合成修饰的Olig2 mRNA及其应用
文献发布时间:2023-06-19 12:18:04
技术领域
本发明涉及干细胞诱导分化技术领域,特别是涉及一种合成修饰的Olig2 mRNA及其应用。
背景技术
少突胶质细胞(oligodendrocyte)是人类大脑中重要的一类胶质细胞,通过形成致密的髓鞘结构包裹于神经元的轴突上,在神经元的信号转导,营养支持,以及维持脑稳态等方面发挥了重要的功能。早在20世纪巴西科学家Hortega即已首次提出少突胶质细胞,但是时至今日,对于少突胶质细胞及其祖细胞的起源及功能上的认识还仍然不足。
在很长的一段时间内,少突胶质细胞的主要功能还是被局限的定于在髓鞘的形成和维持上。然而,越来越多的证据表明,少突胶质细胞对于中枢神经系统的作用具有多样性,比较显著的一个功能便是,少突胶质细胞对神经轴突的功能提供了关键的代谢及营养支持。然而,在大多数神经退行性疾病中,比如多发性硬化等,病理状态下的少突胶质细胞难于自发性的形成髓鞘,髓鞘产生的障碍是导致神经功能丧失的重要原因之一。
通过外源性干/祖细胞植入来达到髓鞘修复的目的越来越得到关注。这些方法的核心是在髓鞘损伤的区域内补充足够的少突胶质前体/祖细胞,以提高脑白质髓鞘再生的能力。近年来,少突胶质前体细胞(oligodendrocyte progenitor cells,OPC)移植已经在一些髓鞘损伤的神经退行性疾病中展现出了能够促进神经功能恢复,以及提高脱髓鞘小鼠的生存率的治疗潜力。但是由于无法大量获得人源OPC进一步制约了OPC移植用于人类疾病治疗的发展。
诱导多能干细胞(induced pluripotent stem cells,iPS)来源的细胞移植,具有治疗个性化,无免疫排斥,细胞来源充足等特点,是临床细胞移植的理想细胞来源。生成iPS技术相对稳定和简单,不需使用卵细胞和胚胎,而是利用人的其他细胞,比如皮肤成纤维细胞和尿液细胞,从而规避了伦理争议和法律难题。因此,如何由iPS诱导分化成大量稳定可靠的OPC成为了当下人源OPC移植的研究热点和难点。
目前将多能干细胞诱导制备少突胶质前体细胞最为广泛的是基于操控转录因子的方法。这种方法分为两步,首先通过SMAD途径双重抑制法(Dual SMAD inhibition)诱导iPS为神经前体细胞。再在神经前体细胞上通过慢病毒介导的调控少突胶质细胞形成和髓鞘再生的转录因子过表达,比如Juan Antonio García-León等在人诱导多能干细胞来源的神经前体细胞上过表达转录因子SOX10,能够在21天内形成大于40%的MBP(Myelin basicprotein)阳性的少突胶质细胞,并且将这些细胞通过侧脑室植入脱髓鞘小鼠的脑内能够有效地促进其髓鞘损伤修复。但是这种方法也存在着明显的缺点。比如,基于慢病毒载体将外源的SOX10整合到神经前体细胞基因组中,从而使经诱导得到的少突胶质细胞可能具有基因整合和插入诱变的风险,这也大大地限制了其临床运用。
为了达到更加安全有效的临床效果,亟需开发一种安全,高效的人诱导多能干细胞来源的少突胶质细胞的方法,能够推进临床试验的快速开展。
发明内容
基于此,有必要针对上述问题,提供一种合成修饰的Olig2 mRNA,将其转染人诱导多能干细胞来源的神经前体细胞,实现了人源诱导多能干细胞向少突胶质细胞的快速且高效的定向分化。
一种合成修饰的Olig2 mRNA,所述Olig2 mRNA翻译的蛋白始终处于去磷酸化状态。
