用于制备卡前列素和卡前列素缓血酸胺的方法和中间物,以及由其制得的卡前列素缓血酸胺
文献发布时间:2023-06-19 18:35:48
技术领域
本发明关于用于制备卡前列素(Carboprost)及卡前列素缓血酸胺的新颖方法及中间物,及由其制得的卡前列素缓血酸胺的新颖高熔点晶体。
背景技术
如以下流程A中所展示的卡前列素及卡前列素缓血酸胺(INN,商品名Hemabate,Tham)两者均为具有催产素特性的PGF2α(特定言的,其为15-甲基-PGF2α)的合成前列腺素类似物。卡前列素及卡前列素缓血酸胺可在妊娠早期诱导宫缩且引发流产,且亦可减少产后出血。
流程A
尽管当前法规愈来愈严格地限制活性医药成分(API)中杂质或不纯物的含量,几乎所有市售卡前列素及卡前列素缓血酸胺仍然含有约3%的5,6-反式异构体与约2%的15(R)-差向异构体。以下流程B展示出卡前列素及其异构体的化学结构,亦即5,6-反式卡前列素及15-差向异构体卡前列素。然而,工业中用于大批量产卡前列素或卡前列素缓血酸胺的方法具有一些待解决的问题,尤其在构筑C5-C6位置处的顺式-双键及卡前列素或卡前列素缓血酸胺的C15位置处的三级醇的空间定向方面。此外,现今用于移除卡前列素或卡前列素缓血酸胺的杂质的纯化方法并非有效且必须进行改良。
流程B
C15-(R/S)-选择性
卡前列素缓血酸胺为Upjohn的原始产物。卡前列素缓血酸胺的首次规模化合成由Upjohn的化学家(Yankee等人,J.Am.Chem.Soc,96(18),5865-5876,1974)揭示。如以下流程C(1)中所展示,15-甲基取代基由式a的苯甲酰基γ-内酯-烯酮与三甲基铝或与甲基溴化镁构筑。然而,在两种情况下15(S)-产物的选择性仅为50%,意谓此方法不具有选择性。WO2008/081191揭示式b的三乙基硅基γ-内酯-烯酮及甲基氯化镁的烷化可获得最高选择性为70%的15(S)-产物,如以下流程C(2)中所展示。WO 2017/093770揭示式c的对苯基苯甲酰基γ-内酯-烯酮及甲基溴化镁的烷化可获得选择性仅为55%的15(S)-产物,且其亦揭示使用各种对掌性添加剂以便增加15(S)-产物的选择性,且发现(S)-Taddol的添加可将选择性增加至70%,如以下流程C(3)中所展示。然而,WO 2017/093770中揭示的最高选择性至多与WO 2008/081191中揭示的选择性相同。
流程C
C5,6-(反式/顺式)-选择性
WO 2008/081191揭示如由Yankee等人所揭示在环境温度下及在二甲亚砜(DMSO)溶剂中进行威悌反应(Wittig reaction)将产生6%至8%的非所需5,6反式-异构体,如以下流程D(1)中所展示。WO 2008/0181191进一步揭示低温(亦即介于5℃至+5℃)下的反应可将5,6反式-异构体的含量降低至约3%,如以下流程D(2)中所展示。鉴于以上,由于所有市售的卡前列素及卡前列素缓血酸胺均含有约3%的5,6-反式异构体,因此似乎改变威悌反应的反应条件对于进一步降低自其产生的5,6-反式异构体的含量方面是没有用处的。
流程D
藉由纯化卡前列素甲酯来移除15(R)-差向异构体及反式异构体
如由Yankee等人及在Eur,J,Pharm,Sci,3,27-38(1995)中所揭示,卡前列素的C15位置处的烯丙基三级醇极不稳定;因此,仅少量酸或极少热就会引起经由差向异构快速地产生大量15(R)-差向异构体。由于此原因,现今于工业中大批量产卡前列素或卡前列素缓血酸胺的方法并不会在前段制程所形成的中间物中构筑烯丙基三级醇的立体化学,此因烯丙基三级醇的立体化学难以维持至最终阶段。因此,几乎现今所有工业中大批量产卡前列素或卡前列素缓血酸胺的方法都直到形成最终产物的结构,或在后段制程所形成的中间物中构筑烯丙基三级醇的立体化学,或在形成最终产物后移除非所需15(R)-差向异构体及反式异构体。
