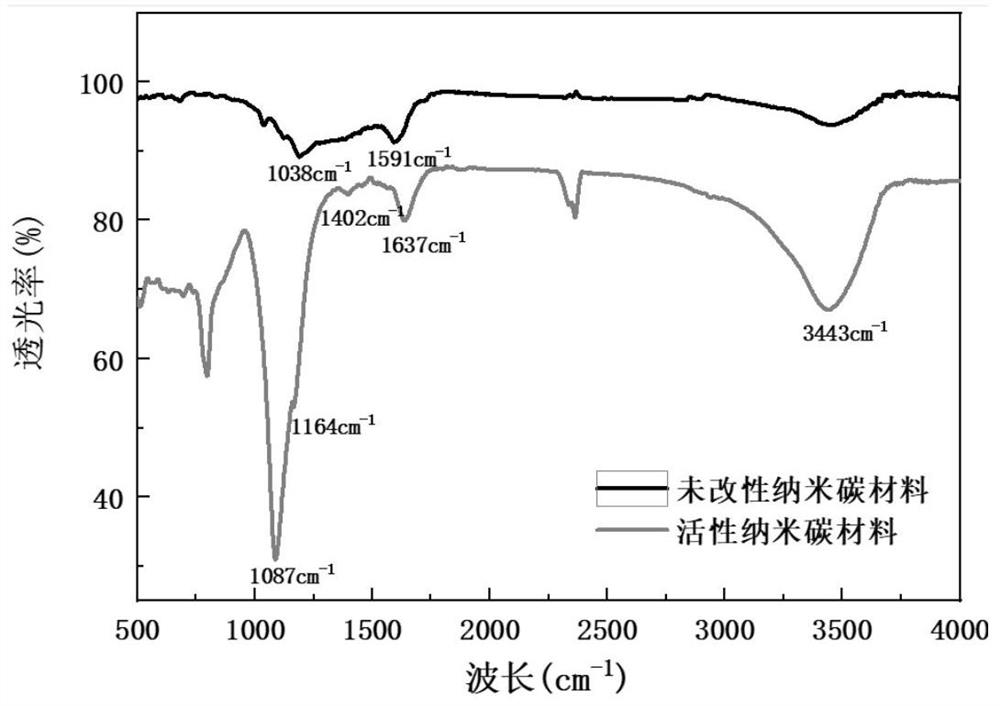
图3是本发明实施例7制备的活性纳米碳材料的傅里叶红外曲线图。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
为了实现上述目的,本发明第一方面提供了一种多接枝位点纳米碳材料,其中,所述多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示的结构单元;
其中,R选自甲苯基、二苯基甲烷基、异佛尔酮基和二环己基甲烷基中的一种或多种。
本发明的发明人意外发现:将式(5)所示的活性纳米碳材料应用于超低渗油藏中,利用活性纳米碳材料软体自润滑、低界面张力、润湿改变能力、分离压效应、高效洗油与兼顾储层微观调控的特点实现大幅度提高超低渗油藏剩余油的动用能力。但是,在将氧化纳米碳材料与活性剂进行酯化反应制备该活性纳米碳材料时,由于氧化纳米碳材料表面羧基反应活性很低,难以直接制备得到该活性纳米碳材料,需要进一步将氧化纳米碳材料表面羧基转变成为反应活性较高的基团。因此,本发明的发明人先将氧化纳米碳材料与接枝剂接触进行接枝反应,使得该氧化纳米碳材料至少部分接枝上式(2)所示的结构单元,释放出CO
根据本发明,所述多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示的结构单元;即,式(1)所示的结构单元的酰基侧链能够接枝上羟基和/或式(2)所示的结构单元,且以式(1)所示的结构单元的酰基侧链的总含量为基准,式(2)所示的结构单元的接枝率为20-100%,换言之,式(1)所示的结构单元中的至少20%的酰基侧链接枝上式(2)所示的结构单元,以及式(1)所示的结构单元中没有接枝上式(2)所示的结构单元的酰基侧链与羟基相连接。
根据本发明,优选情况下,式(2)所示的结构单元的含量占比酰基侧链含量的20-100%;即,式(1)所示的结构单元中的20-100%的酰基位点接枝上式(2)所示的结构单元。
根据本发明,更优选情况下,所述多接枝位点纳米碳材料包括式(1)所示的结构单元和式(2)所示的结构单元;即,式(1)所示的结构单元中的酰基位点都接枝上式(2)所示的结构单元。
根据本发明,所述多接枝位点纳米碳材料的平均粒径为81-170nm,优选为83-189nm。
本发明第二方面提供了一种前述所述的多接枝位点纳米碳材料的制备方法,其中,所述的制备方法包括:
(F
(F
(F
其中,所述氧化纳米碳材料具有式(3)所示的结构:
其中,所述接枝剂选自甲苯二异氰酸酯、二苯基甲烷二异氰酸酯、异佛尔酮二异氰酸酯和二环己基甲烷二异氰酸酯中的一种或多种。
根据本发明,优选情况下,所述接枝剂为二苯基甲烷二异氰酸酯。
根据本发明,所述第一有机溶剂选自甲苯、二甲苯和二氯甲烷中的一种或多种;优选为甲苯。
根据本发明,所述氧化纳米碳材料的平均粒径为50-200nm,优选情况下,平均粒径为80-150nm,更优选情况下,平均粒径为120-140nm。
根据本发明,在步骤(F
根据本发明,在步骤(F
根据本发明,在步骤(F
根据本发明,所述接枝剂与所述第一有机溶剂的用量的重量比为(0.6-2):100,优选为(0.8-1.2):100。
在本发明中,将所述氧化纳米碳材料、所述接枝剂与所述第一有机溶剂的用量的质量比限定为前述范围之内,优点是保证反应试剂可在溶剂中充分分散且氧化纳米碳材料表面羧基可以被充分活化为异氰酸酯基。
根据本发明,所述离心的条件包括:在转速为5000-20000r/min下离心5-17h,优选地,在转速为8000-12000r/min下离心8-14h。
