一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类方法及系统
文献发布时间:2023-06-19 18:27:32
技术领域
本发明涉及一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类方法及系统,属于数据聚类技术领域。
背景技术
Hi-C技术通过消化和重连空间上接近的染色体片段,对其进行高通量测序,可确定染色体不同位点之间的空间交互作用。单细胞Hi-C技术能够用于捕获单个细胞的染色体构象,它使我们能够在单细胞水平上获得染色体相互作用信息,并进一步研究不同细胞类型之间基因组结构的差异,为开展细胞聚类工作提供了数据和技术支持。现有的针对单细胞Hi-C数据且性能较高的聚类方法是scHiCluster,它使用卷积平滑算法和重启随机游走算法来处理单细胞中的染色体接触矩阵,并使用主成分分析(PCA)操作来生成细胞嵌入。最后,scHiCluster基于细胞嵌入使用K-means算法对细胞进行聚类。但是scHiCluster算法还存在以下问题:第一,在处理大规模单细胞Hi-C数据集时聚类效果较差;第二,难以识别出数据集中细胞数量较少的细胞类型。
发明内容
针对现有技术的不足,本发明提供了一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类方法;
本发明还提供了一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类系统;
本发明提出的针对单细胞Hi-C聚类方法性能明显优于现有的聚类方法,大幅提高了处理大规模单细胞Hi-C数据集时的聚类效果,并且可以识别出数据集中细胞数量较少的细胞类型。
术语解释:
1、单细胞Hi-C数据:是指通过单细胞Hi-C技术获取的数据,一般包括细胞特性和交互信息两类数据。其中细胞特性数据中包含细胞的特定信息,如细胞类型、捕获的染色体片段映射到人类全基因序列的百分比等。交互信息数据包含细胞中染色体片段接触的具体信息,如接触片段的起始位置和接触片段的结束位置。其中经过处理后的数据一共包含三列,第一列和第二列是被切割后的染色体片段编号,第三列是两个染色体片段之间的接触数。例如,47 60 5表示47号染色体片段和60号染色体片段之间的接触数为8。
2、染色体相互作用数据,即接触数,两个染色体片段之间的接触数是指两个染色体片段在三维空间结构中接触点的个数。
3、KPCA,核主成分分析,是一种非线性数据处理方法,其核心思想是通过一个非线性映射把原始空间的数据投影到高维特征空间,然后在高维特征空间中进行基于主成分分析(PCA)的数据处理。
本发明的技术方案为:
一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类方法,包括:
(1)数据预处理;以给定分辨率将单细胞Hi-C数据中的染色体相互作用数据标准化为染色体接触矩阵;
(2)基于接触数权重的平滑;染色体接触矩阵中的每一行代表染色体片段与其所有邻居之间的相互作用信息;使用权重矩阵来量化不同邻居片段对目标染色体片段的影响;
(3)重启随机游走平滑;使用重启随机游走算法对步骤(2)基于接触数权重的平滑处理后的染色体接触矩阵进行平滑处理;
(4)接触二值化;对步骤(3)重启随机游走平滑处理后的矩阵进行二值化处理;
(5)主成分提取;使用KPCA进行降维并生成细胞嵌入;