尽管最终产物卡前列素缓血酸胺在室温下为结晶固态,但仍极难以仅藉由结晶纯化同时有效移除15(R)-差向异构体及反式异构体。另外,由于卡前列素的极性极大,因此更难以藉由层析方式自卡前列素移除15(R)-差向异构体及反式异构体。
因此,无法有效地移除在卡前列素或卡前列素缓血酸胺制备期间所产生的异构体,且在先前技术方法中不存在任何适合用于分离且移除异构体的后期制程中间物。为了纯化最终产物,需要两个额外步骤来产生适合用于分离且移除异构体的后期制程中间物。如习知参考文献,例如J,Am.Chem.Soc.,96(18),5865-5876,1974、WO 2008/081191、WO2017/093770、CN 111777537及CN 102816099中所揭示,卡前列素必须经酯化以形成卡前列素甲酯,接着藉由层析移除卡前列素甲酯的15(R)-差向异构体及反式异构体,且将卡前列素甲酯水解成具有更高纯度的卡前列素,但此方法中的产率及纯度极不符合要求。CN1136938 C揭示使用昂贵且特定模拟移动床(simulated moving bed;SMB)层析来分离并移除卡前列素甲酯的异构体,但最终产物的纯度仍然不够好。CN 102816099揭示使用分析级HPLC管柱层析填充(5μm)进行分离,但仍无法完全地移除反式异构体。另外,由于CN102816099揭示方法的分离量较小,因此此方法难以用于工业级大批量产。
发明内容
鉴于以上,需要研究及发展一种用于制备高纯度卡前列素或卡前列素缓血酸胺的方法,其可显著地减少包括15(R)-差向异构体及反式异构体的杂质或异构体的形成,且亦可有效地移除所产生的杂质或异构体。
本发明的目的为提供一种用于形成卡前列素或卡前列素缓血酸胺的有效方法。所述方法包括大环内酯化步骤,其可有效地移除5,6-反式异构体。所述方法亦包括大环内酯-烯酮的甲基化步骤,以用于构筑卡前列素的C15位置处的三级醇的定向。在所有前例中,大环内酯-烯酮的甲基化的15(S)-选择性比γ-内酯-烯酮的甲基化的选择性高得多。本发明亦提供一种可有效地移除所有异构体且可更有效地产生卡前列素及卡前列素缓血酸胺的新颖纯化方法,藉此形成具有高纯度、高熔点及良好稳定性的最终产物。
本发明的一方面提供一种由含有约1%至10%的5,6-反式异构体的式1a化合物制备含有不超过约1%总异构体的高纯度卡前列素或卡前列素缓血酸胺的方法:
其中
本发明另提供一种由含有约1%至10%的5,6-反式异构体的式1b的化合物制备含有不超过约1%总异构体的高纯度卡前列素或卡前列素缓血酸胺的方法,
其中P
本发明另提供一种用于纯化卡前列素的方法,其将含有大量异构体(诸如约1%至10%的5,6-反式异构体与约1%至50%的15(R)-差向异构体)的低纯度卡前列素,有效地纯化为含有不超过约1%总异构体的高纯度卡前列素。
本发明另提供一种式2a'的大环内酯-烯酮中间物,其为用于制备卡前列素或卡前列素缓血酸胺的新颖中间物,
其中P
本发明另提供一种由式2a'的大环内酯-烯酮与甲基化试剂制备式3的大环内酯三级醇的方法,其为用于制备卡前列素或卡前列素缓血酸胺的中间物,
其中P
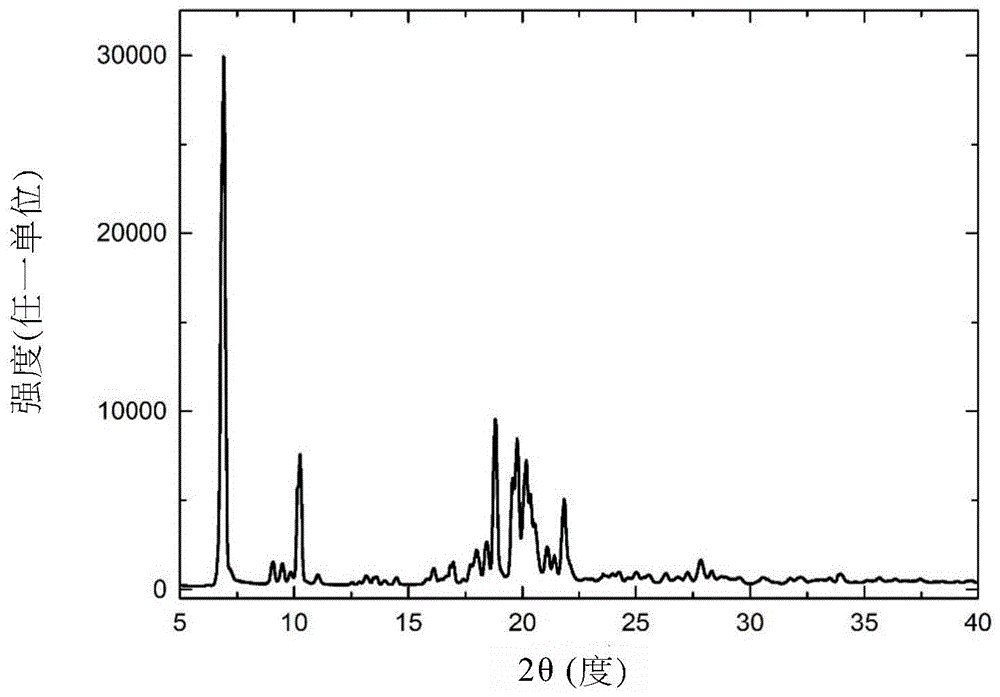
本发明另提供一种卡前列素缓血酸胺的高熔点(106.4±1℃)晶体,其具有展现在以下2θ反射角处的特征峰的X射线粉末绕射(XRPD)图:6.9±0.2°、10.3±0.2°、18.8±0.2°及21.9±0.2°。
