单酚或多元酚的环氧丙基醚产物与其制备方法、及环氧树脂组合物
文献发布时间:2023-06-19 11:37:30
技术领域
本发明是关于单酚或多元酚的环氧丙基醚产物及其制备方法。另外,本发明是关于该单酚或多元酚的环氧丙基醚产物的应用。
背景技术
部分环氧丙基醚及一些环氧丙基醚的一般制备方法已于教科书中描述,例如B.Ellis,“Chemistry and Technology of Epoxy Resins”(多德雷赫,SpringerNetherlands出版社,1993),以及李桂林编着,“环氧树脂与环氧涂料”(北京:化学工业出版社,2003),这些文献基于所有目的而全文并于此以供参考。
环氧丙基醚传统上是通过使用过氧化物的环氧化反应而制备,或者通过环氧卤丙烷(epihalohydrin)与羟基化合物的反应在碱性条件下形成环氧丙基醚。通过环氧卤丙烷与羟基化合物的反应制备环氧丙基醚的方法通常可分为两个主要类型。第一类型是关于聚缩合反应,其中羟基在碱性条件下与环氧卤丙烷反应,从而同时进行开环及合环反应。第二类型是关于使用相转移催化剂来促进羟基化合物与环氧卤丙烷的反应以生成中间产物,然后使用碱来促进合环反应以形成环氧丙基醚的方法。
使用相转移催化剂的方法描述于第2,943,096号美国专利、第GB 897744 A号英国专利公开案、第4,284,573号美国专利、第JP 04353517 A号日本专利公开案、及第CN108299165 A号中国专利公开案,这些文献基于所有目的而全文并于此以供参考。
部分种类的环氧丙基醚可理想地适用于电子元件,例如第60079031 A号日本专利公开案、第JP 62030145 A号日本专利公开案、第JP 62064817 A号日本专利公开案、第DE3635383 A1号德国专利公开案、及第4,876,371 A号美国专利中的描述,这些文献基于所有目的而全文并于此以供参考。
第JP 2004262977号日本专利公开案基于所有目的而全文并于此以供参考,其中尝试改善环氧丙基醚的特性。然而,第JP 2004262977号日本专利公开案大幅地增加了树脂的环氧当量及黏度,这在应用上造成困难。其他公开环氧丙基醚的专利申请案包括第JP2009051937 A号日本专利公开案、第JP 2017155080 A号日本专利公开案、及第WO2018225411 A1号PCT专利公开案,这些文献基于所有目的而全文并于此以供参考。
目前仍持续存在改善环氧丙基醚化合物的需求,因为许多上述文献所描述的化合物及方法仍存在缺陷。
发明内容
本发明的方案是关于单酚或多元酚的环氧丙基醚产物及其制备方法。另外,本发明是关于单酚或多元酚的环氧丙基醚产物的应用。
制造环氧丙基醚化合物的方法通常不会仅生成单一化合物,而是生成多种不同的化合物。以下结构为环氧丙基醚化合物的例示性理论结构,其中R
结构(I)、(II)、及(III)分别代表α-二醇化合物(“α-二醇”)、正环氧丙基醚化合物、及多环氧丙基醚化合物(“MGE”)。
部分归因于使用传统方法所制备的众多环氧丙基醚化合物的复杂性,传统上均认为难以同时获得具低可水解氯(hydrolyzable chlorine)含量、高尺寸安定性、高耐热性、高阶电子材料所需的耐高温性、低水分吸收、及用于复合材料的长适用期(pot life)的环氧丙基醚化合物。
然而,本发明人发现,可通过控制环氧官能基的数目及羟官能基的数目,来获得上述许多特性同时获得改良的本文所公开的单酚或多元酚的环氧丙基醚产物。例如,单酚或多元酚的环氧丙基醚产物合意地包括低量的羟官能基及低量的含卤基团的化合物。
单酚或多元酚的环氧丙基醚产物一般而言包含一环氧当量重量(“EEW”)及一羟值(“HV”),其中环氧当量重量乘以羟值(EEW×HV)的值为1至10当量/100当量(eq/100eq)。在某些情况下,单酚或多元酚的环氧丙基醚产物具有1至9当量/100当量的EEW×HV值。单酚或多元酚的环氧丙基醚产物可具有150至200克/当量(g/eq)的EEW值及/或0.01至0.06当量/100克(eq/100g)的HV值。在至少一实例中,单酚或多元酚的环氧丙基醚产物可包含0.0001至0.11毫当量/克(mEq/g)的α-二醇甘油醚基。另外或替代地,单酚或多元酚的环氧丙基醚产物可包括300ppm或更少的可水解氯。
单酚或多元酚的环氧丙基醚产物可由选自由以下所组成的群组的多元酚形成:间苯二酚、氢醌、2,2-双(4'-羟基苯基)丙烷(即双酚A)、二羟基二苯基甲烷(即双酚F)的异构物的混合物、4,4'-二羟基二苯基环己烷(即双酚Z)、4,4'-二羟基-3,3'-二甲基二苯基丙烷(即双酚C)、4,4'-二羟基二苯甲酮、双(4'-羟基苯基)-1,1-乙烷(即双酚E)、双(4'-羟基苯基)-1,1-异丁烷、双(4'-羟基-三级丁基苯基)-2,2-丙烷、双(羟基-萘基)甲烷、1,5-二羟基萘、三(4-羟基苯基)甲烷、双(4-羟基苯基)醚、双(4-羟基苯基)砜(即双酚S)、酚系酚醛(phenol novolak)、溴化酚系酚醛(brominated phenol novolak)、邻甲酚系酚醛(o-cresol novolak)、间苯二酚系酚醛(resorcin novolak)、溴化间苯二酚系酚醛(brominated resorcin novolak)、三(羟基苯基)甲烷、四酚乙烷酚类树脂(tetraphenolethane phenolic resin)、醛-聚酚类缩合物(aldehyde-polyphenolic condensate)、二环戊二烯-酚类树脂(dicyclopentadiene-phenolic resin)、9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物酚类树脂(9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide(DOPO)phenolic resin)及其组合。