(6)特征融合;通过KPCA生成的细胞嵌入的每一行代表一个细胞中的染色体结构信息;通过细胞嵌入计算细胞之间的欧式距离,并构造距离矩阵作为细胞嵌入的补充;通过行合并的方式合并距离矩阵和步骤(4)处理后得到的矩阵,从而形成新的特征矩阵;
(7)谱聚类;使用谱聚类算法实现细胞聚类。
根据本发明优选的,步骤(1)中,以给定分辨率将单细胞Hi-C数据中的染色体相互作用数据标准化为染色体接触矩阵,包括:
首先,将每条染色体分成n段,n=L/R,L代表染色体长度,R代表分辨率;
其次,将单细胞Hi-C数据表示为染色体接触矩阵A
根据本发明优选的,步骤(2)中,使用权重矩阵P来量化不同邻居片段对目标染色体片段的影响,包括:
根据目标染色体片段的接触信息,生成每个目标染色体片段的所有邻居的权重信息,并将其作为权重矩阵P的行向量;接触数不等于0的两个染色体片段互为邻居,染色体接触矩阵A
式(I)中,k代表平滑的次数,k的取值范围为:1≤k≤6;
染色体接触矩阵A
B
式(II)、式(III)中,S表示平滑后的染色体接触矩阵,A表示染色体接触矩阵A
根据本发明优选的,步骤(3)中,使用重启随机游走算法对步骤(2)基于接触数权重的平滑处理后的染色体接触矩阵进行平滑处理;包括:
将S表示为一个无向加权图,其中,节点代表染色体片段,S
重启随机游走算法的每一步都有随机游走到下一个节点或返回起始节点两种选择,本发明指定重启概率r作为返回起始节点的概率,1-r作为游走到相邻染色体片段的概率;具体实现过程按照式(IV)进行重复计算:
U
式(IV)中,t表示迭代次数,U
根据本发明优选的,步骤(4)中,对步骤(3)重启随机游走平滑处理后的矩阵进行接触二值化处理,包括:
首先,将经过重启随机游走平滑后的矩阵
然后,将所有细胞中s号染色体对应的向量组合成矩阵
对矩阵
最后,通过行合并的方式将23条染色体对应的矩阵
进一步优选的,接触二值化通过设置阈值thr
式(V)中,thr
根据本发明优选的,步骤(5)中,使用KPCA进行降维并生成细胞嵌入,包括:
a、计算出原始数据集的平均值,将原始数据集中所有数据减去平均值;
b、利用KPCA的核函数计算出核矩阵K;
c、计算核矩阵的特征值和特征向量;
d、将特征向量按照其对应特征值的大小按行排列成矩阵,取前k行组成矩阵P;
e、Y=PX,得到降维后的数据Y。
进一步优选的,KPCA的核函数为余弦。
根据本发明优选的,步骤(6)中,把距离矩阵Q中的每一行与细胞嵌入之后矩阵F中的对应行进行拼接,得到最后的特征融合矩阵即新的特征矩阵X。
根据本发明优选的,步骤(7)中,使用谱聚类算法实现细胞聚类,包括:
①输入新的特征矩阵X并使用径向基函数构造相似度矩阵S,其中,新的特征矩阵X的每一行都包含一个细胞的特征信息;
②根据相似度矩阵S构建相似图G,通过相似图G得到邻接矩阵W;
③计算度矩阵D和拉普拉斯矩阵L=D-W;
④计算标准化拉普拉斯矩阵
⑤计算标准化拉普拉斯矩阵L′中最小的k个特征值对应的n维特征向量;
⑥将前k个特征向量组合成一个n行k列的矩阵V
⑦通过式(VI)对矩阵V
式(VI)中,
⑧用k-means算法进行聚类,得到一个包含k个簇的聚类划分。
一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类系统,包括:
数据预处理模块,被配置为;以给定分辨率将单细胞Hi-C数据中的染色体相互作用数据标准化为染色体接触矩阵;