本发明另提供一种用于制备卡前列素缓血酸胺的高熔点晶体的方法,其包含使用无水乙腈的溶剂。
图1展示本发明的卡前列素缓血酸胺晶体的X射线粉末绕射(XRPD)图。
图2展示本发明的卡前列素缓血酸胺晶体的差示扫描热量测定(DSC)热谱图。
图3展示根据本发明的五个不同批次的卡前列素缓血酸胺晶体的X射线粉末绕射(XRPD)图。
图4展示根据本发明的五个不同批次的卡前列素缓血酸胺晶体的差示扫描热量测定(DSC)热谱图。
图5展示由本发明的实例9制备的卡前列素缓血酸胺晶体的差示扫描热量测定(DSC)热谱图。
图6展示由本发明的实例10制备的卡前列素缓血酸胺晶体的差示扫描热量测定(DSC)热谱图。
具体实施方式
定义
当与术语「包含」结合用于申请专利范围及/或说明书中时,使用词语「一(a)」或「一(an)」可意谓「一个(one)」,但其亦与「一或多个」、「至少一个」及「一个或超过一个」的含义相符。尽管本发明支持仅指替代方案及「及/或」的定义,但除非明确指示为仅指替代方案或除非替代方案相互排斥,否则术语「或」在申请专利范围中的使用用于意谓「及/或」。在本申请案通篇中,术语「约」用于指示值包括装置、用以测定所述值的方法的固有误差变化或研究个体当中存在的变化。
如本说明书及申请专利范围中所用,字组「包含(comprising)」(及包含的任何形式,诸如「包含(comprise)」及「包含(comprises)」)、「具有(having)」(及具有的任何形式,诸如「具有(have)」及「具有(has)」)、「包括(including)」(及包括的任何形式,诸如「包括(includes)」及「包括(include)」)或「含有(containing)」(及含有的任何形式,诸如「含有(contains)」及「含有(contain)」为包括性或开放的且并不排除额外未列出的要素或方法步骤。
在本说明书中给定的化合物的描绘中,楔形粗体键
当在本文中使用时,术语「高纯度卡前列素或卡前列素缓血酸胺」、「高纯度卡前列素」或「高纯度卡前列素缓血酸胺」意谓所引述的卡前列素及/或卡前列素缓血酸胺含有不超过约1%总异构体,优选不超过约0.8%总异构体或不超过约0.5%总异构体,且更佳地不超过约0.3%总异构体。本文中所指示的异构体或杂质或不纯物包括5,6-反式异构体、15(R)-差向异构体及任何其他立体异构体。
当在本文中使用时,关于实质上不含5,6-反式异构体或类似者的术语意谓所引述的化合物含有不超过约0.5%、不超过约0.3%、不超过约0.2%、不超过约0.1%、不超过约0.05%或不超过约0.03%的杂质或异构体(诸如5,6-反式异构体),或含有不可侦测含量的杂质或异构体(诸如藉由HPLC量测的5,6-反式异构体),对于其侦测极限不超过约0.03%。
除非另外规定,否则术语「羟基保护基团」具有习知定义于有机合成化学中的含义,亦即能够保护化合物的羟基或部分以抵抗化学反应的攻击的基团。羟基保护基团的实例包括但不限于甲氧基甲基、甲氧基硫代甲基、2-甲氧基乙氧基甲基、双(2-氯乙氧基)甲基、四氢哌喃基、四氢硫代哌喃基、4-甲氧基四氢哌喃基、4-甲氧基四氢硫代哌喃基、四氢呋喃基、四氢硫代呋喃基、1-乙氧基乙基、1-甲基-1-甲氧基乙基、三苯甲基、烯丙基、苯甲基、经取代的苯甲基、乙酰基、经取代的乙酰基、苯甲酰基、经取代的苯甲基及SiRaRbRc,其中Ra、Rb及Rc各自独立地为未经取代或经取代的烷基或未经取代或经取代的芳基,诸如C