根据本发明,所述旋蒸的条件包括:在温度为50-95℃的条件下旋蒸3-8h,优选地,在温度为60-80℃的条件下旋蒸5-7h。
根据本发明,所述研磨的条件包括:研磨次数为3-8次,每次研磨7-15min,优选地,研磨次数为4-6次,每次研磨9-12min。
根据本发明的一种优选的具体实施方式,所述多接枝位点纳米碳材料的制备方法包括:
室温下(20±5℃),在甲苯中先加入氧化纳米碳材料(粒径为120-140nm),在搅拌速率为400-600转/分钟的条件下搅拌10-20min;边搅拌边加入接枝剂,在70-90℃油浴温度下连续搅拌3-5h,得多接枝位点纳米碳材料溶液;将上述得到的溶液经离心、旋蒸和研磨分散,得到多接枝位点纳米碳材料。
本发明第三方面提供了一种活性纳米碳材料的制备方法,其中,所述的制备方法包括:
(F
(F
(F
其中,所述活性剂具有式(4)所示的结构:
其中,R”选自十二烷基、十四烷基、十六烷基和十八烷基中的一种或多种。
根据本发明,所述活性剂为十二烷基羟丙基磺基甜菜碱、十四烷基羟丙基磺基甜菜碱、十六烷基羟丙基磺基甜菜碱和十八烷基羟丙基磺基甜菜碱中的一种或多种;优选情况下,所述活性剂为十四烷基羟丙基磺基甜菜碱和/或十六烷基羟丙基磺基甜菜碱。
根据本发明,在步骤(F
根据本发明,在步骤(F
根据本发明,所述多接枝位点纳米碳材料与所述第二有机溶剂的用量的重量比为(8-22):100,优选为(10-18):100。
根据本发明,所述活性剂与所述第二有机溶剂的用量的重量比为(4-20):100,优选为(11-15):100。
根据本发明,所述催化剂与所述第二有机溶剂的用量的重量比为(0.01-0.5):100,优选为(0.03-0.04):100。
根据本发明,所述催化剂选自二丁基二月桂酸锡、辛酸亚锡和顺丁烯二酸二丁基锡中的一种或多种,优选为二丁基二月桂酸锡。
根据本发明,所述第二有机溶剂选自甲苯、二甲苯和二氯甲烷中的一种或多种,优选为甲苯。
根据本发明,所述离心的条件包括:在转速为6000-25000r/min下离心6-18h,优选地,在转速为10000-12000r/min下离心10-14h。
根据本发明,所述洗涤的条件包括:采用去离子水洗涤2-5次,每次洗涤3-6min,优选为:采用去离子水洗涤3-4次,每次洗涤4-5min。
根据本发明,所述干燥的条件包括:在温度为70-95℃的条件下旋蒸3-8h,优选地,在温度为80-90℃的条件下旋蒸4-7h。
根据本发明,所述研磨的条件包括:研磨次数为3-8次,每次研磨10-20min,优选地,研磨次数为5-7次,每次研磨12-17min。
根据本发明的一种优选的具体实施方式,所述活性纳米碳材料的制备方法包括:
室温下(20±5℃),在甲苯中先加入多接枝位点纳米碳材料,在搅拌速率为400-600转/分钟的条件下搅拌10-20min;边搅拌边加入活性剂,再加入催化剂,40-55℃条件下搅拌2-4h,得活性纳米碳材料分散溶液;将上述得到的分散溶液经离心、洗涤、旋蒸和研磨分散得到活性纳米碳材料。
本发明第四方面提供了一种由前述所述的制备方法制备得到的活性纳米碳材料。
根据本发明,所述的活性纳米碳材料具有式(5)所示的结构:
其中,R
其中,R
根据本发明,优选情况下,所述的活性纳米碳材料的平均粒径为70-200nm,优选为80-195nm。
本发明第五方面提供了一种超低渗油藏用驱油体系,其中,所述驱油体系含有前述所述的活性纳米碳材料。
