染色体三维结构重建方法及装置
文献发布时间:2023-06-19 13:45:04
技术领域
本发明涉及一种染色体三维结构重建方法。
背景技术
人类基因组由23对染色体组成,共含有约60亿个碱基。如果所有染色体首尾相接并充分伸展开,整个基因组的长度将达到2米。然而,众所周知的是,整个基因组被封装在一个狭小而又拥挤的细胞核内,实际上人类基因组所占有的空间尺度不到人的头发丝直径(10μm)的十分之一,这表明基因组并非仅表现为一个简单的一维聚合物,而是折叠成致密的复杂的三维结构。人们逐渐意识到,为了充分理解染色体如何行使各种功能(如基因表达)以及在有丝分裂中如何复制并正确地分离,需要对染色体的三维空间结构有准确而全面的认识。然而,研究三维基因组的传统技术如荧光显微镜、FISH等,不但分辨率非常受限,而且往往只能同时探测少数几个孤立的基因组位点。染色体构象捕获(Chromatinconformation capture,3C)是首个在分子层次上研究染色质物理相互作用的技术
从Hi-C数据重建染色体的三维结构本质上是一个优化问题。与其他应用领域的优化问题一样,一个标准的优化过程需要给出两部分的定义:目标函数和优化算法。对于目标函数,一种策略是基于距离进行定义。也就是说,首先将相互作用频率矩阵转换为空间距离矩阵,然后根据待优化的染色体三维结构模型计算出空间距离矩阵,并最小化这两个距离矩阵之间的差异
无论采用哪种目标函数,最终都会归结为一个非线性、大规模的优化问题。对这类问题,牛顿算法等简单的局部搜索方法并不适用。人们提出了几种全局搜索方案。ChromSDE
经典的多维标度方法(MDS)
通过以上对现有技术的分析可知,ShRec3D算法是现有技术中比较优秀的一种技术,其相比其他算法具有几个数量级的速度优势,因此得到了广泛关注和大量应用;但与其他算法相比,ShRec3D算法重建精度有限,这是由最短路径算法的近似性导致的,现有的各种ShRec3D改进算法也仍未克服这一问题。
发明内容
本发明所要解决的技术问题在于克服现有ShRec3D算法及其改进算法的不足,提供一种染色体三维结构重建方法,在ShRec3D算法基础上进行改进,利用重建出的染色体三维结构的固有距离矩阵对补全后的距离矩阵进行迭代修正,从而继续保持重建速度优势的前提下,大幅提高重建精度。
本发明具体采用以下技术方案解决上述技术问题:
一种染色体三维结构重建方法,包括以下步骤:
将Hi-C相互作用频率矩阵转化为距离矩阵D
将距离矩阵D
按以下方法迭代求解染色体三维结构:
步骤1、根据距离矩阵D
步骤2、根据各基因组位点的空间位置计算出所述染色体三维结构S的固有距离矩阵D
步骤3、利用D
步骤4、满足预设条件后停止迭代并输出当前迭代步所得到的染色体三维结构S。
作为其中一个优选方案,所述利用D
进一步优选地,所述预设条件为:迭代次数达到30次。
进一步优选地,D
优选地,使用最短路径算法将距离矩阵D
基于同一发明构思还可以得到以下技术方案:
一种染色体三维结构重建装置,包括:
距离矩阵转化模块,用于将Hi-C相互作用频率矩阵转化为距离矩阵D
距离矩阵补全模块,用于将距离矩阵D
迭代求解模块,用于按以下方法迭代求解染色体三维结构:
步骤1、根据补全后距离矩阵D
步骤2、根据各基因组位点的空间位置计算出所述染色体三维结构S的固有距离矩阵D
步骤3、利用D
步骤4、满足预设条件后停止迭代并输出当前迭代步所得到的染色体三维结构S。
作为其中一个优选方案,所述利用D
进一步优选地,所述预设条件为:迭代次数达到30次。
进一步优选地,D
优选地,使用最短路径算法将距离矩阵D
相比现有技术,本发明具有以下有益效果:
本发明针对现有ShRec3D算法及其改进算法的不足,对ShRec3D算法进行改进,综合利用基于最短路径算法得到的距离矩阵信息及基于真实的三维空间变换得到的距离矩阵信息,将两者优势互补,达到提高重建精度的效果。本发明方法相对于原生的ShRec3D算法在重建质量上有着本质性的改善,尤其是在强噪声和高信号覆盖度以及弱噪声和低信号覆盖度的数据环境中。同时,与现有的其他算法(如ChromSDE)相比,本发明方法仍然保持了重建速度快的优势。
附图说明
图1a、图1b分别为模拟数据的螺旋结构,以及信号覆盖度sc与最近邻点数K之间的关系;
图2a、图2b分别为RMSD与算法迭代次数的关系曲线,以及D
图3a、图3b、图3c分别为迭代Shrec3D与原生Shrec3D的性能对比热图,算法性能与数据噪声水平的关系曲线,以及不同算法之间的运行时间对比;