可获得的环氧树脂组合物包含硬化剂以及如本文所述的单酚或多元酚的环氧丙基醚产物。适合用于环氧树脂组合物的硬化剂可选自由以下所组成的群组:聚醚胺、异佛尔酮二胺、孟烷二胺、4,4-二胺基二环己基甲烷、4,4'-亚甲基双(2-甲基环己基-胺)(4,4'-methylenebis(2-methylcyclohexyl-amine))、4-甲基-1,3'-环己二胺(4-methyl-1,3'-cyclohexanediamine)、4,4'-亚甲基二苯胺(4,4'-diaminodiphenylmethane)、双氰胺、咪唑、酚系酚醛、二胺基二苯基甲烷、二胺基二苯基砜、间苯二胺、酞酐、四氢酞酐、焦蜜石酸酐、二苯甲酮、四羧酸酐、及其组合。在至少一情况下,硬化剂为异佛尔酮二胺,且其中环氧树脂组合物在固化后的玻璃转移温度系大于137℃。在至少一情况下,硬化剂为异佛尔酮二胺,且其中环氧树脂组合物添加固化剂后在温度30℃下的黏度适用期(viscosity potlife)为40至100分钟。
一种制备单酚或多元酚的环氧丙基醚产物的方法一般而言包括:
(a)在催化剂存在下使环氧卤丙烷与单酚或多元酚反应以制备卤醇醚(halohydrin ether);以及
(b)将该卤醇醚去卤化以形成单酚或多元酚的环氧丙基醚产物。
环氧卤丙烷与单酚或多元酚的反应可于30℃至80℃的温度下进行。在某些情况下,本方法在例如反应步骤期间的环氧卤丙烷与单酚或多元酚的羟基的莫耳比使用3.5:1至11.0:1。较佳地,以环氧卤丙烷的总重量计,于环氧卤丙烷与单酚或多元酚的反应期间所存在的缩水甘油不大于2重量%。另外或替代地,以环氧卤丙烷的总重量计,于环氧卤丙烷与单酚或多元酚的反应期间所存在的水不大于1重量%。
制备单酚或多元酚的环氧丙基醚产物的方法可使用含鎓盐的催化剂。在某些情况下,鎓盐相对于单酚或多元酚的总含量是以1,000至9,000ppm的含量存在。适合的鎓盐包括选自由以下所组成的群组者:苄基三丁基氯化铵、苄基三乙基氯化铵、苄基三甲基氯化铵、四丁基溴化铵、四甲基氯化铵、四丁基铵氢亚硫酸盐、三辛基甲基氯化铵及其组合。
卤醇醚可通过使用碱来去卤化。碱可选自由以下所组成的群组:碱金属或碱土金属的氢氧化物、碳酸盐、碳酸氢盐、及其组合。例如,碱可选自由以下所组成的群组:NaOH、KOH、Na
附图说明
于此将例示性地参考所附图式来描述本技术的实施方式,其中:
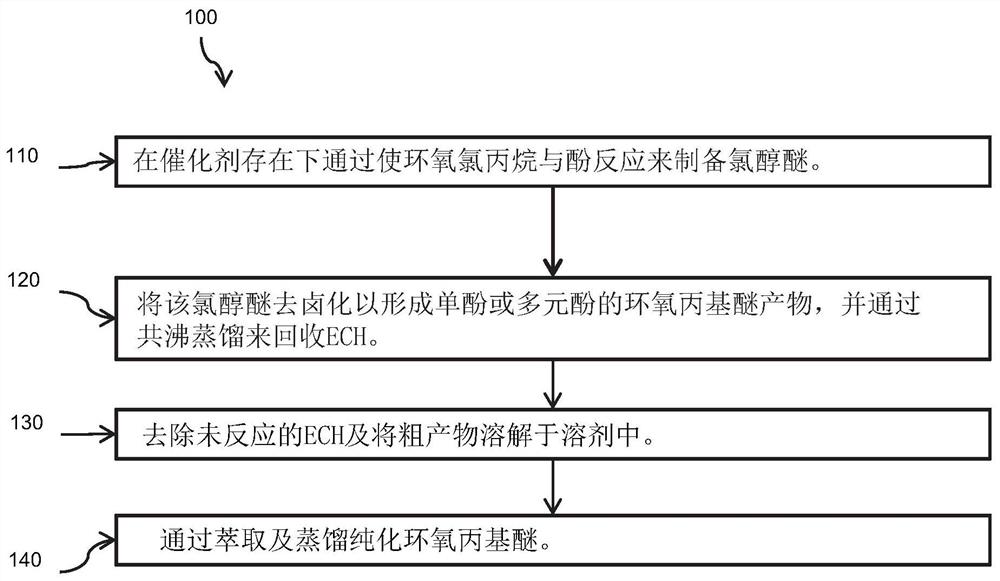
图1为描绘根据本发明的方案的制备单酚或多元酚的环氧丙基醚产物的一例示性方法的流程图;
图2为显示EEW×HV值对根据本发明的方案的例示性环氧树脂与比较环氧树脂的黏度适用期的效果的散布图;以及
图3为显示EEW×HV值对根据本发明的方案的例示性环氧树脂与比较环氧树脂的玻璃转移温度(Tg)的效果的散布图。
应理解各方案并不限于图式所示的安排、手段及特征。
附图标记说明
100:方法
110、120、130、140:步骤。
具体实施方式
本发明的方案是关于单酚或多元酚的环氧丙基醚产物及其制备方法。另外,本发明是关于单酚或多元酚的环氧丙基醚产物的应用。于本文中,单酚或多元酚的环氧丙基醚产物为包含由单酚或多元酚化合物所形成的环氧丙基醚化合物的组合物。多元酚化合物包括具有大于一羟基的酚(例如二羟基苯)、含大于一酚基的化合物(例如双酚A、多元酚等)以及酚树脂(例如酚甲醛树脂)。
根据本发明的一方案,是提供制备酚的环氧丙基醚产物的方法。本发明的方法可有利地制备具有高的环氧丙基相对于羟基的比例的单酚或多元酚的环氧丙基醚产物。
图1描绘制备酚的环氧丙基醚产物的例示性方法100。作为简要概述,方法100包括于步骤110中制备氯醇醚,于步骤120中将该氯醇醚去卤化并通过共沸蒸馏来回收环氧氯丙烷(epichlorohydrin,“ECH”),于步骤130中去除未反应的ECH,并将粗产物(crudeproduct)溶解于溶剂中,以及于步骤140中通过萃取及蒸馏纯化环氧丙基醚。
于步骤110中,在催化剂存在下使ECH与单酚或多元酚反应来制备氯醇醚。该反应可为于液相中进行的偶合反应,较佳伴随混合。反应混合物或溶液的温度可为约20℃至约200℃。例如,反应混合物或溶液的温度在反应期间可为约25℃至约100℃、约30℃至约80℃、约35℃至约75℃、或约40℃至约70℃的温度。
于方法100中所用的ECH可来自新的ECH或回收的来源,其具有小于2重量%的缩水甘油及/或小于1重量%的水。例如,理想上可使用包含至少95重量%的ECH的ECH组合物或来源,较佳至少97重量%、较佳至少98重量%、较佳至少99重量%、较佳至少99.5重量%、或较佳至少99.9重量%的ECH。另外或替代地,氯醇醚较佳是制成一反应混合物,其中基于用于制备氯醇醚的ECH的总重量,具有不大于2重量%、不大于1.5重量%、不大于1重量%、或不大于0.5重量%的缩水甘油,及/或不大于1重量%、不大于0.5重量%、不大于0.3重量%、或不大于0.1重量%的水。虽非旨在限制其任何特定方案,发明人相信反应中的水及缩水甘油可导致产物中更多的水解化合物(例如可水解氯、α-二醇化合物等),且导致酚的聚环氧丙基醚的高环氧当量重量(“EEW”)。
可控制ECH的含量及酚的含量使得ECH与酚类羟基的莫耳比([ECH]/[OH])为3.5:1至11.0:1。在某些情况下,ECH与酚类羟基的莫耳比([ECH]/[OH])为3.7:1至11.0:1、4:1至10.74:1、4:1至10:1、4:1至9:1、4:1至8:1、4.5:1至10.74:1、4.5:1至10:1、4.5:1至9:1、或4.5:1至8:1。一般而言,ECH对酚类羟基([ECH]/[OH])的值越高,产物中的MGE化合物越多且OH基团越少。例如,较大的[ECH]/[OH]值可代表较多环氧丙基醚基团及较低的EEW值。较大的[ECH]/[OH]值也可代表较少量的羟基基团及较低的HV值。本发明人发现[ECH]/[OH]值的增加可降低EEW×HV值或与EEW×HV值的降低相关。
氯醇醚较佳于催化剂存在下制备。催化剂可为鎓盐,例如包含至少一四级铵基团及至少一卤素或磷酸(phosphate)基团。例如,催化剂可为四级铵盐、四级鏻盐、冠醚或其组合。催化剂可包括或形成自包括以下的阳离子:四甲基铵、三甲基-乙基铵、二甲基二乙基铵、三乙基-铵、三丙基-甲基铵、三丁基-甲基铵、三辛基-甲基铵、四乙基铵、三甲基-丙基铵、三甲基苯基铵、苄基三甲基铵、苄基三乙基铵、二烯丙基二甲基铵、正-辛基三甲基铵、硬脂酰基三甲基铵(stearyl trimethyl ammonium)、四丙基铵、十六烷基乙基铵、四丙基铵、四正丁基铵及其组合。另外或替代地,催化剂可包括或形成自例如包括以下的阴离子:CIO