根据本发明,所述驱油体系还含有水。在本发明中,优选情况下,以所述驱油体系的总重量为基准,所述活性纳米碳材料的含量为0.05-0.25重量%,所述水的含量为99.75-99.95重量%。
根据本发明,所述水为去离子水。
根据本发明,所述超低渗油藏的渗透率为0.5-10mD,存在微裂缝发育,模拟油藏温度为90℃。
以下将通过实施例对本发明进行详细描述。
以下实施例和对比例中:
高倍率透射电子显微镜照片参数通过购自日本电子株式会社的型号为JEM-2100的高速显微摄像系统测得。
傅里叶红外曲线图通过购自德国布鲁克的型号为VERTEX70的红外光谱仪测得。
氧化纳米碳材料、甲苯、二甲苯、二氯甲烷、二苯基甲烷二异氰酸酯、异佛尔酮二异氰酸酯、二丁基二月桂酸锡、辛酸亚锡、顺丁烯二酸二丁基锡可购自麦克林公司,十二烷基羟丙基磺基甜菜碱、十四烷基羟丙基磺基甜菜碱、十六烷基羟丙基磺基甜菜碱均可购自上海诺颂实业有限公司。
实施例1
本实施例在于说明采用本发明的方法制备的多接枝位点纳米碳材料。
(F
(F
(F
结果得到粒径为116nm的多接枝位点纳米碳材料,并且,该多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示结构单元,其中60%酰基侧链连接羟基和40%酰基侧链连接式(2)所示的结构单元,其中R基为甲苯基。
实施例2
本实施例在于说明采用本发明的方法制备的多接枝位点纳米碳材料。
(F
(F
(F
结果得到粒径为107nm的多接枝位点纳米碳材料,并且,该多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示结构单元,其中40%酰基侧链连接羟基和60%酰基侧链连接式(2)所示的结构单元,其中R基为二苯基甲烷基。
实施例3
本实施例在于说明采用本发明的方法制备的多接枝位点纳米碳材料。
(F
(F
(F
结果得到粒径为95nm的多接枝位点纳米碳材料,并且,该多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示结构单元,其中20%酰基侧链连接羟基和80%酰基侧链连接式(2)所示的结构单元,其中R基为二苯基甲烷基。
实施例4
本实施例在于说明采用本发明的方法制备的多接枝位点纳米碳材料。
(F
(F
(F
结果得到粒径为83nm的多接枝位点纳米碳材料,并且,该多接枝位点纳米碳材料包括式(1)所示的结构单元和式(2)所示结构单元,其中酰基侧链连接的均为式(2)所示的结构单元,其中R基为二苯基甲烷基;另外,图1为实施例4制备的多接枝位点纳米碳材料的宏观形貌图,从图1能够看出:该多接枝位点纳米碳材料颗粒均匀,有利于后续改性反应。
实施例5
本实施例在于说明采用本发明的方法制备的多接枝位点纳米碳材料。
(F
(F
(F
结果得到粒径为219nm的多接枝位点纳米碳材料,并且,该多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示结构单元,其中80%酰基侧链连接羟基和20%酰基侧链连接式(2)所示的结构单元,其中R基为异佛尔酮基。
实施例6
本实施例在于说明采用本发明的方法制备的多接枝位点纳米碳材料。
(F
(F
(F
结果得到粒径为186nm的多接枝位点纳米碳材料,并且,该多接枝位点纳米碳材料包括式(1)所示的结构单元、羟基和式(2)所示结构单元,其中80%酰基侧链连接羟基和20%酰基侧链连接式(2)所示结构单元,其中R基为二环己基甲烷基。
实施例7
本实施例在于说明采用本发明的方法制备的活性纳米碳材料。
(F
(F
(F
结果得到超低渗油藏活性纳米碳材料,并且,该活性纳米碳材料具有式(5)所示的结构单元,其中,R