图4a、图4b分别为随机游走模型示例以及迭代Shrec3D与原生Shrec3D在随机游走模型上的性能对比热图。
具体实施方式
针对现有ShRec3D算法及其改进算法的不足,本发明的解决思路是在ShRec3D算法基础上进行改进,利用重建出的染色体三维结构的固有距离矩阵对补全后的距离矩阵进行迭代修正,从而继续保持重建速度优势的前提下,大幅提高重建精度。
具体而言,本发明所提出的染色体三维结构重建方法,包括以下步骤:将Hi-C相互作用频率矩阵转化为距离矩阵D
将距离矩阵D
按以下方法迭代求解染色体三维结构:
步骤1、根据距离矩阵D
步骤2、根据各基因组位点的空间位置计算出所述染色体三维结构S的固有距离矩阵D
步骤3、利用D
步骤4、满足预设条件后停止迭代并输出当前迭代步所得到的染色体三维结构S。
为了便于公众理解,在对本发明技术方案进行进一步详细说明之前,先对所涉及的相关现有技术进行简要说明:
一个Hi-C实验产生一个双端读段库。其中每个双端读段代表对应的两个限制性片段之间的相互作用。这些读段被映射到参考基因组,并过滤掉低质量的读段。把高质量读段根据其映射到的基因组位置进行分区间累加,我们就得到一个相互作用频率矩阵F,元素F
给定一个频率矩阵F,染色体三维结构重建的任务是从F出发重建出染色体三维结构X=(x
原生的ShRec3D算法由以下三个步骤组成:
首先,将相互作用矩阵F转换为距离矩阵D。目前所有的重建方法都假定基因组位点之间的相互作用频率与它们之间的三维空间距离满足如下幂律关系:
其中α是转换因子,D
其次,补全距离矩阵D。经典的MDS算法要求所有位点对的距离都是已知的,但由公式(1)得到距离矩阵含有未知元素(即无穷大值)。为了求得这些未知元素的值,我们用一个带权重的图结构表示距离矩阵D。图的结点代表基因组位点,当且仅当D
最后,利用多维标度方法(MDS)从补全的距离矩阵重建出染色体三维结构。MDS是一个基于n个物体两两之间的距离信息推断出它们在k维欧式空间坐标的统计学经典技术
式中tr(.)表示矩阵的迹。为求解上式,可对B做奇异值分解,得到三个最大的特征值λ
对应的代价函数值是:
ψ=λ
由此可以看出,只有当λ
基于以上ShRec3D算法原理可以看出,ShRec3D需要采用最短路径算法补全距离矩阵。最短路径算法给出的距离矩阵(记为D
由Hi-C相互作用频率矩阵F通过公式(1)转化成的(不完整)距离矩阵记为D
所述利用D
为了验证本发明迭代ShRec3D算法的性能,我们模拟Hi-C实验过程,生成模拟数据。模拟数据根据以下形式的螺旋结构产生(见图1a):
x=sin(t),y=cos(t),z=t/10,t=1,…,10π
(5)
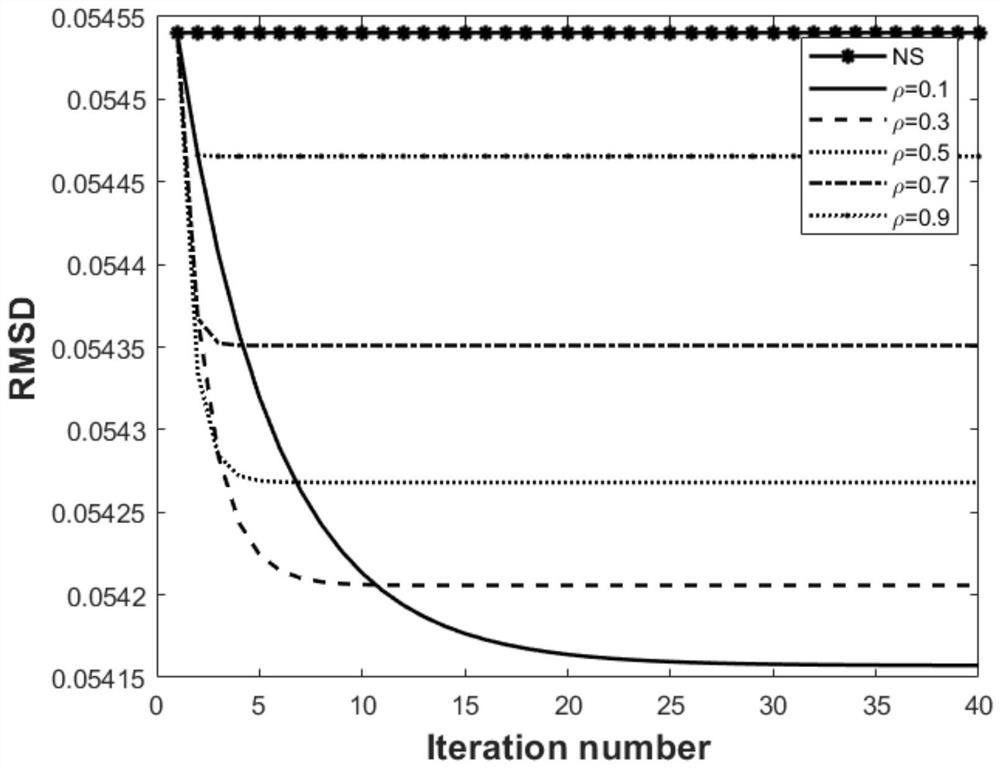
这个螺旋结构由N个点组成,在由上式给出N个点的坐标后,就可计算出对应的N×N距离矩阵D。为了模拟真实的Hi-C相互作用频率矩阵的不完整性,对每个点来说,只保留前K(K 定义信号覆盖度sc∈[0,1]为相互作用频率矩阵中非零元素的百分比。参数K直接决定了所生成相互作用频率矩阵的信号覆盖度,实际上,信号覆盖度sc近似正比于K,图1b显示sc≈K/N。 除模拟螺旋结构外,我们还基于随机游走模型进行三维结构重建 本文采用误差方均根(Root Mean Square Deviation,RMSD)作为评价算法性能的标准。RMSD用来计算两个结构之间的相似性。给定一个基准的三维结构P=(p
在用上式计算RMSD之前,要先对结构Q进行缩放、镜像、平移和旋转操作,使之尽量与基准结构P对齐,具体步骤见文献 本发明的迭代ShRec3D算法首先需要确定迭代次数maxIter和权重ρ的值。图2a(图中的NS表示原生ShRec3D算法)给出了在不同ρ值的情况下,重建结构的RMSD值与迭代次数maxIter的关系曲线。从图中可以看出,在各种权重ρ下,算法都能够达到收敛,即重建结果进入稳定状态,重建精度不再进一步提高,而且在不同权重ρ下收敛后的RMSD都小于原生ShRec3D算法给出的RMSD值,表明我们的算法性能确实优于原生的ShRec3D算法,且这种性能提升是稳健的。当ρ值较大时,算法可以较快地达到收敛,但收敛后的RMSD值较大,表明较大的ρ值给出的重建结果相对于原生的ShRec3D来说改进程度有限。当ρ值较小时,算法收敛速度较慢,但收敛后的RMSD值较小,表明较小的ρ值能够带来高精度的三维结构重建结果。综合算法性能和运算效率,我们优选ρ=0.1,maxIter=30。