一种抗宫炎软胶囊UPLC指纹图谱的建立方法
文献发布时间:2023-06-19 11:40:48
技术领域
本发明涉及抗宫炎软胶囊技术领域,特别是一种抗宫炎软胶囊UPLC指纹图谱的建立方法。
背景技术
抗宫炎软胶囊是由抗宫炎片改剂型而来的中药九类新药,由广东紫珠干浸膏、益母草干浸膏和乌药干浸膏组成,其中广东紫珠具有收敛止血、散瘀、清热解毒的功效;益母草具有活血调经、利尿消肿、清热解毒的功效;乌药具有行气止痛、温肾散寒的功效。抗宫炎系列制剂药效显著,具有清热、祛湿、化瘀、止带的作用,主要用于湿热下注所致的带下病,症见赤白带下、量多臭味、宫颈糜烂。
目前,抗宫炎软胶囊的质量控制是按照国家食品药品监督管理局颁布的国家药品标准实行,以益母草中的盐酸水苏碱的含量测定作为质控指标,而收载于2020版中国药典的抗宫炎系列制剂(片剂、颗粒剂、胶囊)项下含量测定均为其君药广东紫珠中的金石蚕苷和连翘酯苷B。由于中药的多成分、多靶点的整体作用,决定了任何一个单一成分作为指标都不能体现中药的整体质量。
中药指纹图谱是一种能全面反应中药或中药制剂整体性化学特征的质量分析方法,被广泛用于中药及中药制剂的质量控制中。目前仅对抗宫炎系列制剂(片剂、颗粒剂、胶囊)做过HPLC指纹图谱研究外,对抗宫炎软胶囊的指纹图谱和检测方法还未见报道。
发明内容
本发明的目的在于,提供一种抗宫炎软胶囊UPLC指纹图谱的建立方法。本发明方法具有快速、简便、稳定、精密度高、稳定性好、重复性好等优点;用该方法还能全面地反映抗宫炎软胶囊中所含化学成分的种类与数量,并可直接指认去甲异波尔定、盐酸益母草碱、金石蚕苷、连翘酯苷B、毛蕊花糖苷、异毛蕊花糖苷的峰,可以为抗宫炎软胶囊的质量控制提供参考。
本发明的技术方案:一种抗宫炎软胶囊UPLC指纹图谱的建立方法,包括以下步骤:
(1)混合对照品溶液的制备:称取去甲异波尔定、盐酸益母草碱、连翘酯苷B、毛蕊花糖苷、金石蚕苷和异毛蕊花糖苷对照品,置于容器中配制成混合对照品母液,再加40-60%甲醇稀释摇匀,制得混合对照品溶液;
(2)供试品溶液的制备:取抗宫炎软胶囊内容物,称定,置于容器中,加入40-60%甲醇,称定重量,回流,放冷,再称定重量,用40-60%甲醇补足减失的重量,摇匀,滤过,取续滤液,得供试品溶液;
(3)指纹图谱的制作:色谱条件:色谱柱:CAPCELL PAK C18色谱柱,2.1mm×100mm,2μm;流动相:乙腈为A相,0.5%磷酸水溶液为B相,梯度洗脱,流速为0.2mL.min-1;柱温为35℃;进样量为2μL;检测波长:0-10min:280nm;10-35min:332nm;记录色谱图得到抗宫炎软胶囊UPLC指纹图谱;
(4)标准指纹谱图的确认:按照上述提供的方法,对抗宫炎软胶囊建立了UPLC指纹图谱,通过分析比较确定了12个共有峰,这些共有峰构成了抗宫炎软胶囊的指纹特征,作为抗宫炎软胶囊的UPLC指纹图谱。
前述的抗宫炎软胶囊UPLC指纹图谱的建立方法中,所述步骤(1)是精密称取去甲异波尔定、盐酸益母草碱、连翘酯苷B、毛蕊花糖苷、金石蚕苷和异毛蕊花糖苷对照品,置于容量瓶中配制成混合对照品母液,再加50%甲醇稀释摇匀,制得浓度为0.05mg·mL
前述的抗宫炎软胶囊UPLC指纹图谱的建立方法中,所述步骤(2)是取1.0g抗宫炎软胶囊内容物,精密称定,置于圆底烧瓶中,精密加入50mL50%甲醇,称定重量,回流1h,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,取续滤液,得供试品溶液。