前期研究表明,转录因子Olig2是大脑中枢神经系统中少突胶质细胞发育成熟过程中核心的转录因子,在神经前体细胞中,人为地诱导Olig2高表达,可以促进神经前体细胞分化为少突胶质前体细胞,并进一步成熟形成功能性的髓鞘。后续进一步对Olig2蛋白功能的研究表明,Olig2蛋白的磷酸化修饰能够驱使神经前体细胞向运动神经元分化,而非少突胶质细胞。因此,如果能够将Olig2蛋白磷酸化位点(147位丝氨酸)进行适当修饰,阻断其被磷酸化修饰,则可能改变神经前体细胞向运动神经元的命运,继而促进少突胶质细胞的形成。
因此基于以上研究,本发明人在神经前体细胞中转染去磷酸化修饰的Olig2mRNA,结果表明上述去磷酸化修饰的Olig2 mRNA可以高效地促进神经前体细胞向少突胶质细胞的分化。
在其中一个实施例中,所述去磷酸化修饰的Olig2 mRNA编码蛋白中第147位为丙氨酸。
为获得始终处于去磷酸化状态的Olig2 mRNA翻译蛋白,本发明人设计合成了去磷酸化修饰的Olig2 mRNA(Olig2
在其中一个实施例中,所述Olig2 mRNA序列为:
A)如SEQ ID No.1所示序列;或者
B)与A)的核苷酸序列编码相同序列的蛋白质,但因遗传密码的简并性而与A)的核苷酸序列不同的序列。
如SEQ ID No.1中编码蛋白第147位丙氨酸序列为GCC,由于丙氨酸的密码子包括GCC,GCG,GCA或GCU,即序列B)中,与上述丙氨酸序列相对应序列可以为GCG,GCA或GCU,均属序列B)的范畴。
在其中一个实施例中,所述Olig2 mRNA的5’端具有m7G加帽结构,3’端具有多聚腺苷酸poly(A)尾结构。
为了合成的mRNA能最终有效地在体外翻译,在体外转录合成时,掺入5’端的m7G加帽结构和3’端的多聚腺苷酸[poly(A)尾]结构过程如下:上述加帽的过程是通过U7 RNA聚合酶共转录,在mRNA分子5’端掺入诸如ARCA(AnUi-Reverse Cap Analog)。加尾的过程是用Poly(A)聚合酶在mRNA分子3’端,加上poly(A)尾,再用DNase酶消化DNA模板。同时将5-甲基-胞苷(5-MeUhyl-CUP),假尿苷(Pseudo/ψ-UUP)和N端融合3xFLAG标签修饰到mRNA序列中。修饰后mRNA可以降低免疫原性和加强体外转染的稳定性。
本发明还公开了上述的合成修饰的Olig2 mRNA的制备方法,其特征在于,包括以下步骤:
基因编辑:通过定点突变,将Olig2编码区147位丝氨酸密码子UCC突变为丙氨酸的密码子GCC,GCG,GCA或GCU;
体外转录:将上述突变后的DNA经体外转录得到mRNA,即得。
本发明还公开了上述的合成修饰的Olig2 mRNA在促进人诱导多能干细胞向少突胶质细胞分化中的应用。
本发明合成修饰的Olig2 mRNA表现出比野生型效率更高的诱导分化能力。利用本发明所述方法可以实现在诱导多能干细胞快速分化为少突胶质细胞。
本发明还公开了一种促进人诱导多能干细胞向少突胶质细胞分化的试剂盒,包括上述的合成修饰的Olig2 mRNA。
本发明还公开了一种促进人诱导多能干细胞向少突胶质细胞分化的方法,包括以下步骤:
单细胞培养:进行人诱导多能干细胞单细胞传代培养;
