一种采用双波长一测多评法测定银翘散中7种成分含量的方法
文献发布时间:2023-06-19 11:44:10
技术领域
本发明属于中药质量控制与分析技术领域,具体涉及一种采用双波长一测多评法测定银翘散中7种成分含量的方法。
背景技术
中药质量是保障临床用药有效性和安全性的前提,是保证中药发挥其疗效的关键因素,也是中药现代化、科学化的基本保障,因此提升中药质控标准对于提高中药质量尤为重要。中药复方在临床应用上具有整体性、系统性,而采用单一的组分来评价中药质量缺乏全面性。银翘散作为治疗温病的代表性组方,具有辛凉解表、清热解毒的功效,以治疗风温、温热病以及某些杂病属于邪在卫分、上焦而闻名,在临床上应用广泛。银翘散由金银花、连翘、薄荷、荆芥、桔梗、淡竹叶、牛蒡子、淡豆豉、甘草、芦根10味中药材组成,处方中以金银花、连翘为君药,且入药量较大。目前HPLC单独测定银翘散中绿原酸、连翘苷、牛蒡苷的文献报道较多,在2020版的《中国药典》中,银翘散仅以牛蒡苷为质量控制指标性成分,指标单一,单一成分的含量测定难以表达银翘散的物质特征。
为了更好地控制银翘散的质量、确保临床药效,不使用过多价格昂贵的对照品、节约检测成本,在2020版《中国药典》的基础上,提升银翘散的质量控质方法具有重要意义。
发明内容
针对现有技术中存在的检测银翘散质量控制指标性成分单一,无法对处方中以金银花、连翘为君药成分进行综合评价的问题,本发明提供了一种采用双波长一测多评法测定银翘散中7种成分含量的方法,通过一测多评技术,选用连翘酯苷A作为内参物,建立该成分与连翘苷、牛蒡苷、新绿原酸、绿原酸、异绿原酸A、异绿原酸C的相对校正因子,得到银翘散中7种成分的含量。
本发明通过以下技术方案实现:
一种采用双波长一测多评法测定银翘散中7种成分含量的方法,包括以下步骤:
(1)对照品溶液的制备:分别取新绿原酸、绿原酸、连翘酯苷A、异绿原酸A、异绿原酸C、连翘苷、牛蒡苷对照品,以甲醇为溶剂,配置成浓度分别为1~10、50~150、200~350、20~100、5~20、20~40、50~150μg/ml的混合对照品溶液;
(2)供试品溶液的制备:称取待测银翘散0.4~0.6g,用30~100%甲醇水溶液定容,超声10~40min,补足失重,滤膜过滤,得供试品溶液;
(3)相对校正因子的计算:取步骤(1)中的对照品溶液,注入高效色谱仪中,以连翘酯苷A为内参物,在检测波长为237nm下,分别计算牛蒡苷和连翘苷的相对校正因子ƒ
相对校正因子计算公式如下:
其中,
(4)测定:取步骤(1)中的连翘酯苷A对照品溶液、步骤(2)中的供试品溶液,注入高效液相色谱仪中,记录237nm下,连翘酯苷A、连翘苷、牛蒡苷的峰面积,327nm下,连翘酯苷A、新绿原酸、绿原酸、异绿原酸A、异绿原酸C的峰面积,结合步骤(3)得到的相对校正因子的计算连翘酯苷A、连翘苷、牛蒡苷、新绿原酸、绿原酸、异绿原酸A、异绿原酸C 7种成分的含量。
进一步地,步骤(3)和步骤(4)中所述的高效液相色谱仪的色谱条件为:
检测器:UV-DAD检测器;色谱柱:C18柱,4.6×250 mm,5 μm;流动相:乙腈-0.1%磷酸水溶液,梯度洗脱;双波长同时采集,流速:1ml/min;进样量:3~15 μl;柱温:25~35 ℃;分析时间:130min;
所述的梯度洗脱条件如下表所示:
进一步地,步骤(1)中所述的混合对照品溶液中新绿原酸、绿原酸、连翘酯苷A、异绿原酸A、异绿原酸C、连翘苷、牛蒡苷浓度分别为7.7、96、296、65、15.2、27.5、113μg/ml。
进一步地,步骤(2)中所述的供试品溶液的浓度为0.02g/ml。
进一步地,步骤(2)中所述的待测银翘散的取样量为0.5g;所述的提取溶剂为50%甲醇水溶液,常温超声,超声时间为30min。
进一步地,步骤(2)中所述的微滤采用0.45μm有机系微孔滤膜。
进一步地,步骤(3)和步骤(4)中所述的C18色谱柱为Thermo Syncronis C18柱、Hypersil ODS-2 C18柱中的一种;所述的梯度洗脱条件如下表所示:
进一步地,步骤(3)和步骤(4)中所述的进样量为5μl,色谱柱柱温为30℃。
进一步地,步骤(1)中的甲醇为50%色谱甲醇。
连翘散由金银花、连翘、薄荷、荆芥、桔梗、淡竹叶、牛蒡子、淡豆豉、甘草、芦根10味中药组成,金银花、连翘为君药。本发明提供了一种操作简单,检测灵敏度高,成本低廉,准确高效,且可同时定量分析银翘散中君臣药中连翘酯苷A、连翘苷、牛蒡苷、新绿原酸、绿原酸、异绿原酸A、异绿原酸C等多种活性成分的质量控制方法(一测多评法)检测方法,解决了2020版《中国药典》银翘散中仅以牛蒡苷为质量控制指标性成分、指标单一的情况,提升了银翘散的质量控质方法,既保障了银翘散临床用药有效性和安全性,也保证了其疗效的发挥。
有益效果
(1)本发明突破了测定一个成分含量就需要一种对照品、一种供试品溶液、一种色谱条件、一种检测波长的传统高效液相色谱含量测定方式,建立了采用同一种供试品溶液,在两个最大吸收波长下,完成7个成分的含量测定,提高了检测效率,降低了检测成本;
(2)在2020版《中国药典》的基础上,提升银翘散的质量控质方法,本发明提供了一种操作简单,检测灵敏度高,成本低廉,准确高效,且可同时定量分析银翘散中君臣药中连翘酯苷A、连翘苷、牛蒡苷、新绿原酸、绿原酸、异绿原酸A、异绿原酸C等多种活性成分的质量控制方法(一测多评法)检测方法。
附图说明
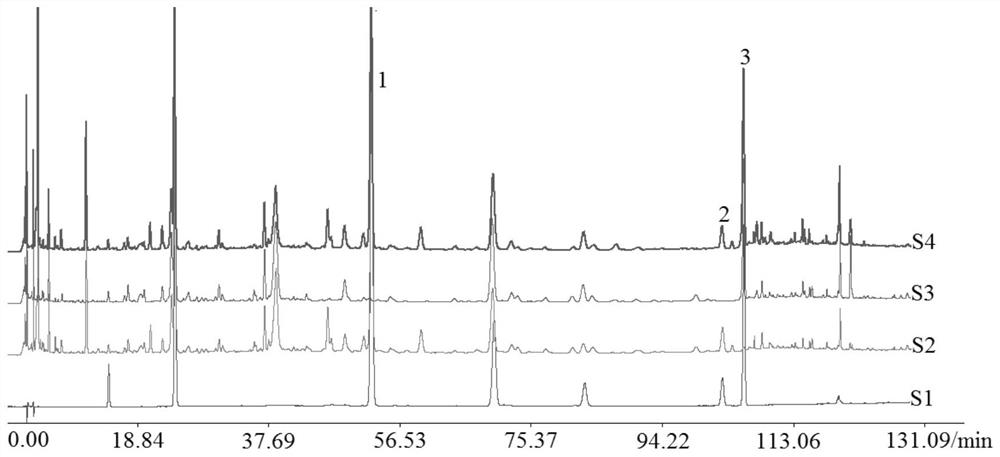
图1为实施例1 在237nm下银翘散样品、混合对照品溶液和阴性样品溶液的高效液相色谱图,其中1.连翘酯苷A,2.连翘苷,3.牛蒡苷,S1.混合对照品,S2.缺牛蒡子阴性,S3.缺连翘阴性,S4.银翘散;
图2为实施例1 在327nm下银翘散样品、混合对照品溶液和阴性样品溶液的高效液相色谱图,其中1.新绿原酸,2.绿原酸,3.连翘酯苷A,4.异绿原酸A,5.异绿原酸C,S1.混合对照,S2.缺金银花和牛蒡子阴性,S3.缺连翘阴性,S4.缺金银花阴性,S5.银翘散;
图3为实施例2 十批银翘散在237nm下的高效液相液相色谱图;
图4为实施例2 十批银翘散在327nm下的高效液相液相色谱图。
具体实施方式
为了使本领域的人员更好地理解本发明的技术方案,下面对本发明的技术方案进行清楚、完整的描述,基于本申请中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的其它类同实施例,都应当属于本申请保护的范围。
实施例1
(1)对照品溶液的制备:精密称取新绿原酸、绿原酸、连翘酯苷A、异绿原酸A、异绿原酸C、连翘苷、牛蒡苷对照品,用50%色谱甲醇配置成浓度分别为7.7、96、296、65、15.2、27.5、113μg/ml的混合对照品溶液;
(2)供试品溶液的制备:精密称取银翘散0.5g,50%甲醇水溶液定容,称重,超声30min,放凉,补足失重,过滤,取续滤液过0.45μm有机系微孔滤膜得待测样品溶液,浓度为0.02g/ml;同法分别制备缺金银花、连翘、牛蒡苷单一药材的阴性样品、缺金银花和牛蒡子两味药材的阴性样品;
(3)色谱条件:色谱柱Thermo Syncronis(4.6×250 mm,5 μm)C18 柱,流动相乙腈-0.1%磷酸水梯度洗脱(梯度洗脱条件见下表1),检测波长:237nm、327nm,双波长同时采集,流速:1 ml/min,进样量:5 μl,柱温:30 ℃,分析时间:130min;
表1 梯度洗脱条件
(3)相对校正因子的计算:
相对校正因子的计算:取步骤(1)混合对照品溶液,按照步骤(3)色谱条件,注入到高效液相色谱仪中进行测定,并记录各成分在237nm、327nm下的峰面积,以连翘酯苷A为内参物,在检测波长为237nm下,分别记录连翘酯苷A、牛蒡苷和连翘苷的峰面积,并计算牛蒡苷和连翘苷的相对校正因子;在检测波长为327nm下,分别记录连翘酯苷A、新绿原酸、绿原酸、异绿原酸A、异绿原酸C的峰面积,并计算新绿原酸、绿原酸、异绿原酸A、异绿原酸C的相对校正因子
经计算,牛蒡苷、连翘苷、新绿原酸、绿原酸、异绿原酸A、异绿原酸C的相对校正因子如下表2所示;