前述的抗宫炎软胶囊UPLC指纹图谱的建立方法中,所述梯度洗脱的程序是0-20min,12%A→17%A;20~35min,17%A→35%A;35~37min,30%A→90%A;37~40min,90%A→12%A。
前述的抗宫炎软胶囊UPLC指纹图谱的建立方法中,所述12个共有峰中,以5号峰连翘酯苷B为参照峰,则1号峰为去甲异波尔定,3号峰为盐酸益母草碱,5号峰为连翘酯苷B,6号峰为毛蕊花糖苷,7号峰为金石蚕苷,9号峰为异毛蕊花糖苷。
为研究本抗宫炎软胶囊UPLC指纹图谱的建立方法,发明人进行了大量的试验,部分试验记录如下:
实验例:抗宫炎软胶囊UPLC指纹图谱研究
1仪器与材料
1.1仪器
超高效液相色谱仪,万分之一电子天平,超声波清洗器,纯水仪。
1.2试剂与样品
广东紫珠干浸膏、益母草干浸膏、乌药干浸膏(贵州汇正制药有限责任公司);对照品去甲异波尔定(批号P10M9L61140,UPLC峰面积法测得纯度≥98%);盐酸益母草碱(批号R04N8F47273,UPLC峰面积法测得纯度≥98%);连翘酯苷B(批号R14N9F74963,UPLC峰面积法测得纯度≥98%);金石蚕苷(批号H29M3X1,UPLC峰面积法测得纯度≥98%);毛蕊花糖苷(批号W14O10C100217,UPLC峰面积法测得纯度≥98%);异毛蕊花糖苷(批号W17J10C90785,UPLC峰面积法测得纯度≥98%)均购自上海源叶生物科技有限公司。
抗宫炎软胶囊(由贵州汇正制药有限公司提供,批号200101(S1)、200102(S2)、200303(S3)、200501(S4)、200502(S5)、200503(S6)、200504(S7)、200602(S8)、200603(S9)、200701(S10)、200702(S11)、200704(S12)、200705(S13)、200801(S14)、200805(S15)、200806(S16)、200902(S17)、200903(S18)、20101(S19)、201002(S20)。乙腈为色谱纯(默克);水为屈臣氏双蒸水;其余试剂为分析纯。
2实验方法
2.1溶液的制备
2.1.1混合对照品溶液
精密称取去甲异波尔定、盐酸益母草碱、连翘酯苷B、毛蕊花糖苷、金石蚕苷、异毛蕊花糖苷适量,置容量瓶中,加50%甲醇稀释,摇匀,制得浓度分别为0.05(去甲异波尔定)、0.15(盐酸益母草碱)、0.15(连翘酯苷B)、0.15(毛蕊花糖苷)、0.15(金石蚕苷)、0.15(异毛蕊花糖苷)mg·mL
2.1.2供试品溶液
取约1.0g抗宫炎软胶囊内容物,精密称定,置于圆底烧瓶中,精密加入50mL50%甲醇,称定重量,回流1h,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,取续滤液即得
2.1.3阴性对照溶液
分别制备缺广东紫珠干浸膏、益母草干浸膏、乌药干浸膏的阴性样品,按“2.1.2”项下方法制备,即得阴性供试品溶液。
2.1.4色谱条件
采用CAPCELL PAK C
3结果
3.1指纹图谱方法学考察
3.1.1精密度试验
取同一供试品,连续进样6次,记录峰面积。以5号峰连翘酯苷B为参照峰,计算各共有峰的相对保留时间RSD≤0.13%,相对峰面积的RSD≤4.2%,表明仪器精密度良好,符合指纹图谱测定要求。
3.1.2重复性试验