可使用的鎓盐的实例包括四甲基氯化铵、四乙基氯化铵、四丁基氯化铵、十六烷基-三甲基氯化铵、四甲基溴化铵、四乙基溴化铵、四丁基溴化铵、十六烷基-三甲基溴化铵物、四苯基氯化鏻、三苯基甲基氯化鏻、三苯基乙基氯化鏻、三苯基丙基氯化鏻、三苯基丁基氯化鏻、苄基三苯基氯化鏻、四苯基溴化鏻、三苯基甲基溴化鏻、三苯基乙基溴化鏻、三苯基乙基碘化鏻、三苯基丁基碘化鏻、苄基三苯基碘化鏻、苄基三甲基氯化铵、苄基三甲基溴化铵、苄基三甲基氢氧化铵、苄基三乙基氯化铵、苄基三乙基溴化铵、四丁基氢氧化铵、四丁基铵亚硫酸氢(tetrabutylammonium hydrogen sulfite)、四辛基氯化铵、四丁基氯化鏻、四丁基溴化鏻、四丁基碘化鏻、乙基三苯基氯化鏻、乙基三苯基溴化鏻、乙基三苯基碘化鏻、苄基三丁基氯化铵、三辛基甲基氯化铵、氯化胆碱(chlorine chloride)及其组合。适合的鎓盐包括但不限于苄基三甲基氯化铵、苄基三乙基氯化铵、四乙基氯化铵及其组合。在某些情况下,催化剂是选自或包括苄基-三乙基氯化铵、苄基-三甲基氯化铵或其组合。
催化剂(例如鎓盐)相对于单酚或多元酚的总含量可以1,000至9,000ppm的含量存在。一般而言,据信催化剂量太多可能导致颜色加深,而催化剂量太少可能降低反应效率。例如,相对于单酚或多元酚的总含量,催化剂可以约1,500至约8,500ppm、约2,000至约7,000ppm、约2,500至约6,500ppm、或约3,000至约6,000ppm的含量存在。另外或替代地,催化剂相对于羟基的含量可以约1,000至约12,000ppm的含量存在。在某些情况下,催化剂含量相对于羟基含量可为约1,500至约9,500ppm、约2,000至约9,000ppm、约2,500至约8,500ppm、或约3,000至约8,000ppm。
所制备的氯醇醚可视需要地为单酚或多元酚的相应的氯丙醇醚(propylenechlorohydrin ether)。ECH与单酚或多元酚之间的反应可生成根据下式的产物:
在某些情况下,R为氢、烷基、芳基、(羟基芳基)烷基((hydroxyar)alkyl)、(羟基芳基)巯基((hydroxyar)thiol)、或双(羟基芳基)烷基。该氯丙醇醚又可接着与第二当量的环氧氯丙烷反应以生成少量1,3-二氯丙醇及该酚的环氧丙基醚产物。
于步骤120中,将氯醇醚去卤化以形成单酚或多元酚的环氧丙基醚产物。氯醇醚可使用碱来去卤化。碱是选自碱金属或碱土金属的氢氧化物、碳酸盐、碳酸氢盐及其组合。例如,碱可包括以下一种或多种:NaOH、KOH、LiOH、Ca(OH)
另外,于步骤120中,水与ECH是例如于去卤化期间通过共沸蒸馏来回收。基于以下所述的实施例,使用本领域技术人员已知的仪器及方法,水与ECH的共沸物可被冷凝,然后成为有机相及水相。有机相可回收至反应系统中。较佳抛弃水相。
于步骤130中,在减压下去除未反应的ECH。较佳地,粗制的环氧丙基醚溶解于溶剂中。
于步骤140中,将该粗制的环氧丙基醚溶液用水洗涤。在减压下从所得混合物滤除溶剂以获得环氧丙基醚。
根据本发明的另一方案,提供一种单酚或多元酚的环氧丙基醚产物。该单酚或多元酚的环氧丙基醚产物一般而言包含一环氧当量重量(“EEW”)及一羟值(“HV”),其中环氧当量重量乘以羟值(EEW×HV)的值为1至10当量/100当量。例如,单酚或多元酚的环氧丙基醚产物的EEW×HV值可为:1至9当量/100当量、1.5至9当量/100当量、2至9当量/100当量、2.5至9当量/100当量、或3至9当量/100当量;1至8.5当量/100当量、1.5至8.5当量/100当量、2至8.5当量/100当量、2.5至8.5当量/100当量、或3至8.5当量/100当量;1至8当量/100当量、1.5至8当量/100当量、2至8当量/100当量、2.5至8当量/100当量、或3至8当量/100当量;1至7.5当量/100当量、1.5至7.5当量/100当量、2至7.5当量/100当量、2.5至7.5当量/100当量、或3至7.5当量/100当量;或1至7当量/100当量、1.5至7当量/100当量、2至7当量/100当量、2.5至7当量/100当量、或3至7当量/100当量。
单酚或多元酚的环氧丙基醚产物可具有150至200克/当量的EEW值及/或0.01至0.06当量/100克的HV值。于部分情况中,EEW值可为:150至200克/当量、155至200克/当量、160至200克/当量、或165至200克/当量;150至195克/当量、155至195克/当量、160至195克/当量、或165至195克/当量;150至190克/当量、155至190克/当量、160至190克/当量、或165至190克/当量;或者150至185克/当量、155至180克/当量、160至185克/当量、165至185克/当量。HV值可为:0.01至0.06当量/100克、0.02至0.06当量/100克、0.03至0.06当量/100克;0.01至0.05当量/100克、0.02至0.05当量/100克、或0.03至0.05当量/100克;或者0.01至0.04当量/100克、0.02至0.04当量/100克、0.03至0.04当量/100克。
单酚或多元酚的环氧丙基醚产物可包含0.0001至0.11毫当量/克的α-二醇甘油醚基。例如,α-二醇甘油醚基的含量可为:0.0001至0.11毫当量/克、0.001至0.11毫当量/克、0.01至0.11毫当量/克、或0.05至0.11毫当量/克;0.0001至0.9毫当量/克、0.001至0.9毫当量/克、0.01至0.9毫当量/克、或0.05至0.9毫当量/克;0.0001至0.7毫当量/克、0.001至0.7毫当量/克、0.01至0.7毫当量/克、或0.05至0.7毫当量/克;0.0001至0.5毫当量/克、0.001至0.5毫当量/克、或0.01至0.5毫当量/克;或者0.0001至0.3毫当量/克、0.001至0.3毫当量/克、0.01至0.3毫当量/克。另外或替代地,单酚或多元酚的环氧丙基醚产物可包括300ppm或更少的可水解氯。在某些情况下,存在于单酚或多元酚的环氧丙基醚产物中的可水解氯的含量较佳为280ppm或更少、较佳260ppm或更少、较佳240ppm或更少、较佳220ppm或更少、较佳200ppm或更少、较佳180ppm或更少、较佳160ppm或更少、较佳140ppm或更少、较佳120ppm或更少、或较佳100ppm或更少。