表2 237nm、327nm下银翘散中各成分相对校正因子
(5)测定:分别取步骤(2)制备的待测样品溶液和阴性样品溶液注入到高效液相色谱仪中、按步骤(3)色谱条件进行含量测定,在237nm和327nm下的高效液相色谱图分别如图1和图2所示,图1中峰1、2、3分别为连翘酯苷A、连翘苷和牛蒡苷的色谱峰,S1表示为混合对照品,S2表示为缺牛蒡子阴性样品,S3表示为缺连翘阴性样品,S4表示为银翘散样品;图2中峰1、2、3、4、5分别为新绿原酸、绿原酸、连翘酯苷A、异绿原酸C、异绿原酸C的色谱峰,S1表示为混合对照品,S2表示为缺金银花和牛蒡子阴性样品,S3表示为缺连翘阴性样品,S4表示为缺金银花阴性,S5表示为银翘散样品。
(6)相对校正因子考察:改变步骤(3)中的色谱条件,分别考察ThermoUltiMate3000-1、Thermo UltiMate3000-2两台高效液相色谱仪和 Hypersil ODS-2 (250mm × 4. 6 mm,5 μm)及Thermo Syncronis C 18(250 mm × 4. 6 mm,5 μm)两种色谱柱,记录并计算相对校正因子。在不同仪器、不同色谱柱条件下,相对校正因子具有良好的重现性(表3)。
表3 不同仪器、不同色谱柱下各成分的相对校正因子
(7)线性关系考察
按照实施例1步骤(1)方法制备混合对照品溶液,按照步骤(3)中的色谱条件,分别进样5、7、10 μl;取混合对照品溶液1ml,稀释5倍,分别进样3、5、15 μl,分别在检测波长为237nm和327nm下的色谱图并记录其峰面积。以各对照品峰面积(Y,A)与其对应的进样量(X,μg)进行线性回归,得各对照品的线性回归方程(见表4),各线性回归方程r>0.9995,表明各对照品线性关系良好。
表4 7种对照品的线性关系分析
(8)精密度的考察
按照步骤(3)中的色谱条件,取混合对照品溶液连续进样6针,分别在检测波长为237nm和327nm下的色谱图并记录7种成分的峰面积,进行精密度分析,结果见表5。
(9)稳定性的考察
精密称取银翘散0.5g,按照步骤(2)的方法制备供试品溶液,按照步骤(3)中的色谱条件,于0、3、6、9、12、24h进样,分别在检测波长为237nm和327nm下的色谱图并记录7种成分的峰面积,进行稳定性分析,分析结果见表5。
(10)重复性的考察
取同一份银翘散,精密称取6份,每份0.5g,按照步骤(2)的方法制备供试品溶液,按照步骤(3)中的色谱条件进行进样,分别在检测波长为237nm和327nm下的色谱图并记录7种成分的峰面积,并计算各成分的含量,进行重复性分析,分析结果见表5。
(11)加样回收率的考察
取一份已知含量的银翘散样品,精密称取0.25g,平行6份,加入适量的对照品,按照步骤(2)方法制备供试品溶液,记录不同波长下各成分的峰面积,按照实施例1步骤(3)中的色谱条件进行进样,分别在检测波长为237nm和327nm下的色谱图并记录7种成分的峰面积,并计算各成分的含量,进行加样回收率分析,分析结果见表5。
表5 方法学考察结果分析
实施例2
取10批银翘散样品,按照实施例1步骤(2)方法制备供试品溶液,按实施例步骤(3)色谱条件进行进样,记录237nm下,连翘酯苷A、连翘苷、牛蒡苷的峰面积,327nm下,连翘酯苷A、新绿原酸、绿原酸(C)、异绿原酸A、异绿原酸C的峰面积。
采用实施例1中相对校正因子,分别采用外标一点法(ESM)和一测多评法(QAMS),计算银翘散各成分含量,ESM和QAMS两种方法测定结果的相对偏差在-0.35%~0.11%之间,均小于3%(见表6、表7)。
表6 237nm下ESM和QAMS含量测定结果比较
表7 327nm下ESM与QAMS计算结果比较
由检测结果比较可知,两种方法测得的银翘散中7种成分的含量没有显著性差异,表明一侧多评法用于银翘散多种成分质量控制是可行的。
- 一种采用双波长一测多评法测定银翘散中7种成分含量的方法
- 一种一测多评法测定茯苓中多种三萜类成分含量的方法