取同一批号的抗宫炎软胶囊6份,每份1.0g,平行制得供试品溶液6份,进行测定,记录峰面积。以5号峰连翘酯苷B为参照峰,计算各共有峰的相对保留时间RSD≤0.27%,相对峰面积的RSD≤4.4%,表明本方法重复性良好。
3.1.3稳定性试验
取同一供试品溶液,分别于0、2、4、8、12、24h进样,记录峰面积。以5号峰连翘酯苷B为参照峰,计算各共有峰的相对保留时间RSD≤0.25%,相对峰面积的RSD≤4.8%,表明供试品溶液在24h稳定。
3.1.4UPLC特征指纹图谱的生成与相似度评价
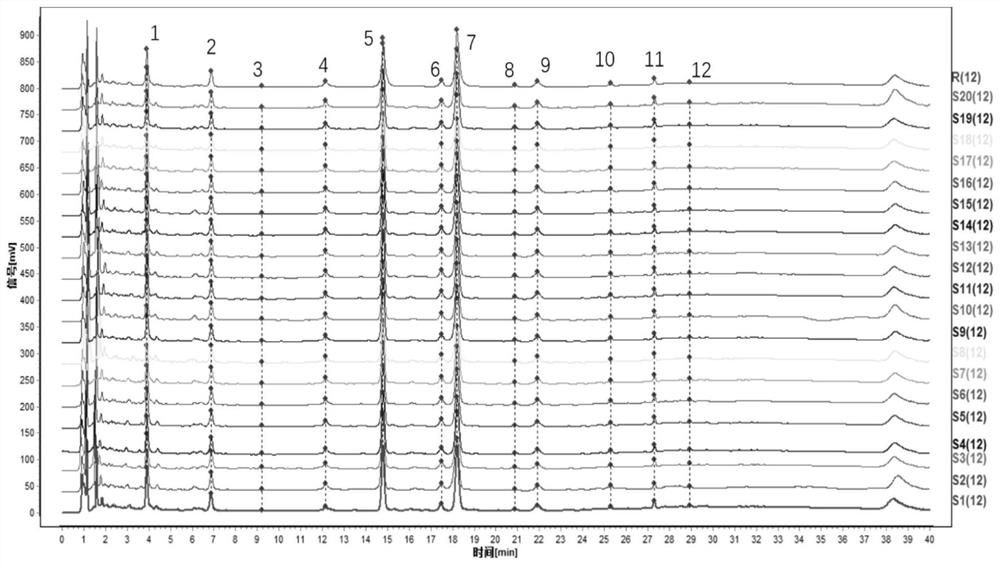
分别精密称取编号为S1~S20的抗宫炎软胶囊供试品内容物1.0g,并采用《中药指纹图谱相似度评价系统(2012A版)》对各样品图谱的AIA格式文件进行分析。以S1为参照图谱,对色谱峰进行多点校正,标记好共有峰,随后进行Mark峰匹配,生成20批抗宫炎软胶囊的特征指纹图谱匹配图以及对照特征指纹图谱(图1)。共标记12个共有峰,选择5号峰(连翘酯苷B)为参照峰。经软件计算,得出S1~S20号样品特征指纹图谱与对照特征指纹图谱的相似度,结果见表1,20批抗宫炎软胶囊指纹图谱共有峰的相对保留时间结果见表2。从相似度结果来看,各批次抗宫炎软胶囊相似度均大于0.9,说明各批次样品间较稳定、一致。
表1 20批抗宫炎软胶囊相似度计算
表2 20批抗宫炎软胶囊指纹图谱共有峰的相对保留时间结果
3.1.5缺味药材与抗宫炎软胶囊的相关性分析
分别取抗宫炎软胶囊及缺味样品,通过与阴性供试品图谱对照,可以指认1号峰存在于乌药药材中,2号峰存在于广东紫珠和益母草药材中,3号峰存在于益母草药材中,4~12号峰均存在于广东紫珠药材中。通过与对照品比对(图6),共指认出其中的6个峰,其中1号峰为去甲异波尔定,3号峰为盐酸益母草碱,5号峰为连翘酯苷B,6号峰为毛蕊花糖苷,7号峰为金石蚕苷,9号峰为异毛蕊花糖苷,结果见图2-6。
与现有技术相比,本发明具有如下的有益效果:
1、目前对抗宫炎制剂的指纹图谱研究仅限于HPLC,分析时长为115min,本发明提出的抗宫炎软胶囊UPLC指纹图谱,将分析时间缩短至40min,本发明方法更加快速便捷。
2、本发明建立的抗宫炎软胶囊UPLC指纹图谱方法,精密度好,重复性好,稳定性好,可系统、全面的评价抗宫炎软胶囊的质量。
3、本发明的提出,为优化抗宫炎软胶囊的样品处理方法,用正交实验考察了提取方法、提取时间、提取溶剂,最后选择了50%甲醇加热回流1h的最佳处理方法。(部分考察记录是:考察了回流提取1h和超声提取20min以及超声提取40min,结果发现回流1h时的峰面积响应最高,故选择回流1h。其次,考察了以甲醇、50%甲醇以及50%乙醇为提取溶剂时的影响,结果表明,以50%甲醇为溶剂时的峰面积最高,故选择以50%甲醇为提取溶剂。还比较了提取回流30min、60min和90min,结果表明回流60min和90min时的峰面积响应差别不大,考虑到时间成本,故选择了回流提取1h。)
4、本发明的研究过程中,发现抗宫炎软胶囊在制备浸膏时采用水煎煮提取法,其中的成分极性较大,并由于在益母草和乌药中主要为生物碱类,在分离时拖尾导致与其他峰粘连,影响测定结果,给指纹图谱方法的建立造成了困难。通过比较了资生堂CAPCELLPAK C18 IF2(100×2.1,2μm)、Thermo Hypersil GOLD(150×2.1,1.9μm)、ACE Excel2C18-Amide(100×2.1,2μm)这3款不同厂家的色谱柱,发现虽然使用Thermo HypersilGOLD(150×2.1,1.9μm)柱分离检测时,在流动相里面加入一定量的三乙胺可以达到较好的分离效果,但是由于三乙胺能够与固定相中的硅羟基结合,阻碍样品中碱性化合物与硅羟基的作用,减少峰拖尾,改善峰型,对色谱柱的伤害较大。