单酚或多元酚的环氧丙基醚产物可由单酚、多元酚、或酚类树脂来形成。酚类化合物可通过生物转化或化学转化而获得。有用的酚类化合物包括例如酚、邻甲酚、间甲酚、对甲酚、对-三级丁基酚、对-三级辛基酚、对苯基酚、对枯基酚(p-cumylphenol)、对异丙基酚、对壬基酚、2,3-二甲基酚、2,4-二甲基酚、2,5-二甲基酚、2,6-二甲基酚、3,4-二甲基酚、3,5-二甲基酚、邻乙基酚、间乙基酚、对乙基酚、2,3,4-三甲基酚、2,3,5-三甲基酚、3,4,5-三甲基酚、腰果酚(cardanol)及愈创木酚(guaiacol)或其混合物。
用于制备单酚或多元酚的环氧丙基醚产物的酚可为经取代或未经取代的双酚化合物,例如双酚、2,2-双(4-羟基苯基)丙烷(即双酚A)、1,1-双(4-羟基苯基)-1-苯基-乙烷(即双酚AP)、2,2-双(4-羟基苯基)六氟丙烷(即双酚AF)、2,2-双(4-羟基苯基)丁烷(即双酚B)、双(4-羟基苯基)二苯基甲烷(即双酚BP)、2,2-双(3-甲基-4羟基苯基)丙烷(即双酚C)、双(4-羟基苯基)-2,2-二氯乙烯(即双酚C2)、1,1-双(4-羟基苯基)乙烷(即双酚E)、双(4-羟基苯基)甲烷(即双酚F)、2,2-双(4-羟基-3-异丙基-苯基)丙烷(即双酚G)、1,3-双(2-(4-羟基苯基)-2-丙基)苯(即双酚M)、双(4-羟基苯基)砜(即双酚S)、1,4-双(2-(4-羟基苯基)-2-丙基)苯(即双酚P)、5,5'-(1-甲基伸乙基)-双[1,1'-(双苯基)-2-醇]丙烷(即双酚PH)、1,1-双(4-羟基苯基)-3,3,5-三甲基-环己烷(即双酚TMC)、1,1-双(4-羟基苯基)-环己烷(即双酚Z)、四甲基双酚A、四甲基双酚F、四甲基双酚S、四甲基双酚Z、卤化双酚A、四溴化双酚A、四氯化双酚A、二羟基二苯基硫化物、4,4-硫基双(3-甲基-6-三级丁基酚)或其混合物。
在某些情况下,用于制备单酚或多元酚的环氧丙基醚产物的酚为未经取代的二酚化合物。经取代或未经取代的二酚化合物可为例如儿茶酚(catechol)、间苯二酚、甲基间苯二酚、氢醌、单甲基氢醌、二甲基氢醌、三甲基氢醌、单-三级丁基氢醌、二-三级丁基氢醌、二羟基萘、二羟基甲基萘或其混合物。
单酚或多元酚的环氧丙基醚产物可由来自酚类化合物与醛的反应所获的原料或化合物来形成。可用于形成酚类化合物或原材料的醛包括例如甲醛、乙醛、丙醛、丁醛、戊醛、己醛、苯甲醛、氯醛、溴醛、乙二醛、丙二醛、琥珀醛、戊二醛、己二醛、环己醛、萜醛(terpaldehyde)、丙烯醛、巴豆醛、柳醛、糠醛、羟基苯甲醛、富马醛、己-2,4-二烯二醛、辛-2,4,6-三烯二醛、苯基乙二醛、对酞醛、酞醛、异酞醛、萘二醛或其混合物。有用的酚类树脂包括例如酚系酚醛、溴化酚系酚醛、邻甲酚系酚醛、间苯二酚系酚醛、溴化间苯二酚系酚醛、三(羟基苯基)甲烷、四酚乙烷酚类树脂或其组合。其他酚类树脂包括第10,138,325B2号美国专利的醛-聚酚类缩合物、第0148817号欧洲专利的二环戊二烯-酚类树脂、第6,984,716号及第8,426,547号美国专利的9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物酚类树脂,这些文献基于所有目的而全文并于此以供参考。
在某些情况下,单酚或多元酚的环氧丙基醚产物可由选自以下所组成的群组的多酚来形成:间苯二酚、氢醌、2,2-双(4'-羟基苯基)丙烷(即双酚A)、二羟基二苯基甲烷(即双酚F)的异构物的混合物、4,4'-二羟基二苯基环己烷(即双酚Z)、4,4'-二羟基-3,3'-二甲基二苯基丙烷(即双酚C)、4,4'-二羟基二苯甲酮、双(4'-羟基苯基)-1,1-乙烷(即双酚E)、双(4'-羟基苯基)-1,1-异丁烷、双(4'-羟基-三级丁基苯基)-2,2-丙烷、双(羟基-萘基)甲烷、1,5-二羟基萘、三(4-羟基苯基)甲烷、双(4-羟基苯基)醚、双(4-羟基苯基)砜(即双酚S)、酚系酚醛、溴化酚系酚醛、邻甲酚系酚醛、间苯二酚系酚醛、溴化间苯二酚系酚醛、三(羟基苯基)甲烷、四酚乙烷酚类树脂、醛-聚酚类缩合物、二环戊二烯-酚类树脂、9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物酚类树脂及其组合。
单酚或多元酚的环氧丙基醚产物可由选自以下所组成的群组的环氧卤丙烷来形成:环氧氯丙烷、环氧溴丙烷、环氧碘丙烷、甲基环氧氯丙烷、甲基环氧溴丙烷、甲基环氧碘丙烷及其组合。环氧卤丙烷的来源可为新的或回收的环氧卤丙烷,例如于步骤140所分离得到。环氧卤丙烷可具有杂质,包括例如水、反应中所用的溶剂、缩水甘油、甘油、1-卤-丙醇、2-卤-丙醇、2-卤-烯丙基醇、1,3-二卤-2-丙醇、2,3-二卤-2-丙醇及1-卤-2,3-丙二醇。
如上所述,单酚或多元酚的环氧丙基醚产物的ECH与酚类羟基的莫耳比([ECH]/[OH])较佳为3.5:1至11.0:1。在某些情况下,ECH与酚类羟基的莫耳比([ECH]/[OH])为3.5:1至10.9:1、3.7:1至11:1、4:1至10.74:1、4:1至10:1、4:1至9:1、4:1至8:1、4.5:1至10.74:1、4.5:1至10:1、4.5:1至9:1、或4.5:1至8:1。
本发明的单酚或多元酚的环氧丙基醚产物可于反应期间使用溶剂溶解反应物而制备,或可不使用溶剂而制备。溶剂的用量可变化,但一般而言相对于100重量份的固含量是以20重量份至95重量份的含量存在。在某些实例中,溶剂存在于环氧树脂中的含量为相对于100重量份的固含量的30重量份至90重量份、40重量份至85重量份、或50至80重量份。
于本文中可用的溶剂并无特别限制,只要其对于羧酸酯化反应为惰性溶剂即可。溶剂可为脂族烃溶剂、酯溶剂、醚溶剂、有机溶剂、及/或酮溶剂。溶剂的实例包括甲苯、二甲苯、乙基苯、四甲基苯、己烷、辛烷、癸烷、石油醚、白汽油(white gasoline)、溶剂石脑油、或其组合。适合的溶剂包括:乙酸烷酯,例如乙酸乙酯、乙酸丙酯、及乙酸丁酯;环酯,例如γ-丁内酯;乙二醇单甲基醚乙酸酯、二乙二醇、甲基醚单乙酸酯、二乙二醇单乙基醚单乙酸酯、三乙二醇单乙基醚;单烷二醇单烷基醚单乙酸酯,例如单乙酸酯、二乙二醇单丁基醚单乙酸酯、丙二醇单甲基醚乙酸酯、及丁二醇单甲基醚乙酸酯;及/或聚烷二醇单酯,例如单烷基醚单乙酸酯、二烷基戊二酸酯、二烷基琥珀酸酯、及二烷基己二酸酯。