诱导转染:以无血清神经诱导培养基进行诱导培养,得神经前体细胞,再将上述的合成修饰的Olig2 mRNA递送至所述神经前体细胞中,转染得到神经前体细胞,在胶质诱导培养基中进行诱导培养;
分化培养:将胶质诱导培养基更换为分化培养基,连续诱导分化培养,即得少突胶质细胞。
本发明设计将Olig2编码区147位丝氨酸突变为丙氨酸,使得合成的Olig2 mRNA翻译的蛋白始终处于去磷酸化状态(Olig2
在其中一个实施例中,所述诱导转染步骤中,采用阳离子脂质体将合成修饰的Olig2mRNA递送至所述神经前体细胞中,连续4天转染诱导得到的神经前体细胞。
在其中一个实施例中,所述单细胞培养步骤中,使用Cellartis DEF-CS进行人诱导多能干细胞单细胞传代培养;
所述诱导转染步骤中,待细胞生长融合至60%-80%(优选70%-80%)时,加入神经诱导培养基,连续培养5-9天,诱导培养得神经前体细胞,随后进行转染,转染后将培养基更换为胶质诱导培养基,每1-2天更换培养基,持续2-6天;每天以合成修饰的Olig2 mRNA转染神经前体细胞操作后更换胶质诱导培养基,持续2-6天;
所述分化培养步骤中,每1-3天更换分化培养基,连续分化培养14-20天。
在其中一个实施例中,所述单细胞培养步骤中,具体方法如下:
1)使用含有钙镁离子的D-PBS按1/20比例稀释COAT-1包被液,每孔1ml包被6孔板,包被后置于37℃恒温培养箱中孵育至少半小时;
2)用移液枪吸弃包被液,将诱导多能干细胞吹散成单细胞悬液,按每孔3×10
3)单细胞状态下的人诱导多能干细胞在Cellartis DEF-CS培养基中生长,并补充营养因子DEF-CS GF-1、GF-2和GF-3。
上述DEF-CS GF-1按照3μl/1ml基础培养基的的浓度使用,GF-2按照1μl/1ml基础培养基的浓度使用,GF-3按照1μl/1ml基础培养基的的浓度使用。
在其中一个实施例中,所述神经诱导培养基的培养液成分包括:N2B27基础培养基和神经诱导化合物;所述神经诱导化合物包括:SB431542和LDN193189。
在其中一个实施例中,所述SB431542在所述神经诱导培养基中的终浓度为10±3μM,LDN193189在神经诱导培养基培养液中的终浓度为250±50nM。
在其中一个实施例中,以温和的Accutase酶消化神经前体细胞,按照每孔3×10
在其中一个实施例中,所述胶质诱导培养基成分包括:N2B27基础培养基,1±0.2wt%的青霉素,1±0.2wt%的链霉素和1±0.2wt%的谷氨酰胺,1±0.2μM Smoothened受体激动剂(SAG),10±2ng/mL重组人血小板衍生生长因子-AA(PDGF-AA),10±2ng/mL重组人neurotropin 3(NT3),10±2ng/mL重组类胰岛素一号生长因子(IGF-I),200±50μM L-抗坏血酸(AA),0.1wt%的微量元素B(Trace Elements B)和10±2ng/mL三碘甲状腺原氨酸(T3)。
在其中一个实施例中,所述分化培养基成分包括:N2B27基础培养基,1±0.2wt%的青霉素,1±0.2wt%的链霉素和1±0.2wt%的谷氨酰胺(penicillin/streptomycin/gluta分钟e),60±10ng/mL三碘甲状腺原氨酸(T3),10±2ng/mL重组人neurotropin 3(NT3),10±2ng/mL重组类胰岛素一号生长因子(IGF-I),200±50μM L-抗坏血酸(AA),1:1,000微量元素B(Trace Elements B),和100±20μM二丁酰环腺苷酸(dbcAMP)。