我们用皮尔森系数度量D 由于本发明是对ShRec3D算法的一个重要改进,我们主要关注于将该算法与原生的ShRec3D算法做对比分析。我们知道,现有的染色体三维结构重建方法的重建结果的好坏都严重依赖于数据的噪声和有效数据的稀疏程度。我们预期本发明提出的迭代ShRec3D算法相对于原生ShRec3D算法的改进程度也与这两个因素有关。把原生ShRec3D与迭代ShRec3D算法的RMSD之差记为ΔR,即 ΔR=RMSD(原生ShRec3D)-RMSD(迭代ShRec3D) (7) ΔR反映了迭代ShRec3D算法相对于原生ShRec3D算法的性能改进程度,ΔR>0表明迭代ShRec3D算法的性能优于原生ShRec3D算法,且ΔR越大这种优越性就越显著;反之,ΔR<0则表明迭代ShRec3D算法的性能相对于原生ShRec3D算法来说表现更差。图3a所示的热图显示了ΔR与模拟数据的噪声水平S和最近邻点数K之间的变化关系。从图中可以看出,几乎在所有情况下都有ΔR>0,表明我们的迭代ShRec3D算法在几乎所有的噪声环境和信号稀疏度下都能改进原生ShRec3D算法的性能,因而我们的这种算法性能改进策略是稳健的。令人感兴趣的是,在低噪声水平和低信号覆盖度以及高噪声水平和高信号覆盖度这两种情况下,迭代ShRec3D算法相对于原生ShRec3D算法的性能改进更加显著,在高噪声水平和低信号覆盖度以及低噪声水平和高信号覆盖度这两种情况下改进则不够明显。对此一个直观的解释是,对于低噪声且有效信号足够稠密的数据来说,原生ShRec3D算法的性能已经足够好,事实上,当距离矩阵不存在缺失值且无噪声时,ShRec3D算法能够严格地重建出基准的三维结构;另一方面,当噪声很强且有效信号过于稀疏时,表明数据质量较差,这时ShRec3D算法不能有效地重建出基准的三维结构,这种数据质量过差导致的算法失效犹如“病入膏肓,无可救药”,即使改进算法也难以达到提高重建精度的效果;在其他情况下,则表明数据质量中等,ShRec3D算法的重建效果还有改进的空间。 图3b给出了RMSD值随数据噪声变化的曲线。我们设置数据模拟程序的参数为α=1.0,N=100,K=30,实际上与图3a中热图的第三列相对应。作为对比,图3b还显示了ChromSDE算法的性能曲线。可以看出,随着噪声水平的增大,两个算法的RMSD值都逐渐变大,说明染色体三维结构的重建质量随着噪声的增加而下降。在整个噪声水平区间[0,1],我们的算法都显著优于原生的ShRec3D算法。图3b还表明,在强噪声环境下原生ShRec3D和迭代ShRec3D的性能都优于ChromSDE算法,但在低噪声环境下两者性能都不如ChromSDE算法,这就证实了文献[15]中的结论。 既然我们的迭代ShRec3D算法与原生ShRec3D算法相比多了一个迭代过程,那么可以预期前者比后者的时间复杂度会更高一些。尽管如此,图3c显示出我们的迭代ShRec3D算法的运行时间仍然远远小于ChromSDE算法,这表明迭代ShRec3D算法相对于原生ShRec3D来说以较小的时间代价就获得了显著的性能提升。 我们还利用模拟的随机游走模型来检验我们的算法,所得结论与模拟的螺旋结构类似(见图4b)。 根据以上验证试验可以看出,本发明所提出的迭代ShRec3D算法相对于原生的ShRec3D算法在重建质量上有着本质性的改善,尤其是在强噪声和高信号覆盖度以及弱噪声和低信号覆盖度的数据环境中。同时,与现有的其他算法(如ChromSDE)相比,我们的迭代ShRec3D算法仍然保持了重建速度快的优势。 参考文献 [1]DEKKER J,RIPPE K,DEKKER M,et al.Capturing Chromosome Conformation[J].Science,295(2002)1306-1311. [2]SIMONIS M,KLOUS P,SPLINTER E,et al.Nuclear Organization of Activeand Inactive Chromatin Domains Uncovered by Chromosome Conformation Capture-on-chip(4C)[J].Nat.Genet,38(2006)1348-1354. [3]ZHAO Z,TAVOOSIDANA G, [4]DOSTIE J,RICHMOND T A,ARNAOUT R A,et al.Chromosome ConformationCapture Carbon