醚溶剂的实例包括:烷基醚,例如二乙基醚及乙基丁基醚;二醇醚,例如乙二醇二甲基醚、乙二醇二乙基醚、二丙二醇二甲基醚、二丙二醇二乙基醚、三乙二醇二甲基醚、三乙二醇二乙基醚;及环醚,例如四氢呋喃。适合的酮溶剂包括丙酮、甲基乙基酮、甲基丙基酮、甲基异丁基酮、环己酮、及/或异佛尔酮。
若存在溶剂,则溶剂可为选自醇类的有机溶剂,例如一级醇、二级醇、乙醇、1-丙醇、1-丁醇、1-戊醇、1-己醇、异丙醇、2-丁醇、2-戊醇、3-戊醇、2-己醇、环己醇、2-庚醇、3-庚醇、三级丁醇及三级戊醇。其他有机溶剂的实例包括乙二醇、乙二醇单甲基醚、乙二醇单乙基醚、乙二醇单正丙基醚、乙二醇单丁基正丁基醚、乙二醇单苯基醚、二乙二醇、二乙二醇单甲基醚、二伸乙基缩水甘油(diethylene glycidyl)、单乙基醚、二乙二醇单正丙基醚、二乙二醇单正丁基醚、三乙二醇、三乙二醇单甲基醚、三乙二醇单丁基正丁基醚、丙二醇、丙二醇单甲基醚、丙二醇单乙基醚、丙二醇单正丙基醚、丙二醇单正丁基醚、丙二醇单苯基醚、二丙二醇、二丙二醇单甲基醚、二丙二醇单乙基醚、二丙二醇单正丙基醚、二丙二醇单正丁基醚、三丙二醇、三丙二醇单甲基醚及三丙二醇单正丁基醚。在至少一情况下,有机溶剂是选自脂族及芳族烃、脂族二级醇、卤化脂族烃、脂族醚、脂族腈、环醚、酮、酰胺、亚砜及其组合,例如戊烷、己烷、辛烷、甲苯、二甲苯、甲基乙基酮、甲基异丁基酮、N,N-二甲基甲酰胺、二甲基亚砜、四氢呋喃、1,4-二恶烷、二氯甲烷、乙二醇二甲基醚、N,N-二甲基乙酰胺、乙腈、异丙醇、异丁醇、丙二醇单甲基醚及其组合。
根据本发明的另一方案,提供一种环氧树脂组合物,其包含硬化剂及本文所记载的单酚或多元酚的环氧丙基醚产物。硬化剂可选自由以下所组成的群组:聚醚胺、异佛尔酮二胺、孟烷二胺、4,4-二胺基二环己基甲烷、4,4'-亚甲基双(2-甲基环己基-胺)、4-甲基-1,3'-环己二胺、4,4'-亚甲基二苯胺、双氰胺、咪唑、酚系酚醛、二胺基二苯基甲烷、二胺基二苯基砜、间苯二胺、酞酐、四氢酞酐、焦蜜石酸酐、二苯甲酮、四羧酸酐及其组合。
硬化剂可经选择及/或此硬化剂的用量可经控制使得环氧树脂组合物在固化后具有大于137℃的玻璃转移温度。在某些情况下,玻璃转移温度系大于138℃、大于139℃、大于140℃、大于141℃、大于142℃、大于143℃、大于144℃、大于145℃、大于146℃、大于147℃、大于148℃、大于149℃、大于150℃、大于151℃或大于152℃。在至少一情况下,硬化剂为异佛尔酮二胺,且其中环氧树脂组合物在固化后的玻璃转移温度大于137℃。另外或替代地,硬化剂可经选择及/或此硬化剂的用量可经控制使得环氧树脂组合物添加固化剂后在温度30℃下的黏度适用期为40至100分钟。例如在温度30℃下的黏度适用期可为:40至100分钟、45至100分钟、50至100分钟、55至100分钟、60至100分钟、70至100分钟、或75至100分钟;40至85分钟、45至85分钟、50至85分钟、55至85分钟、60至85分钟、70至85分钟、或75至85分钟;或40至75分钟、45至75分钟、50至75分钟、55至75分钟、或60至75分钟。在特定情况下,硬化剂为异佛尔酮二胺,且其中环氧树脂组合物在温度30℃下的黏度适用期为40至100分钟。
实施例
以下实施例主要提供以用于阐述本发明各方案所达成的优势及优点。
实施例1至4及比较例1至4
于实施例1至4中,4,4'-(丙烷-2,2-二基)二酚的环氧丙基醚产物系由获自长春人造树脂厂的4,4'-(丙烷-2,2-二基)二酚(以下称为“BPA”)来制备。BPA的比较环氧丙基醚产物是根据比较例1至4来制备。实施例1至4及比较例1至4所制备的BPA的环氧丙基醚产物的性质提供于下文表1。
[实施例1]
使用装备有如下装置的3公升-4颈反应器来制备BPA的环氧丙基醚产物:用于控制及显示温度及压力的装置、以及用于冷凝水与环氧氯丙烷(本文也称为“ECH”)的共蒸馏混合物并将该共蒸馏混合物分离为有机相及水相的装置。具体而言,将300克的BPA、900克的ECH、以及作为偶合反应的催化剂的2.7克的苄基三乙基氯化铵(相对于BPA为9,000ppm)添加至3公升-4颈反应器。ECH与来自BPA的OH基的莫耳比为3.7([ECH]/[OH]=3.7)。环氧氯丙烷的纯度为超过99重量%,且缩水甘油及水在环氧氯丙烷中的含量分别为0.31重量%及0.15重量%。于大气压力下及40℃下搅拌该混合物以形成均质溶液,然后将温度于50小时期间逐渐从40℃升至75℃,然后将温度维持于75℃下16小时。
接着进行第一去氢卤化步骤,其中于60℃的温度下以固定速率以6小时将205克49.5重量%氢氧化钠水溶液添加至该混合物,同时将反应系统中所含有的水于100托(torr)的绝对压力下共沸地蒸馏并冷凝。将冷凝的共沸物分离为有机相及水相。将有机相(主要为ECH)回收至反应器中且抛弃水相。在完成前述205克的氢氧化钠水溶液(氢氧化钠浓度49.5重量%)的添加之后,将系统维持于相同条件下1小时。完成第一去氢卤化步骤,并将未反应的ECH于减压下馏除。
再进行额外的去氢卤化步骤,其中于80℃的温度及大气压力下以2小时添加4.0克49.5重量%氢氧化钠水溶液。然后,将所得的粗制的环氧树脂中所含有的氯化钠溶解于甲苯及去离子水中,然后用水洗涤。将有机溶剂于减压下从所得混合物中滤除以获得环氧丙基醚。
[实施例2]
使用与实施例1所述相同的方法合成BPA的环氧丙基醚产物,但是:环氧氯丙烷的用量为1,120克;ECH与来自BPA的OH基的莫耳比为4.61([ECH]/[OH]=4.61);缩水甘油及水在环氧氯丙烷中的含量分别为0.44重量%及0.04重量%;以及苄基三乙基氯化铵的含量为1.35克(相对于BPA为4,500ppm)。
[实施例3]
使用与实施例1相同的方法合成BPA的环氧丙基醚产物,但是:环氧氯丙烷的用量为1,650克;ECH与来自BPA的OH基的莫耳比为6.79([ECH]/[OH]=6.79);缩水甘油及水在环氧氯丙烷中的含量分别为0.15重量%及0.01重量%;以及苄基三乙基氯化铵的含量为0.6克(相对于BPA为2,000ppm)。