基于接触数权重的平滑模块,被配置为;染色体接触矩阵中的每一行代表染色体片段与其所有邻居之间的相互作用信息;使用权重矩阵来量化不同邻居片段对目标染色体片段的影响;
重启随机游走平滑模块,被配置为;使用重启随机游走算法对基于接触数权重的平滑处理后的染色体接触矩阵进行平滑处理;
接触二值化模块,被配置为;对重启随机游走平滑处理后的矩阵进行二值化处理;
主成分提取模块,被配置为;使用KPCA进行降维并生成细胞嵌入;
特征融合模块,被配置为;通过KPCA生成的细胞嵌入的每一行代表一个细胞中的染色体结构信息;通过细胞嵌入计算细胞之间的欧式距离,并构造距离矩阵作为细胞嵌入的补充;通过行合并的方式合并距离矩阵和使用KPCA进行降维并生成细胞嵌入处理后得到的矩阵,从而形成新的特征矩阵;
谱聚类模块,被配置为;使用谱聚类算法实现细胞聚类。
本发明的有益效果为:
1、由于目前Hi-C数据的采集技术还不够成熟,导致Hi-C数据中的接触数量与实际接触数量之间存在差异。因此,本发明提出了一种新的方法,通过降低不同类型细胞中染色体结构的特异性来减少差异。现有的聚类方法利用线性相邻染色体片段的接触信息或空间相邻染色体片段的接触信息对目标染色体片段的接触信息进行平滑处理。然而,这些处理方法在平滑过程中忽略了空间相邻染色体片段之间的权重差异。而本发明提出的新的基于接触数权重的平滑方法给不同接触数的邻居片段赋予不同的权重,用权重来量化拥有不同接触数的邻居片段对目标染色体片段的影响,从而能够生成具有更精确细胞特征的细胞嵌入。
2、本发明提出的特征融合方法将KPCA降维生成的细胞嵌入矩阵与生成的不同细胞之间的欧式距离矩阵进行行合并,能够实现将细胞内的染色体结构信息与细胞间的距离信息进行融合的效果,从而补充细胞的特征信息。
3、本发明所使用的谱聚类算法是由图论发展而来的一种算法,已被广泛应用于各类数据的聚类。与聚类方法scHiCluster使用的传统K-Means算法相比,谱聚类对数据分布的适应性更强,聚类效果也十分优异。
附图说明
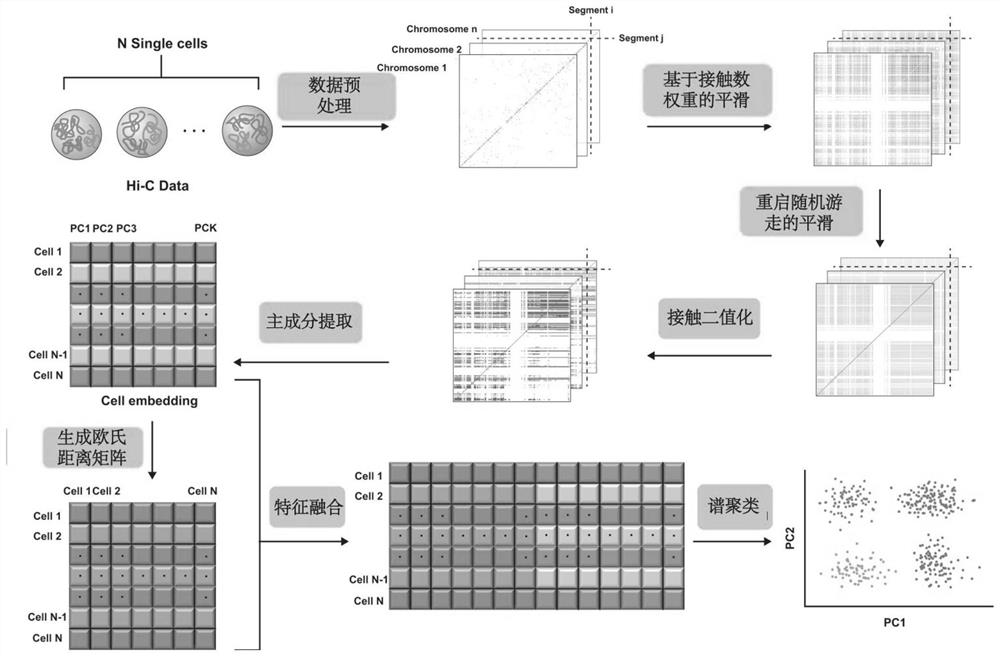
图1为本发明基于接触数权重平滑和特征融合的单细胞Hi-C聚类方法的流程框图;
图2为五种聚类方法在ML1和ML3的626个人类细胞数据上的聚类性能比较示意图;
图3为五种聚类方法在包含2655个人类细胞的Ramani数据集上的聚类性能比较示意图;
图4(a)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于ARI评价指标的性能比较示意图;
图4(b)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于AMI评价指标的性能比较示意图;
图4(c)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于HM评价指标的性能比较示意图;
图4(d)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于FM评价指标的性能比较示意图。
具体实施方式
下面结合说明书附图和实施例对本发明作进一步限定,但不限于此。
实施例1
一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类方法,如图1所示,包括:
(1)数据预处理;以给定分辨率将单细胞Hi-C数据中的染色体相互作用数据标准化为染色体接触矩阵;
(2)基于接触数权重的平滑;现有聚类方法中的平滑方法通过线性相邻染色体片段或空间相邻染色体片段的接触信息平滑目标染色体片段的接触信息。然而,这些处理方法都忽略了平滑过程中空间相邻染色体片段之间的权重差异。因此,为了获得更准确的接触信息,本发明提出了一种新的基于接触数权重的平滑方法。染色体接触矩阵中的每一行代表染色体片段与其所有邻居之间的相互作用信息;由于不同的邻居片段和目标片段之间的接触数不同,不同邻居片段对目标片段的影响也是不同的。因此,需要使用权重矩阵来量化不同邻居片段对目标染色体片段的影响;
(3)重启随机游走平滑;使用重启随机游走算法对步骤(2)基于接触数权重的平滑处理后的染色体接触矩阵进行平滑处理;
(4)接触二值化;对步骤(3)重启随机游走平滑处理后的矩阵进行二值化处理;
(5)主成分提取;使用KPCA进行降维并生成细胞嵌入;
(6)特征融合;通过KPCA生成的细胞嵌入的每一行代表一个细胞中的染色体结构信息;通过细胞嵌入计算细胞之间的欧式距离,并构造距离矩阵作为细胞嵌入的补充;由于细胞嵌入及其相关的欧式距离矩阵具有相同的行数,通过行合并的方式合并距离矩阵和步骤(4)处理后得到的矩阵,从而形成新的特征矩阵;在该过程中,通过融合细胞内的染色体结构信息和细胞间的距离信息,进一步补充细胞的特征信息。
(7)谱聚类;使用谱聚类算法实现细胞聚类。
实施例2
根据实施例1所述的一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类方法,其区别在于:
步骤(1)中,以给定分辨率将单细胞Hi-C数据中的染色体相互作用数据标准化为染色体接触矩阵,包括:
首先,将每条染色体分成n段,n=L/R,L代表染色体长度,R代表分辨率;
其次,将单细胞Hi-C数据表示为染色体接触矩阵A
步骤(2)中,使用权重矩阵P来量化不同邻居片段对目标染色体片段的影响,包括:
根据目标染色体片段的接触信息,生成每个目标染色体片段的所有邻居的权重信息,并将其作为权重矩阵P的行向量;接触数不等于0的两个染色体片段互为邻居,染色体接触矩阵A
式(I)中,k代表平滑的次数,k的取值范围为:1≤k≤6;
染色体接触矩阵A
B
式(II)、式(III)中,S表示平滑后的染色体接触矩阵,A表示染色体接触矩阵A
步骤(3)中,使用重启随机游走算法对步骤(2)基于接触数权重的平滑处理后的染色体接触矩阵进行平滑处理;包括:
重启随机游走算法有助于从全局角度推断节点之间的关系。因此,本发明使用重启随机游走算法从图的整体结构信息中捕获两个节点之间的多方面关系。矩阵S描述了染色体片段之间的联系,因此,将S表示为一个无向加权图,其中,节点代表染色体片段,S
重启随机游走算法的每一步都有随机游走到下一个节点或返回起始节点两种选择,本发明指定重启概率r作为返回起始节点的概率,1-r作为游走到相邻染色体片段的概率;具体实现过程按照式(IV)进行重复计算:
U
式(IV)中,t表示迭代次数,U
步骤(4)中,对步骤(3)重启随机游走平滑处理后的矩阵进行接触二值化处理,包括:
首先,为了获得细胞的整体信息,将经过重启随机游走平滑后的矩阵
然后,将所有细胞中s号染色体对应的向量组合成矩阵
为了统一不同细胞中染色体接触数的尺度,对矩阵
最后,通过行合并的方式将23条染色体对应的矩阵
接触二值化通过设置阈值thr
式(V)中,thr