除非另外规定,否则术语「羰基保护基团」具有习知地定义于有机合成化学中的含义,亦即能够保护化合物的羰基或部分以抵抗化学反应的攻击的基团。羰基保护基团的实例包括但不限于二烷基缩酮、二芳烷基缩酮、二乙酰基缩酮、二硫基缩酮、1,3-二
上文所提及的基团中的每一者(诸如烷基及芳基)可视情况经一或多个选自由以下组成的群的取代基取代:卤素、烷基、芳基、烷氧基、芳氧基、硫代烷氧基、硫代芳氧基、烷胺基、芳胺基、氰基、烷氧羰基、芳基羰基、芳基胺基羰基、烷胺基羰基及羰基,或选自由以下组成的群的杂环基:吡啶基、噻吩基、呋喃基、咪唑基、吗啉基、
由式1a或式1b的已知前列腺素中间物合成高纯度卡前列素
根据本发明,高纯度卡前列素或卡前列素缓血酸胺可根据流程1及流程2中所展示的反应来制备:
流程1
流程2
流程1中的式1a化合物,其中
如流程1的步骤(1)及流程2的步骤(1)中展示,大环内酯化反应可涉及羧基或/及羟基官能基的活化。在此途经中,大环内酯化反应包含用适合的试剂初始形成硫酯,所述试剂包括但不限于氯甲硫代酸S-吡啶-2-基酯、2,2'-二吡啶基二硫化物/三苯基膦或4-三级丁基-2-(2-(4-三级丁基-1-异丙基-1H-咪唑-2-基)二硫基)-1-异丙基-1H-咪唑/三苯基膦。
大环内酯化反应可替代地用于在存在或不存在碱或刘易斯酸(Lewis acid)的情况下用适合试剂初始形成混合酸酐。用于形成混合酸酐的适合试剂包括但不限于2,4,6-三氯苯甲酰氯、2-硝基-6-硝基苯甲酸酸酐、对硝基氟甲基苯甲酸酐、对硝基苯甲酸酐及其类似者。适合碱的实例包括4-(二甲胺基)吡啶、吡啶并吡啶、三乙胺、N,N-二异丙基乙胺及异丙基二乙胺。适合路易酸的实例包括Sc(OTf)
大环内酯化反应亦可藉由在适当溶剂中使用缩合试剂及碱来实现。适合的缩合试剂包括但不限于N,N'-二环己基碳二亚胺、2-氯-1-甲基-吡啶鎓碘化物、2-氯-4,5-二氢-1,3-二甲基-1H-咪唑鎓氯化物、N,N-二笨基氯苯基亚甲基氯化铵、三聚氯化氰、1,3-二甲基-2-氯咪唑鎓氯化物、N,N,N,N-四甲基氯甲脒氯化物及其类似者。适合碱的实例包括吡啶、三乙胺、二异丙基乙胺、4-二甲胺基吡啶(DMAP)及其类似者。用于缩合反应的适合溶剂包括二氯甲烷、四氢呋喃及1,2-二氯乙烷及其混合物。
在藉由HPLC或UPLC分析式2a、2a'、2b、3或4的所得化合物时,出乎意料地发现式2a、2a'、2b、3或4的所得化合物含有不超过约0.1%或更少的5,6-反式异构体,此证实大环内酯化反应展现顺式-选择性;亦即式1a或1b的5,6-顺式化合物在大环内酯化反应中占主导性,而式1a或1b的5,6-反式化合物几乎不经历大环内酯化反应。
流程1的步骤(2)涉及藉由移除ω-侧链处的P
举例而言,将其中P
流程1的步骤(2)亦涉及式2a化合物(其中
此外,流程2的步骤(2)涉及藉由移除ω-侧链处的P
流程1的步骤(3)展示其中P
出乎意料地发现,大环内酯-烯酮及甲基格林纳试剂(Grignard reagent)作为甲基化试剂的反应展现高达约65%的15(S)-选择性。然而,如流程A(1)中所展示,γ-内酯-烯酮及甲基格林纳试剂的反应的15(S)-选择性仅为50%。似乎大环内酯的结构比γ-内酯有助于15(S)-选择性。另外,本发明出人意料地发现,使用低成本且更适宜的MeLi作为甲基化试剂,大环内酯-烯酮的甲基化可具有约75%或更大的15(S)-选择性,其为极高的且即使在γ-内酯反应中使用对掌性添加剂亦无法实现的选择性(WO 2017/093770,使用(S)-Taddol,最高选择性仅为70%)。