[实施例4]
使用与实施例1相同的方法合成BPA的环氧丙基醚产物,但是:环氧氯丙烷的用量为850克;ECH与来自BPA的OH基的莫耳比为3.5([ECH]/[OH]=3.5);缩水甘油及水在环氧氯丙烷中的含量分别为0.15重量%及0.01重量%;苄基三乙基氯化铵的含量为1.2克(相对于BPA为4,000ppm);以及于偶合反应中,将温度于48小时期间逐渐从45℃升至60℃,然后将温度维持于60℃下8小时。
[比较例1]
根据第6,001,873号美国专利的方法合成BPA的环氧丙基醚产物,该文献基于所有目的而全文并于此以供参考。例如,将300克的BPA、680克的环氧氯丙烷、及100克的乙酸异丙酯添加至3公升-4颈反应器,该3公升-4颈反应器装备有用于控制及显示温度及压力的装置以及用于冷凝水的共蒸馏混合物的装置。ECH与来自BPA的OH基的莫耳比为2.8([ECH]/[OH]=2.8)。将环氧氯丙烷与溶剂分离为有机相及水相。
于大气压力下搅拌混合物以形成均质溶液,然后于60mm Hg的绝对压力下加热至70℃。在达到压力及温度的平衡后,以固定速率以5小时将205克49.5重量%氢氧化钠水溶液添加至该混合物,同时将反应系统中所含有的水共沸地蒸馏并冷凝。将冷凝的共沸物分离为有机相及水相。将有机相(主要为ECH)回收至反应器中且抛弃水相。在完成反应之后,将未反应的环氧氯丙烷及溶剂于减压下滤除。将所得的粗制的环氧丙基醚混合物中所含有的氯化钠溶解于甲苯及去离子水中,然后用水洗涤。将有机溶剂于减压下从所得混合物中滤除以获得BPA的环氧丙基醚产物。
[比较例2]
根据实施例2所述的方法合成BPA的环氧丙基醚产物,但是:环氧氯丙烷的用量为730克;ECH与来自BPA的OH基的莫耳比为3.0([ECH]/[OH]=3.0);以及缩水甘油及水在ECH中的含量分别为0.65重量%及1.34重量%。
[比较例3]
比较例3为D.E.R.
[比较例4]
比较例4为D.E.R.
表1
根据ASTM D1726的方法对实施例1至4及比较例1至4进行评断以测定可水解氯(本文也称为“HyCl”)。实施例1至4及比较例1至4的黏度(本文也称为“VIS”)是根据ISO 3219的方法评断。环氧当量重量(本文也称为“EEW”)是根据ASTM D1652的方法评断。α-二醇是依JIS K7146量测。
羟值(本文也称为“HV值”)是根据ASTM E222的方法(方法C)及使用下式计算:
其中:
C=0.5毫当量/毫升,NaOH标准溶液,
EEW=样品的环氧当量重量(克/当量),
Vo=空白测试所需的NaOH标准溶液的毫升数,
Vs=样品所需的NaOH标准溶液的毫升数,以及
W=所用样品的克数。
使用Waters 600仪器、Waters 2487双波长吸收侦测器、及Waters XTerra RP184.6毫米x250毫米,5微米管柱对实施例1至4及比较例1至4进行HPLC分析。所用的侦测波长为254奈米;管柱温度为40℃;移动相为80分钟期间水与乙腈的比为40:60至0:100(体积:体积)的溶液;以及移动相的流速为1.0毫升/分钟。
如表1所示,实施例1至4具有低的α-二醇及低的HV值。另外,实施例1至4具有高含量的n=0及MGE(多环氧丙基醚,结构(III))。参照以下所示的结构,n=0的量越高,则EEW越低,此代表较高纯度的环氧丙基醚产物。
上方结构为理论的BPA二环氧丙基醚结构,如上所述,于实际形成单酚或多元酚的环氧丙基醚产物的方法期间,制备了具有类似于以上所呈现的理论性代表结构的变化结构的多种化合物。实施例1至4有利地具有小于10的EEW与HV的乘积值(EEW×HV),其为OH基与环氧丙基醚基之比。相反地,比较例1至4具有大于10的EEW与HV的乘积值。发明人发现,一般而言ECH对酚类羟基([ECH]/[OH])的值越高,则产物中越多MGE化合物及越少OH基。例如,较大的[ECH]/[OH]值可代表较多的MGE化合物(结构(III))及较低的EEW值。较大的[ECH]/[OH]值也可代表较少量的羟基及较低的HV值。本发明人发现[ECH]/[OH]值的增加可降低EEW×HV值或与EEW×HV值的降低相关。
实施例5至7及比较例5至7
在实施例5至7中,双酚F的环氧丙基醚产物系由获自长春人造树脂厂的双酚F(以下称为“BPF”)来制备。BPF的比较环氧丙基醚产物是根据比较例5至7来制备。实施例5至7及比较例5至7所制备的环氧丙基醚的性质提供于下文表2。
[实施例5]
根据实施例1所述的方法合成BPF的环氧丙基醚产物,但是使用具有85.2%的二聚物含量的BPF(获自长春人造树脂厂)替代实施例1所用的BPA来制备环氧丙基醚。另外,环氧氯丙烷的用量为1,360克。ECH与来自BPF的OH基的莫耳比为4.88([ECH]/[OH]=4.88)。缩水甘油及水在环氧氯丙烷中的含量分别为0.12重量%及小于0.01重量%。苄基三乙基氯化铵的用量为1.35克(相对于BPF为4,500ppm),以及49.5重量%氢氧化钠水溶液的用量为240克。
[实施例6]
根据实施例5所述的方法合成BPF的环氧丙基醚产物,但是:环氧氯丙烷的用量为1,850克,ECH与来自BPF的OH基的莫耳比为6.33([ECH]/[OH]=6.33);苄基三乙基氯化铵的用量为0.45克(相对于BPF为1,500ppm),以及49.5重量%氢氧化钠水溶液的用量为255克。
[实施例7]
根据实施例5所述的方法合成BPF的环氧丙基醚产物,但是使用获自本州化学公司(Honshu Chemical)的具有90.1重量%的二聚物含量的BPF替代具有85.2重量%的二聚物含量的BPF。
[比较例5]
根据实施例5所述的方法合成BPF的环氧丙基醚产物,但是环氧氯丙烷的用量为836克,且ECH与来自BPF的OH基的莫耳比为3.0([ECH]/[OH]=3.0)。
[比较例6]
根据比较例1所述的方法合成BPF的环氧丙基醚产物,但是:使用具有GPC积分面积85.2%的二聚物含量的BPF替代比较例1所用的BPA;环氧氯丙烷的用量为1,360克,ECH与来自BPF的OH基的莫耳比为4.88([ECH]/[OH]=4.88);缩水甘油及水在环氧氯丙烷中的含量分别为0.12重量%及小于0.01重量%;以及49.5重量%氢氧化钠水溶液的用量为240克。制备此BPF的环氧丙基醚产物的方法不包括鎓盐催化剂。