在其中一个实施例中,所述单细胞培养、mRNA转染和诱导分化、培养步骤中,所用细胞培养条件为:37℃,5%CO
本发明还公开了上述的促进人诱导多能干细胞向少突胶质细胞分化的方法制备得到的人源少突胶质细胞。
本发明还公开了上述的少突胶质细胞在制备用于髓鞘损伤的神经退行性疾病细胞移植治疗中的应用。
与现有技术相比,本发明具有以下有益效果:
本发明设计的一种合成修饰的Olig2 mRNA,基于去磷酸化修饰设计得到,将其转染人诱导多能干细胞来源的神经前体细胞,实现了人源诱导多能干细胞向少突胶质细胞的快速且高效的定向分化,且设计修饰的Olig2 mRNA表现出比野生型效率更高的诱导分化能力。
利用本发明所述方法可以实现将诱导多能干细胞来源的神经前体细胞快速分化为少突胶质细胞,且可以避免以往基于慢病毒介导的转录因子SOX10过表达产生的少突胶质细胞存在着基因被整合的风险,更有利于临床实验的开展。
本发明利用一种基于合成修饰的mRNA的技术促进人诱导多能干细胞向少突胶质细胞分化的方法,实现了人源少突胶质细胞快速诱导,且为今后少突胶质前体细胞移植治疗髓鞘损伤类神经退行性疾病提供了技术支撑。
附图说明
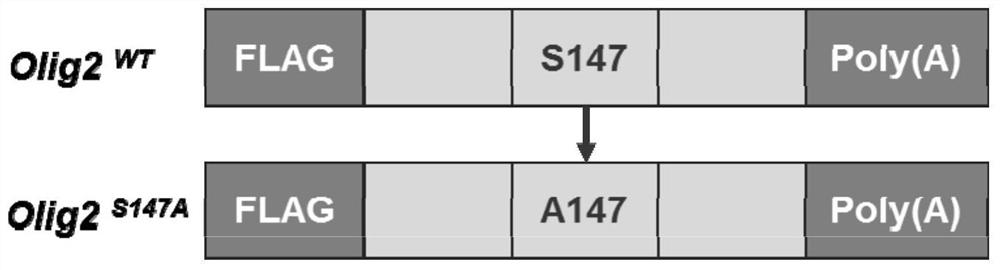
图1为合成的mRNA编码Olig2野生型(Olig2
箭头:Olig2蛋白编码序列147位丝氨酸突变为丙氨酸。
图2为单细胞培养下诱导多能干细胞明场图(比例尺为100μm)。
图3为诱导型神经前体细胞单细胞传代后第一代明场图(比例尺为100μm)。
图4为人源诱导多能干细胞分化的神经前体细胞免疫荧光检测表达PAX6(比例尺为100μm)
图5为PAX6阳性细胞占所有细胞(DAPI阳性)的比例统计图。
图6为合成修饰的Olig2 mRNA调控的人诱导多能干细胞向少突胶质细胞分化策略。
图7为人源诱导型少突胶质细胞明场图(比例尺为100μm)。
图8为荧光定量PCR检测诱导多能干细胞来源的少突胶质细胞谱系标志物基因的表达情况。
图9为免疫荧光鉴定诱导多能干细胞来源的少突胶质细胞及前体细胞蛋白标志物A2B5,NG2,MBP。
图10为A2B5,NG2,MBP阳性细胞占所有细胞(DAPI阳性)的比例统计图。
具体实施方式
为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
以下实施例所用试剂,如非特别说明,均为市售可得;以下实施例所用方法,如非特别说明,均为常规操作可实现。
实施例1
一种合成修饰的Olig2 mRNA,其策略如图1所示,通过以下方法制备得到:
1、基因编辑。
通过定点突变,采用常规方法,将Olig2蛋白编码区147位丝氨酸密码子UCC突变为丙氨酸的密码子GCC,GCG,GCA或GCU。
2、体外转录。
将上述突变后的DNA经体外转录合成Olig2
使用LR reaction protocol试剂盒(赛默飞,美国),将野生型Olig2和突变的Olig2的基因开放阅读框克隆到载体PCR2-UTR-R1R2中,使用限制性内切酶处理,将其线性化,得到目的基因的开放阅读框。以线性化后的载体为模板,用5′端T7聚合酶启动子引物和3′端UTR长T尾引物进行tail-PCR反应。PCR产物经凝胶回收后作为体外转录的模板。体外转录反应包括修饰抗逆转cap类似物(ARCA)以及一些核苷酸,比如5-甲基胞苷-5-三磷酸和假尿苷-5--三磷酸,这些核苷酸的修饰可以减少由于细胞转染激活固有免疫反应而产生的细胞毒性。最后对获得的mRNA进行大小验证,并将其分装成小份保存在-80℃下使用。