但是,在同样的条件下,资生堂CAPCELL PAK C18 IF2(2.1×100,2μm)色谱柱可以不通过加三乙胺的方式就能对各成分进行良好的分离。故选择资生堂CAPCELL PAK C18 IF2(2.1×100,2μm)色谱柱。同时,对流动相也做了考察,先后使用了甲醇(A)-0.5%磷酸水溶液(B)、乙腈(A)-0.1%磷酸水溶液(B)、乙腈(A)-0.5%磷酸水溶液(B)种不同的流动相系统,结果表明使用乙腈(A)-0.5%磷酸水溶液(B)为流动相梯度洗脱时效果最佳。此外,也考察了在流速为0.1mL
5、本发明建立的抗宫炎软胶囊UPLC指纹图谱,共有峰12个,各批次间相似度均高于0.9;并确认了6个共有峰,5号峰连翘酯苷B峰面积较大,并且分离度良好,为参照峰,则1号峰为去甲异波尔定,3号峰为盐酸益母草碱,5号峰为连翘酯苷B,6号峰为毛蕊花糖苷,7号峰为金石蚕苷,9号峰为异毛蕊花糖苷。
综上所述:本发明方法具有快速便捷、精密度高、稳定性好、重复性好、可系统、全面的评价抗宫炎软胶囊的质量。本发明方法以5号峰连翘酯苷B为参照峰,可直接找到去甲异波尔定、盐酸益母草碱、连翘酯苷B、毛蕊花糖苷、金石蚕苷和异毛蕊花糖苷的峰。
附图说明
图1是20批抗宫炎软胶囊UPLC特征指纹图谱(S1~S20)及其对照特征指纹图谱(R);
图2是抗宫炎软胶囊UPLC色谱图;
图3是缺广东紫珠阴性样品UPLC色谱图;
图4是缺益母草阴性样品UPLC色谱图;
图5是缺乌药阴性样品UPLC色谱图;
图6是抗宫炎软胶囊特征指纹图谱及混合对照品的UPLC图谱。
附图6中附图标记为:1-去甲异波尔定;3-盐酸益母草碱;5-连翘酯苷B;6-毛蕊花糖苷;7-金石蚕苷;9-异毛蕊花糖苷。
具体实施方式
下面结合实施例对本发明作进一步的说明,但并不作为对本发明限制的依据。
实施例。一种抗宫炎软胶囊UPLC指纹图谱的建立方法,建立步骤为:
(1)混合对照品溶液的制备:精密称取去甲异波尔定、盐酸益母草碱、连翘酯苷B、毛蕊花糖苷、金石蚕苷、异毛蕊花糖苷适量,置容量瓶中,加50%甲醇稀释,摇匀,制得浓度分别为0.05(去甲异波尔定)、0.15(盐酸益母草碱)、0.15(连翘酯苷B)、0.15(毛蕊花糖苷)、0.15(金石蚕苷)、0.15(异毛蕊花糖苷)mg·mL
(2)供试品溶液的制备:取1.0g抗宫炎软胶囊内容物,精密称定,置于圆底烧瓶中,精密加入50mL50%甲醇,称定重量,回流1h,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,取续滤液,得供试品溶液;
(3)指纹图谱的制作:色谱条件:色谱柱:CAPCELL PAK C
(4)标准指纹谱图的确认:按照上述提供的方法,对抗宫炎软胶囊建立了UPLC指纹图谱,通过分析比较确定了12个共有峰,这些共有峰构成了抗宫炎软胶囊的指纹特征,作为抗宫炎软胶囊的UPLC指纹图谱;所述12个共有峰中,以5号峰连翘酯苷B为参照峰,则1号峰为去甲异波尔定,3号峰为盐酸益母草碱,5号峰为连翘酯苷B,6号峰为毛蕊花糖苷,7号峰为金石蚕苷,9号峰为异毛蕊花糖苷。
- 一种抗宫炎软胶囊UPLC指纹图谱的建立方法
- 一种抗宫炎软胶囊的检测方法