[比较例7]
比较例7的性质数据是来自日本专利JP 2017/155080A的实施例1,该文献基于所有目的而全文并于此以供参考。日本专利JP 2017/155080A的实施例1中,使用装备有回流冷凝器、搅拌器、氮进气管、油-水分离器、及真空装置的玻璃的可分离烧瓶来制备双酚F型液态树脂。将100重量份BPF-1、231.3重量份的具有莫耳比ECH/OH=以每莫耳的双酚F的酚类羟基计2.5的环氧氯丙烷、及1.6重量份水填充至前述设备中。BPF-1的二聚物(2核种)GPC积分面积含量为97.2%。于氮气气氛下将温度升至60℃以溶解所添加的成分。
接着,在维持反应系统中的温度于60℃的同时,将反应系统缓慢减压以使环氧氯丙烷及水共沸,将水通过油-水分离器自上层移除,并将下层的环氧氯丙烷回收至系统。在维持此状态的同时,将73.5重量份浓度49重量%的氢氧化钠水溶液以150分钟逐滴添加,该氢氧化钠水溶液具有莫耳比NaOH/OH=以每莫耳的双酚F的酚类羟基计0.9的氢氧化钠。在此期间,反应系统维持于60℃至65℃的温度,同时真空度维持于约100至约140mmHg,以及系统中的水含量为2.6至3.2重量%。
在完成逐滴添加之后,将回流的环氧氯丙烷自系统中移除,然后减压程度及温度缓慢增加。于150℃的温度及5mmHg的压力下将环氧氯丙烷回收并移除以获得粗制环氧丙基醚。的后,反应系统回复至常压,添加300重量份的甲苯以溶解该粗制环氧丙基醚。另外,将10重量份浓度20重量%的氢氧化钠水溶液添加至该粗制环氧丙基醚溶液中。然后于80℃的温度下进行反应1.5小时。添加500重量份水以分离并移除副产物盐。之后,以300重量份水进行水洗数次,并重复至水洗水变为中性。将此溶液于5mmHg的减压下加热至150℃的温度以去除甲苯,从而获得双酚F型液态树脂。
表2
实施例5至7及比较例5至6的BPF的环氧丙基醚产物中的二聚物结构的含量使用凝胶渗透层析测定。具体而言,使用Waters 717自动样品注射器(Autosampler)及Waters 515泵以及Waters 2487双波长吸收侦测器以用于凝胶渗透层析。侦测波长为254奈米,且侦测器温度为35℃。
如表2所示,来自实施例5至7的BPF的环氧丙基醚产物具有较低的α-二醇、低OH值、及低黏度。另外,来自实施例5及6的BPF的环氧丙基醚产物如凝胶渗透层析所测定具有较多的双官能环氧丙基醚结构。尤其,尽管相较于用于制备实施例5及6的BPF的环氧丙基醚产物,比较例7由具有显著较大量的二聚物结构的进料BPF来制备,来自实施例5及6的BPF的环氧丙基醚产物相较于比较例7的BPF的环氧丙基醚产物系具有较大量的二聚物结构。
实施例8至10及比较例8至10
于实施例8中,羟基苯甲醛酚类树脂的环氧丙基醚产物是通过使用获自长春人造树脂厂的羟基苯甲醛酚类树脂(以下称为“PF1250”)来制备。于实施例9中,根据本发明的方案来制备乙二醛-酚缩合物的环氧丙基醚产物。于实施例10中,二环戊二烯酚树脂的环氧丙基醚产物是通过使用获自长春人造树脂厂的二环戊二烯酚树脂(以下称为“PF9110”)来制备。比较例8至10可商购自长春人造树脂厂的环氧丙基醚。比较例8至10所用的原材料分别与实施例8至10一致。实施例8至10及比较例8至10所制备的环氧丙基醚的性质提供于以下表3。
[实施例8]
根据实施例2所述的方法合成PF1250的环氧丙基醚产物,但是:使用PF1250替代实施例2所用的BPA;环氧氯丙烷的用量为1,850克;ECH与来自PF1250的OH基的莫耳比为6.53([ECH]/[OH]=6.53);缩水甘油及水在环氧氯丙烷中的含量分别为0.12重量%及小于0.01重量%;以及49.5重量%氢氧化钠水溶液的用量为220克。
以下提供实施例8及PF1250的环氧丙基醚的理论上的例示性结构,以显示实施例8的环氧丙基醚的理论上的核心重复单元。
以上结构为理论性代表,且如前文所述,于实际形成实施例8的环氧丙基醚产物的方法期间,制备了具有类似于上方所呈现的理论性代表的变化结构的多种化合物。
[实施例9]
根据实施例2所述的方法合成乙二醛-酚缩合物的环氧丙基醚产物,但是使用乙二醛-酚缩合物替代实施例2所用的BPA。乙二醛-酚缩合物是根据第1,0138,325号美国专利的实施例2所述的方法合成。另外,此实施例所用的环氧氯丙烷的用量为1,480克。ECH与来自乙二醛-酚缩合物的OH基的莫耳比为6.67([ECH]/[OH]=6.67)。缩水甘油及水在环氧氯丙烷中的含量分别为0.12重量%及小于0.01重量%。另外,于偶合反应期间,温度自50℃的起始温度升温。49.5重量%氢氧化钠水溶液的用量为190克。
以下提供实施例9及多酚乙烷酚醛的环氧丙基醚的理论上的例示性结构,以显示实施例9的环氧丙基醚的理论上的核心重复单元。
以上结构为理论性代表,且如前文所述,于实际形成实施例9的环氧丙基醚产物的方法期间,制备了具有类似于上方所呈现的理论性代表的变化结构的多种化合物。
[实施例10]
根据实施例1所述的方法合成PF9110的环氧丙基醚产物,但是:使用PF9110替代实施例1所用的BPA;环氧氯丙烷的用量为1,550克;ECH与来自PF9110的OH基的莫耳比为10.75([ECH]/[OH]=10.75);缩水甘油及水在环氧氯丙烷中的含量分别为0.12重量%及小于0.01重量%;以及49.5重量%氢氧化钠水溶液的用量为220克。
以下提供实施例10及PF9110的环氧丙基醚的理论上的例示性结构,以显示实施例10的环氧丙基醚的理论上的核心重复单元。
以上结构为理论性代表,且如前文所述,于实际形成实施例10的环氧丙基醚产物的方法期间,制备了具有类似于上方所呈现的理论性代表的变化结构的多种化合物。
[比较例8至10]
比较例8至10分别为获自长春人造树脂厂的商购产品TFE1250、TNE190及DNE260。
表3
实施例8及比较例8制备了具有核心重复单元为多酚甲烷的环氧丙基醚产物。实施例9及比较例9制备了具有重复单元为多酚乙烷的环氧丙基醚。实施例10及比较例10制备了具有重复单元为DCPD-酚系酚醛的环氧丙基醚。如表3所示,实施例8至10具有低的α-二醇、低OH值及低的ICI黏度性质。比较例8至10的EEW×HV值系大于10当量/100当量,而实施例8至10的EEW×HV值小于10当量/100当量。