另外,在90℃条件下,采用该活性纳米材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%的该体系能够降低油水界面张力,使得油水界面张力为5.37×10
该超低渗油藏用活性纳米碳材料驱油体系的宏观形貌如图2所示,从图2能够看出:该超低渗油藏用活性纳米碳材料能够均匀分散在水溶液中,没有团聚现象,能够组成均匀的超低渗油藏用活性纳米碳材料驱油体系。
另外,该超低渗油藏用活性纳米碳材料的红外曲线见图3所示,从图3能够看出:表面活性剂的活性基团(即,十四烷基羟丙基磺基甜菜碱和十六烷基羟丙基磺基甜菜碱)能够成功接枝在多接枝位点纳米碳材料,并进一步形成了具有界面活性的超低渗油藏用活性纳米碳材料。
实施例8
本实施例在于说明采用本发明的方法制备的活性纳米碳材料。
按照与实施例7相同的方法制备超低渗油藏用活性纳米碳材料,所不同之处在于:将实施例1制备的多接枝位点纳米碳材料(粒径为116nm)替换为实施例4制备的多接枝位点纳米碳材料(粒径为83nm)。
结果得到的活性纳米碳材料具有式(5)所示的结构单元,其中,R
结果在90℃条件下,采用该得到的超低渗油藏用活性纳米碳材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%时能够降低油水界面张力,使得油水界面张力为3.81×10
实施例9
本实施例在于说明采用本发明的方法制备的活性纳米碳材料。
(F
(F
(F
实施例10
本实施例在于说明采用本发明的方法制备的活性纳米碳材料。
按照与实施例9相同的方法制备超低渗油藏活性纳米碳材料,所不同之处在于:将实施例3制备的多接枝位点纳米碳材料(粒径为95nm)替换为实施例4制备的多接枝位点纳米碳材料(粒径为83nm)。
结果得到的活性纳米碳材料具有式(5)所示的结构单元,其中,R
结果在90℃条件下,采用该得到的超低渗油藏用活性纳米碳材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%时能够降低油水界面张力,使得油水界面张力为6.55×10
实施例11
本实施例在于说明采用本发明的方法制备的活性纳米碳材料。
(F
(F
(F
另外,在90℃条件下,采用该活性纳米材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%的该体系能够降低油水界面张力,使得油水界面张力为9.7×10
实施例12
本实施例在于说明采用本发明的方法制备的活性纳米碳材料。
按照与实施例11相同的方法制备超低渗油藏活性纳米碳材料,所不同之处在于:实施例6制备的多接枝位点纳米碳材料(粒径为186nm)替换为实施例4制备的多接枝位点纳米碳材料(粒径为83nm)。
结果得到的活性纳米碳材料具有式(5)所示的结构单元,其中,R
对比例1
该对比例没有采用多接枝位点纳米碳材料,而是直接将氧化纳米碳材料与活性剂进行酯化反应。
(1)室温下(20℃),在81g甲苯中先加入13.2g粒径为140nm的氧化纳米碳材料,在搅拌速率为400转/分钟的条件下搅拌10min;
(2)边搅拌边加入3g十四烷基羟丙基磺基甜菜碱和2.8g十六烷基羟丙基磺基甜菜碱,再加入0.32g二丁基二月桂酸锡催化剂,40℃条件下搅拌2h,得超低渗油藏活性纳米碳材料分散溶液;
(3)将上述得到的溶液依次用去离子水洗涤3次,每次洗涤4min;在转速为10000r/min条件下离心10h;在80℃条件下旋蒸4h;研磨分散5次,每次研磨12min得到超低渗油藏活性纳米碳材料。