实施例2
一种促进人诱导多能干细胞向少突胶质细胞分化的方法,包括以下步骤:
1、人诱导多能干细胞单细胞传代培养
1.1使用含有钙镁离子的D-PBS按1/20比例稀释COAT-1包被液,每孔1ml包被6孔板,包被后置于37℃恒温培养箱中孵育至少半小时;
1.2人诱导多能干细胞在Cellartis DEF-CS单细胞培养基中培养,使用TrypLEExpress Enzyme消化,在37℃恒温培养箱中孵育5分钟;
1.3消化完成后使用培养基终止消化,并离心洗涤,重悬在Cellartis DEF-CS培养基中,按照3×10
1.4单细胞状态下的人诱导多能干细胞在Cellartis DEF-CS培养基中生长,并补充营养因子DEF-CS GF-1(1:333),GF-2(1:1000)和GF-3(1:1000)。
2、诱导培养和转染
2.1人源诱导型神经前体细胞的诱导。
待上述诱导多能干细胞生长融合至70%-80%时(如图2所示),移液枪移去培养基并更换至无血清神经诱导培养基,每天更换神经诱导培养基,连续培养8天。
上述神经诱导培养基培养液成分包括N2B27基础培养基和神经诱导化合物。所述的神经诱导化合物为SB431542和LDN193189。其中SB431542在神经诱导培养基培养液中的终浓度为10μM,LDN193189在神经诱导培养基培养液中的终浓度为250nM。
2.2人源诱导型神经前体细胞的单细胞传代,扩增培养。
诱导多能干细胞经神经诱导培养基诱导8天后,吸弃旧培养基,用预热的DPBS洗涤两遍,之后用温和的细胞消化液Accutase在37℃环境中孵育5-7分钟,孵育结束后加入1mlDMEM/F12终止消化。
细胞经洗涤离心后按照1:6比例重新接种于基质胶包被的6孔板中,使用神经前体维持培养基进行神经前体细胞扩增培养,并补充10μM的Y-27632,以提高神经前体细胞单细胞存活率。
单细胞传代后如图3所示,细胞经免疫荧光鉴定62.23%±2.34的第一代神经前体细胞表达神经前体细胞标志物PAX6,结果如图4-5所示。
2.3转染。
1)取实施例1中经单细胞传代后诱导的神经前体细胞,提前一天接种于12孔板中。
2)转染时以细胞汇合度为30%为宜,转染时完全培养基总量为450μL。
3)取100ng的Olig2
4)取1.5ul的Screenfect mRNA kit,然后加入24ul OPTI-MEM,充分混匀,制成Screenfect mRNA kit稀释液,终体积为25μl。室温静置5分钟。
5)将Screenfect mRNA kit稀释液和mRNA稀释液充分混合(可用振荡器振荡或用移液枪反复吹吸10次以上)混合,室温静置15分钟。
6)将50μl转染复合物滴加到有450μL完全培养基的细胞上,前后移动培养板,使mRNA与阳离子脂质体复合物充分混合均匀。
7)转染后6小时后加入胶质诱导培养基,同样的操作连续转染mRNA 4天。
3、分化培养。
4天后将胶质诱导培养基更换至分化培养基继续培养17天,每两天更换一次分化培养基.