实施例11至16及比较例11至15
由实施例1至3及比较例1至3的BPA环氧丙基醚以及实施例5至7及比较例5及6的BPF环氧丙基醚来形成树脂。
具体而言,将实施例1至3及比较例1至3的BPA的环氧丙基醚产物以及实施例5至7及比较例5及6的BPF的环氧丙基醚产物与异佛尔酮二胺(以下称为“IPDA”,即购自巴斯夫(BASF)的
黏度适用期(viscosity pot life)是针对树脂及硬化剂的混合物于黏度增加至例如1,500cp的期间测定,通过使用TA仪器的Discovery系列混合流变仪HR-2与25毫米ETC铝平行板进行峰值保持模式测试(peak hold mode test)。将环氧丙基醚保持于30℃±0.5℃的温度,以及标定的剪切频率为1001/秒,标定的应变幅度为5%且间隙为1000微米。环氧丙基醚树脂保持等温直至其硬化。玻璃转移温度(以下称为“Tg”)的样品是于80℃固化12小时。根据ASTM E1356的方法以TA Q200装置进行均匀固化树脂(neat curing resin)的Tg结果。
表4显示由实施例11至13及比较例11至13的BPA环氧丙基醚所形成的树脂的性质。表5显示由实施例14至16及比较例14及15的BPF环氧丙基醚所形成的树脂的性质。由图2及图3可见,EEW×HV值的增加与黏度适用期及玻璃转移温度的增加相关。令人惊讶地,虽然实施例1与比较例3具有相似的EEW值,相较于比较例,本发明的环氧丙基醚在固化后具有较高的Tg且具有较长的黏度适用期。
表4
表5
本发明人发现,对于相同种类的环氧丙基醚产物,一般而言环氧丙基醚的EEW越低,其固化树脂的Tg越高。另外,据信对于具有相似的EEW量的环氧丙基醚产物,“HPLC n=0”及“MGE结构”导致Tg的增加(参见例如实施例1与比较例3的环氧丙基醚产物)。在不受限于任何特定理论之下,本发明人相信,环氧丙基醚中的α-二醇基及OH基的含量(HV值)将增进固化速率。因此,α-二醇及OH值的含量含量越低,黏度适用期越长。
另外,异构物及寡聚物含量的量可降低黏度及Tg。如表5所示,实施例及比较例之间的BPF环氧丙基醚的Tg差异并不如表4所示的大。据信此差异的原因是由于BPF环氧丙基醚的n=0的二聚物含量(表2)远小于BPA环氧丙基醚的n=0的二聚物含量(表1)。然而,实施例5至7的低EEW×HV值仍增进了Tg以及增加黏度适用期。
实施例17至18及比较例16至17
由实施例1、实施例5、比较例3、及比较例6的环氧丙基醚与IPDA/D230溶液来形成树脂。IPDA/D230溶液含有约25重量%的IPDA(AHEW=43)及75重量%的D230(AHEW=43),该两种都是购自巴斯夫,且混掺物IPDA/D230的AHEW为55.22。质量比是基于环氧丙基醚的环氧基的当量以及IPDA/D230的胺氢当量。IPDA/D230溶液与实施例1、实施例5、比较例3、及比较例6的环氧丙基醚的混合物是于80℃的温度下固化8小时、于50℃的温度下退火16小时,然后于标准大气下调节(condition)24小时以形成固化树脂。
由实施例1的BPA的环氧丙基醚产物所形成的树脂与由实施例5的BPF的环氧丙基醚产物所形成的树脂显示相似的EEW值,且因此在各自种类的环氧丙基醚中所用的硬化剂的量是接近的,以避免使硬化剂的效应遮盖掉例示性环氧树脂的改善。如表6所示,实施例17及18的热膨胀系数(以下称为“CTE”)及水分吸收低于比较例16及17,其中,热膨胀系数是根据ISO 11359-2的方法进行测试,水分吸收是根据ISO 62(方法2)进行测试。
表6
实施例19至20及比较例18至19
由实施例1、实施例5、比较例3、及比较例6的环氧丙基醚与获自长春人造树脂厂的1,4-丁二醇二环氧丙基醚(以下称为“BDGE125”)来形成树脂。质量比为85重量%的环氧丙基醚与15重量%的BDGE125(EEW=125.5)。用作硬化剂的IPDA/D230具有25对75的质量比,且混掺物IPDA/D230的AHEW为55.22。
硬化剂与BDGE125及特定环氧丙基醚的质量比是基于环氧丙基醚的环氧基的当量以及硬化剂的胺氢当量。将树脂于80℃的温度下固化8小时、于50℃的温度下退火16小时,然后于标准大气下调节24小时。由实施例1、实施例5、比较例3、及比较例6所形成的固化树脂的机械性质提供于下表7,其中,拉伸测试是根据ASTM D638的方法进行测试,弯曲测试是根据ASTM D790的方法进行测试。
表7
相较于由比较例3及6所形成的固化树脂,由实施例1及5所形成的固化树脂不仅在拉伸(tensile)及弯曲测试中展现较高的强度及模数,也展现较佳的伸长率性质。图2提供散布图,针对实施例环氧树脂11至16及比较例环氧树脂11至15显示EEW×HV值对黏度适用期的效果。图3提供散布图,针对实施例环氧树脂11至16及比较例环氧树脂11至15显示EEW×HV值对玻璃转移温度(Tg)的效果。
如本文中所用,所提供的所有范围均旨在包括该给定范围之内的所有特定范围及其间的次范围的组合。另外,除非另外说明,本文所提供的所有范围均包括此范围的端点。因此,1至5的范围具体而言包括1、2、3、4、及5、以及如2至5、3至5、2至3、2至4、1至4等次范围。
于本说明书中所引用的所有文献及专利申请案均并于此以供参考,且基于任何及所有目的,如同特定及分别地指出各自文献或专利申请案并于此以供参考。在本发明与任何并于此以供参考的文献或专利申请案之间存在不一致的情况下,以本发明为准。
如本文中所用,术语“包含”、“具有”、及“包括”以开放式、非限定性的方式使用。术语“一”及“该”应理解为包含复数以及单数。用语“一或多”是指“至少一”,且因此可包括单独特征或混合物/组合物。
除了在操作实施例中之外,或另外说明之外,所有显示成分及/或反应条件的数量的数字均可在所有情况下由术语“约”修饰,其代表所指定的数字的+/-5%内。本文所用的术语“实质上无”或“本质上无”是指特定特征为小于约2%。本发明的权利保护范围如权利要求书所列。
- 单酚或多元酚的环氧丙基醚产物与其制备方法、及环氧树脂组合物
- 一种双酚S二缩水甘油醚环氧树脂的制备方法及高环氧值双酚S二缩水甘油醚环氧树脂