该活性纳米碳材料具有式(5)所示的结构单元,其中,R
另外,在90℃条件下,采用该活性纳米材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%的该体系能够降低油水界面张力,使得油水界面张力为1.7mN/m。
对比例2
按照与实施例7相同的方法制备活性纳米碳材料,所不同之处在于:将活性剂“十四烷基羟丙基磺基甜菜碱和十六烷基羟丙基磺基甜菜碱”替换为“月桂酰胺丙基羟磺基甜菜碱”。
该活性纳米碳材料具有式(5)所示的结构单元,其中,R
另外,在90℃条件下,采用该活性纳米材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%的该体系能够降低油水界面张力,使得油水界面张力为5.2mN/m。
对比例3
按照与实施例7相同的方法制备活性纳米碳材料,所不同之处在于:多接枝位点纳米碳材料的制备方法是按照与实施例4相同的方法制备的,所不同之处在于:将接枝剂“二苯基甲烷二异氰酸酯”修改为“单异氰酸酯”,结果得到的多接枝位点纳米碳材料具有式(5)所示的结构单元,其中,R
另外,在90℃条件下,采用该活性纳米材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%的该体系能够降低油水界面张力,使得油水界面张力为1.9mN/m。
对比例4
按照与实施例7相同的方法制备活性纳米碳材料,所不同之处在于:
(F
(F
该活性纳米碳材料具有式(5)所示的结构单元,其中,R
另外,在90℃条件下,采用该活性纳米材料与水配制超低渗油藏用活性纳米碳材料驱油体系;结果浓度为0.2wt%的该体系能够降低油水界面张力,使得油水界面张力为7.2mN/m。
应用例
将实施例7-12和对比例1-4制备的超低渗油藏用活性纳米碳材料驱油体系应用于超低渗油藏提高采收率测试中,实验中使用的模拟油90℃下粘度4.6mPa·s,地层模拟水矿化度为10000mg/L,实验温度:90℃;岩心基本参数:长×直径=10cm×2.5cm;在90℃下,将上述岩心抽真空饱和水、饱和油,水驱至98%后,注入浓度为0.2%的上例活性纳米碳材料驱油体系,注入量为岩心孔隙体积的30%,测试注入体系驱和后续水驱的采收率增值,结果如表1所示。
表1
通过表1的结果可以看出,采用本发明的实施例7-12具有较好的提高采收率效果,注入体系驱采收率和后续水驱采收率之和均大于30%,明显好于对比例1-4。
对比例1由于没有采用多接枝位点纳米碳材料,由于氧化纳米碳材料表面羧基反应活性较低,导致其无法与活性剂中的羟基较为完全的进行酯化反应,会造成活性纳米碳材料表面活性基团含量较少,进而结果不好。
对比例2由于活性剂采用的是月桂酰胺丙基羟磺基甜菜碱,由于月桂酰胺丙基羟磺基甜菜碱降低油水界面张力能力有限,导致活性纳米碳材料降低油水界面张力能力下降,进而结果不好。
对比例3由于接枝剂采用的是单异氰酸酯,由于单异氰酸酯活化羧基反应能力没有二异氰酸酯强,导致后续反应中活性纳米碳材料表面活性基含量下降,进而结果不好。
对比例4由于搅拌条件以及反应条件不在本发明所限定的范围之内,由于接枝反应和活性化反应进行不充分,导致活性纳米碳材料表面活性基团含量较低,进而结果不好。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
- 多接枝位点纳米碳材料和活性纳米碳材料及其制备方法和超低渗油藏用驱油体系
- 多接枝位点纳米碳材料和活性纳米碳材料及其制备方法和超低渗油藏用驱油体系