总体诱导分化方案如图6所示,通过上述诱导方案诱导分化的少突胶质细胞明场如图7所示。
实施例3
荧光定量PCR和细胞免疫荧光鉴定诱导多能干细胞来源的少突胶质细胞及前体细胞的标志物基因和相应的表面蛋白标志物。
1、荧光定量PCR鉴定
分别提取mRNA转染后的第4天,第14天,第21天诱导细胞的总RNA,反转录成cDNA,荧光定量PCR分别检测少突胶质细胞及前体细胞标志物基因ST8SIA1(A2B5),CSPG4(NG2),和PLP1。上述A2B5于第4天检测,NG2于第14天检测,PLP1于第21天检测。
具体方法为:
1.1细胞总RNA提取
除去旧培养基,每孔加入500μL裂解液,用移液枪反复吹打十次,裂解细胞。再加入等体积的无水乙醇,充分混匀液体;
混匀后,将裂解产物加入至离心柱中,于4000×g,室温,离心1分钟;
离心结束后,向离心柱中添加500μL洗涤缓冲液。将离心柱于12000×g,室温,离心1分钟;
离心结束后,将离心柱转移至新的PCR管中,打开PCR管盖,晾干2分钟。向离心柱中加入20μL洗脱缓冲液,室温放置2分钟。将离心柱于12000×g,室温,离心1分钟;
将洗脱缓冲液加回离心柱中,室温静置5分钟,将离心柱于12000×g,室温,离心1分钟,离心产物即为RNA;
NanoDrop
1.2RNA反转录为cDNA与gDNA的去除
反转录PCR反应体系如下:
表1.反转录PCR反应体系
混匀后,于PCR仪中反应,反应参数条件如下。
表2.反转录反应的孵育参数
1.3三步法荧光定量PCR
将上一步反转录产物cDNA用无核酸酶的去离子水稀释5倍;再按下列反应体系制备荧光定量PCR体系;
表3.荧光定量PCR体系的溶液配制
每组设置3个复孔,分别加入19μL配制好的荧光定量PCR体系于荧光定量PCR仪中;使用三步法设置荧光定量PCR运行参数。荧光定量PCR的参数如下表所示。
表4.荧光定量PCR反应的循环参数
荧光定量PCR所用的引物如下表所示。
表5.荧光定量PCR所用引物
结果如图8所示,图8为荧光定量PCR检测诱导后少突胶质前体细胞基因标志物mRNA转录水平。
结果显示,相比于野生物Olig2,经修饰过的Olig2 mRNA(Olig2
2、细胞免疫荧光鉴定
免疫荧光分别检测mRNA转染后的第4天,第14天,第21天的诱导细胞的表面蛋白标志物A2B5,NG2,MBP的表达情况。
具体方法为:
采用4%多聚甲醛室温固定15分钟,用PBS缓冲液清洗3次,每次五分钟;
然后用10%驴血清,37℃孵育封闭半小时;
加入稀释过的一抗4℃孵育过夜。所用一抗规格如下表所示。
表6.细胞免疫荧光标志物一抗
结果如图9-10所示。图9为免疫荧光鉴定诱导多能干细胞来源的少突胶质细胞及前体细胞表面蛋白标志物A2B5,NG2,以及MBP。图10为A2B5,NG2,MBP阳性细胞占所有细胞(DAPI阳性)的比例统计图。
结果表明,本发明相比于转染野生型Olig2 mRNA,利用去磷酸化修饰的Olig2mRNA介导人源少突胶质细胞的分化,虽然A2B5(98.03%±1.6vs 97.58%±2.08)阳性的胶质前体细胞并没有明显差别,但是能够高效且快速诱导人源诱导多能干细胞向少突胶质细胞及前体细胞分化,表达少突胶质前体细胞标志物NG2(68.58%±7.02vs 50.92%±2.51),以及表达成熟的标志物MBP(55.55%±0.58vs 34.33%±2.60)的少突胶质细胞。
综上,调控转录因子表达在细胞再编程中发挥了越来越重要的作用,而病毒介导的基因调控是目前最成功和最有效的基因操控手段之一,但是由于存在严重的安全问题,寻找新的基因调控手段十分必要。此制备方法的创新性在于使用了化学修饰的mRNA,并以阳离子脂质体作为基因递送载体进行转染,从而不仅避免了使用病毒载体带来的细胞基因被整合的风险,也极大地提高了人源诱导型细胞的诱导效率以及细胞治疗的安全性。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以作出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
序列表
<110> 中山大学•深圳
中山大学
<120> 合成修饰的Olig2 mRNA及其应用
<160> 8
<170> SIPOSequenceListing 1.0
<210> 1
<211> 972
<212> DNA/RNA
<213> 人工序列(Artificial Sequence)
<400> 1
auggacucgg acgccagccu gguguccagc cgcccgucgu cgccagagcc cgaugaccuu 60
uuucugccgg cccggaguaa gggcagcagc ggcagcgccu ucacuggggg caccgugucc 120
ucguccaccc cgagugacug cccgccggag cugagcgccg agcugcgcgg cgcuaugggc 180
ucugcgggcg cgcauccugg ggacaagcua ggaggcagug gcuucaaguc auccucgucc 240
agcaccucgu cgucuacguc gucggcggcu gcgucgucca ccaagaagga caagaagcaa 300
augacagagc cggagcugca gcagcugcgu cucaagauca acagccgcga gcgcaagcgc 360
augcacgacc ucaacaucgc cauggauggc cuccgcgagg ucaugccgua cgcacacggc 420
ccuucggugc gcaagcuugc caagaucgcc acgcugcugc uggcgcgcaa cuacauccuc 480