接触二值化后的矩阵D和矩阵F是二进制矩阵,染色体片段之间的联系只用0和1表示,这统一了不同细胞的接触矩阵中染色体接触数的尺度。此过程有助于保留染色体上最重要的结构信息。
步骤(5)中,使用KPCA进行降维并生成细胞嵌入,包括:
a、计算出原始数据集的平均值,将原始数据集中所有数据减去平均值;
b、利用KPCA的核函数计算出核矩阵K;
c、计算核矩阵的特征值和特征向量;
d、将特征向量按照其对应特征值的大小按行排列成矩阵,取前k行组成矩阵P;
e、Y=PX,得到降维后的数据Y。
接触二值化后的二进制矩阵F的特征维度为
KPCA的核函数为余弦。
步骤(6)中,KPCA降维生成的细胞嵌入矩阵F中,每一行都代表了一个细胞中的染色体结构信息,我们根据欧氏距离计算公式计算出每个细胞之间的欧氏距离进而生成距离矩阵Q。又因为距离矩阵Q和细胞嵌入之后的矩阵F具有相同的行数,因此把距离矩阵Q中的每一行与细胞嵌入之后矩阵F中的对应行进行拼接,得到最后的特征融合矩阵即新的特征矩阵X。这样特征融合矩阵X就融合了细胞内的染色体结构信息和细胞间的距离信息,进一步补充了细胞的特征信息。
步骤(7)中,使用谱聚类算法实现细胞聚类,包括:
①输入新的特征矩阵X并使用径向基函数(RBF)构造相似度矩阵S,其中,新的特征矩阵X的每一行都包含一个细胞的特征信息;
②根据相似度矩阵S构建相似图G,通过相似图G得到邻接矩阵W;
③计算度矩阵D和拉普拉斯矩阵L=D-W;
④计算标准化拉普拉斯矩阵
⑤计算标准化拉普拉斯矩阵L′中最小的k个特征值对应的n维特征向量;
⑥将前k个特征向量组合成一个n行k列的矩阵V
⑦通过式(VI)对矩阵V
式(VI)中,
⑧用k-means算法进行聚类,得到一个包含k个簇的聚类划分。
将本发明提出的聚类方法scHiCSC与现有的4种聚类聚类方法(包括scHiCluster、Raw PCA、HiCRep+MDS、Decay)进行了性能对比。在对比过程中,使用了Adjusted RandIndex(ARI)、Adjusted Mutual Information(AMI)、homogeneity(HM)和Fowlkes Mallows(FM)这五个常用的聚类评价指标来评估5种聚类方法的性能;使用的数据集是下载自GEO数据库的Ramani数据集。Ramani数据集是一个典型的Hi-C数据集,该数据集共包含ML1、ML2、ML3、ML4、PL1和PL2六个部分。六个部分中共包含GM12878、HAP1、HeLa和K562四种人类细胞系,经过筛选后共包含2655个细胞的信息。从Ramani数据集中提取出了七个不同规模的子数据集,并将它们作为对比实验的数据集,它们所包含的不同类型细胞的数量如表1所示。
表1
Ramani数据集的ML1和ML3部分被scHiCluster用于人类细胞的聚类分析,因此,首先使用ML1和ML3中包含的人类细胞数据比较五种聚类方法的性能。在数据预处理后,从ML1和ML3中共筛选出626个人类细胞。图2为五种聚类方法在ML1和ML3的626个人类细胞数据上的聚类性能比较示意图,从图2可以看出,本发明在AMI、ARI、HM和FM四个指标上都超过了其他四种聚类方法,表现出卓越的性能。此外,scHiCSC的ARI、AMI、HM和FM分别为0.872、0.878、0.818和0.92,比次优的聚类方法scHiCluster高出5.4%、6.7%、6.6%、2%和4.2%。