Copy (5C):A Massively Parallel Solution for MappingInteractions between Genomic Elements[J].Genome Res,16(2006)1299-1309. [5]LIEBERMAN-AIDEN E,VAN BERKUM N L,WILLIAMS L,et al.ComprehensiveMapping of Long-range Interactions Reveals Folding Principles of the HumanGenome[J].Science,326(2009)289-293. [6]ZHANG Z,LI G,TOH K C,SUNG WK.3D Chromosome Modeling with Semi-Definite Programming and Hi-C Data[J].J Comput Biol,2013,20:831-46. [7]VAROQUAUX N,AY F,NOBLE W S,VERT J P.A Statistical Approach forInferring the 3D Structure of the Genome[J].Bioinformatics,2014,30:i26-33. [8]YAFFE E,TANAY A.Probabilistic modeling of Hi-C Contact MapsEliminates Systematic Biases to Characterize Global Chromosomal Architecture[J].Nat Genet,2011,43:1059-65. [9]HU M,DENG K,QIN Z,DIXON J,SELVARAJ S,FANG J,et al.BayesianInference of Spatial Organizations of Chromosomes[J].PLoS Comput Biol,2013,9:1002893. [10]TORGERSON WS.Multidimensional Scaling:I.Theory and Method[J].Psychometrika,1952,17:401-19. [11]WACHTER A,BIEGLER L T.On the Implementation of an Interior-PointFilter Line-Search Algorithm for Large-Scale Nonlinear Programming[J].MathProgram,2006,106:25-57. [12] [13]SERRA F, [14]RUSSEL D,LASKER K,WEBB B,et al.SCHNEIDMAN-DUHOVNY D,et al.Puttingthe Pieces Together:Integrative Modeling Platform Software for StructureDetermination of Macromolecular Assemblies[J].PLoS Biol,2012,10:e1001244. [15]ZOU C,ZHANG Y,OUYANG Z.HSA:Integrating Multi-track Hi-C Data forGenome-scale Reconstruction of 3D Chromatin Structure[J].Genome Biol,2016,17:40. [16]ROUSSEAU M,FRASER J,Ferraiuolo M,et al.Three-dimensional Modelingof Chromatin Structure from Interaction Frequency Data Using Markov ChainMonte Carlo Sampling[J].BMC Bioinform,2011,12:414. [17]METROPOLIS N,ROSENBLUTH A W,ROSENBLUTH M N,et al.Equation ofState Calculation by Fast Computing Machines[J].J Chem Phys,1953,21:1087-92. [18]PENG C,FU L Y,DONG P F,et al.The Sequencing