augcucacca acucgcugga ggagaugaag cgacugguga gcgagaucua cgggggccac 540
cacgcuggcu uccacccguc ggccugcggc ggccuggcgc acuccgcgcc ccugcccgcc 600
gccaccgcgc acccggcagc agcagcgcac gccgcacauc accccgcggu gcaccacccc 660
auccugccgc ccgccgccgc agcggcugcu gccgccgcug cagccgcggc uguguccagc 720
gccucucugc ccggauccgg gcugccgucg gucggcucca uccguccacc gcacggccua 780
cucaagucuc cgucugcugc cgcggccgcc ccgcuggggg gcgggggcgg cggcaguggg 840
gcgagcgggg gcuuccagca cuggggcggc augcccugcc ccugcagcau gugccaggug 900
ccgccgccgc accaccacgu gucggcuaug ggcgccggca gccugccgcg ccucaccucc 960
gacgccaagu ga 1004
<210> 2
<211> 972
<212> DNA/RNA
<213> 人工序列(Artificial Sequence)
<400> 2
auggacucgg acgccagccu gguguccagc cgcccgucgu cgccagagcc cgaugaccuu 60
uuucugccgg cccggaguaa gggcagcagc ggcagcgccu ucacuggggg caccgugucc 120
ucguccaccc cgagugacug cccgccggag cugagcgccg agcugcgcgg cgcuaugggc 180
ucugcgggcg cgcauccugg ggacaagcua ggaggcagug gcuucaaguc auccucgucc 240
agcaccucgu cgucuacguc gucggcggcu gcgucgucca ccaagaagga caagaagcaa 300
augacagagc cggagcugca gcagcugcgu cucaagauca acagccgcga gcgcaagcgc 360
augcacgacc ucaacaucgc cauggauggc cuccgcgagg ucaugccgua cgcacacggc 420
ccuucggugc gcaagcuuuc caagaucgcc acgcugcugc uggcgcgcaa cuacauccuc 480
augcucacca acucgcugga ggagaugaag cgacugguga gcgagaucua cgggggccac 540
cacgcuggcu uccacccguc ggccugcggc ggccuggcgc acuccgcgcc ccugcccgcc 600
gccaccgcgc acccggcagc agcagcgcac gccgcacauc accccgcggu gcaccacccc 660
auccugccgc ccgccgccgc agcggcugcu gccgccgcug cagccgcggc uguguccagc 720
gccucucugc ccggauccgg gcugccgucg gucggcucca uccguccacc gcacggccua 780
cucaagucuc cgucugcugc cgcggccgcc ccgcuggggg gcgggggcgg cggcaguggg 840
gcgagcgggg gcuuccagca cuggggcggc augcccugcc ccugcagcau gugccaggug 900
ccgccgccgc accaccacgu gucggcuaug ggcgccggca gccugccgcg ccucaccucc 960
gacgccaagu ga 1004
<210> 3
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 3
gtcctctgtt ggctctacat ct 22
<210> 4
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 4
ccccgtcata ccacatgctc 20
<210> 5
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 5
ctttgaccct gactatgttg gc 22
<210> 6
<211> 19
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 6
tgcaggcgtc cagagtaga 19
<210> 7
<211> 19
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 7
acctatgccc tgaccgttg 19
<210> 8
<211> 21
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 8
tgctggggaa ggcaatagac t 21
- 合成修饰的Olig2 mRNA及其应用
- RNA合成-用于反向合成RNA的亚磷酰胺,以及在3’-末端合成RNA的配体、发色团和修饰物的方便引入中的应用