为了对比五种聚类方法在大规模数据集上的性能,从Ramani数据集的六个部分中筛选了2655个人类细胞的数据,并在该数据集上进行了对比,图3为五种聚类方法在包含2655个人类细胞的Ramani数据集上的聚类性能比较示意图。
为了验证scHiCSC在不同尺度数据集上的泛化性,从Ramani数据集中随机选择了800、1000、1200、1400和1600个细胞的数据,并将五种聚类方法的泛化能力进行了对比。图4(a)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于ARI评价指标的性能比较示意图;图4(b)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于AMI评价指标的性能比较示意图;图4(c)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于HM评价指标的性能比较示意图;图4(d)为五种聚类方法在800、1000、1200、1400、1600细胞数上基于FM评价指标的性能比较示意图。从图4(a)至图4(d)中可以看出,与其他四种聚类方法相比,本发明提出的聚类方法scHiCSC具有出色的泛化能力。
不同类型的细胞数量在实际聚类应用中是不同的。细胞数量较多的细胞类型具有明显的特征并且易于识别,而细胞数量较少的细胞类型则难以识别。因此,比较了本发明提出的聚类方法scHiCSC与其他四种聚类方法对细胞数量较少的细胞类型的识别能力。
表2为五种聚类方法在不同尺度数据集上对GM12878细胞的识别效果,从表2中可以看出,现有的四种聚类方法scHiCluster、Raw_PCA、HiCRep+MDS和Decay都无法有效地从HAP1、K562和HeLa中识别出GM12878。那是因为GM12878中的细胞数量远远小于其他三种类型的细胞,当细胞总数较多时,GM12878容易与其他三种类型的细胞混淆。然而,本发明提出的聚类方法scHiCSC可以在一定程度上识别出GM12878。这表明scHiCSC对细胞数目较少的细胞类型也具有较好的识别效果。
表2
实施例3
一种基于接触数权重平滑和特征融合的单细胞Hi-C聚类系统,包括:
数据预处理模块,被配置为;以给定分辨率将单细胞Hi-C数据中的染色体相互作用数据标准化为染色体接触矩阵;
基于接触数权重的平滑模块,被配置为;染色体接触矩阵中的每一行代表染色体片段与其所有邻居之间的相互作用信息;使用权重矩阵来量化不同邻居片段对目标染色体片段的影响;
重启随机游走平滑模块,被配置为;使用重启随机游走算法对基于接触数权重的平滑处理后的染色体接触矩阵进行平滑处理;
接触二值化模块,被配置为;对重启随机游走平滑处理后的矩阵进行二值化处理;
主成分提取模块,被配置为;使用KPCA进行降维并生成细胞嵌入;
特征融合模块,被配置为;通过KPCA生成的细胞嵌入的每一行代表一个细胞中的染色体结构信息;通过细胞嵌入计算细胞之间的欧式距离,并构造距离矩阵作为细胞嵌入的补充;通过行合并的方式合并距离矩阵和使用KPCA进行降维并生成细胞嵌入处理后得到的矩阵,从而形成新的特征矩阵;
谱聚类模块,被配置为;使用谱聚类算法实现细胞聚类。
- 一种基于闪电接触过程算法的特征选择方法及系统
- 一种基于多特征融合的空间图像检索系统及检索方法
- 一种融合多种特征权重的短文本聚类方法
- 基于自适应权重特征的多源降水数据动态融合方法及系统