Bias RelaxedCharacteristics of Hi-C Derived Data and Implications for Chromatin 3DModeling[J].Nucleic Acids Res,2013,41:183–3. [19]OLUWADARE O,ZHANG Y,CHENG J.A Maximum Likelihood Algorithm forReconstructing 3D Structures of Human Chromosomes from Chromosomal ContactData[J].BMC Genomics,2018,19:161. [20]TRIEU T,CHENG J.MOGEN:a Tool for Reconstructing 3D Models ofGenomes from Chromosomal Conformation Capturing Data[J].Bioinformatics,2016,32:1286-92. [21]TRIEU T,CHENG J.Large-scale reconstruction of 3D structures ofhuman chromosomes from chromosomal contact data[J].Nucleic Acids Res 2014,42(7):e52. [22]TRIEU T,CHENG J.3D Genome Structure Modeling by LorentzianObjective Function[J].Nucleic Acids Res,2017,45:1049-58. [23]LESNE A,RIPOSO J,ROGER P,et al.3D Genome Reconstruction fromChromosomal Contacts[J].Nat Methods,2014,11:1141-43. [24]LI J,ZHANG W,LI X.3D Genome Reconstruction with ShRec3D+and Hi-CData[J].IEEE/ACM Trans Comput Biol Bioinform,2018,15:460-8. [25]KAPILEVICH V,SENO S,MATSUDA H,TAKENAKA Y.Chromatin 3DReconstruction from Chromosomal Contacts Using a Genetic Algorithm[J].IEEE/ACM Trans Comput Biol Bioinform,2019,16(5):1620-1626. [26]LI F-Z,LIU Z-E,LI X-Y,et al.,Cai-Ming Zhang.Chromatin 3DStructure Reconstruction with Consideration of Adjacency Relationship amongGenomic Loci[J],BMC Bioinformatics,2020,21:272. [27]OLUWADARE1 O,HIGHSMITH1 M,CHENG J.An Overview of Methods forReconstructing 3-D Chromosome and Genome Structures from Hi-C Data[J].Biological Procedures Online,2019,21(1):7. [28]彭城,李国亮,张红雨,阮一骏.染色质三维结构重建及其生物学意义[J].中国科学:生命科学.2014(08):794-802. [29]BUJA A,SWAYNE D F,LITTMAN M L,et al.Data Visualization WithMultidimensional Scaling[J].Journal of Computational and GraphicalStatistics,2007,17:444-72. [30]ARUN K S,HUANG T S,BLOSTEIN S D.Least-squares Fitting of Two 3-dPoint Sets[J].IEEE Trans Pattern Anal Mach Intell,1987,9:698-700.
- 染色体三维结构重建方法及装置
- 三维结构简化重建方法、装置、计算机设备和存储介质