流程1的步骤(4)及流程2的步骤(3)涉及式3的大环内酯三级醇的纯化,以供移除15(R)-差向异构体。通常,分离卡前列素的异构体在大批量产卡前列素中为最昂贵及耗时的步骤。然而,分离本发明的异构体的成本比先前技术技艺低得多,例如J,Am.Chem.Soc.,96(18),5865-5876,1974;WO 2008/081191;WO 2017/093770;CN 111777537及CN102816099A。原因如以下列出:
(a)在先前技术技艺中,卡前列素必须经酯化以形成中间物卡前列素甲酯,其适合用于移除异构体,且接着卡前列素甲酯的异构体可藉由层析纯化移除。然而,出乎意料地发现,式3的后段制程中间物为适合用于移除异构体的中间物;因此在本发明中不必使用额外酯化反应。
(b)当使用层析纯化来移除卡前列素中间物的5,6-反式异构体及15(R)-差向异构体时,5,6-反式异构体(HPLC,0.93)通常比15(R)-差向异构体(HPLC,RRT 0.88)更难以分离。CN 102816099甚至使用利用填料(5μm)的分析级HPLC来移除卡前列素甲酯的5,6-反式异构体(RRT 0.93)。相反,在大环内酯化后形成的式3的中间物以及式2a'的化合物实质上不含5,6-反式异构体,因此,使用工业大批量产的一般硅胶管柱层析就可简单用于分离更易于移除的15(R)-差向异构体。
(c)本发明的大环内酯-烯酮的甲基化展现更高的选择性,因此所产生15(R)-差向异构体的量较少且可更易于移除。
在流程1的步骤(4)及流程2的步骤(3)中,层析纯化可使用酯类、醚类、酮类或卤代溶剂或类似者或其混合物来进行。对于使用二氯甲烷及丙酮的混合物,亦可用乙酸乙酯、乙酸异丙酯、甲基三级丁基醚、丙酮、甲基乙基酮或其混合物实现良好分离。藉由层析,非所需15(R)-差向异构体的量可减小至规定极限,亦即<约0.5%、<约0.3%、<约0.2%、<约0.1%或更小。
如流程1的步骤(5)及流程2的步骤(4)中所展示,藉由用氢氧化锂溶液处理以将式3的大环内酯三级醇水解成含卡前列素的甲醇溶液。酸化以获得卡前列素必须快速地执行,以避免酸性培养基中的差向异构化反应。
因此,本发明提供一种用于制备含有不超过1%总异构体的卡前列素的方法,所述方法包含以下步骤:
(1)使含有1%至10%的5,6-反式异构体的式1a化合物大环内酯化
其中
其中
(2)当
其中P
(3)用甲基化试剂使式2a'化合物甲基化,以形成式3化合物:
其中P
(4)层析分离式3化合物以移除异构体;
(5)水解式3化合物,以形成式4化合物:
其中P
(6)当P
本发明亦提供一种用于制备含有不超过1%总异构体的高纯度卡前列素的方法,所述方法包含以下步骤:
(1)使含有1%至10%的5,6-反式异构体及1%至50%的15(R)-差向异构体的式1b化合物大环内酯化,
其中P
其中P
(2)当P
(3)层析分离式3a化合物,其中P
(4)水解式3a化合物,以形成式4化合物:
其中P
(5)当P
纯化含有过量异构体的低纯度卡前列素
自习知反应或由于差向异构化反应的过度反应获得的粗制卡前列素通常含有大量或过量异构体。本案发明人发现,含有过量异构体的卡前列素可藉由使用本发明的方法进一步纯化。本发明的方法包含提供含有至少约1%至10%的5,6-反式异构体及约1%至50%的15(R)-差向异构体的低纯度卡前列素;使低纯度卡前列素大环内酯化以形成大环内酯三级醇;层析分离大环内酯三级醇的过量异构体;及水解大环内酯三级醇,以形成含有不超过约1%总异构体的高纯度卡前列素。
因此,本发明提供一种用于纯化卡前列素的方法,其包含以下步骤:
(1)使含有1%至10%的5,6-反式异构体及1%至50%的15(R)-差向异构体的低纯度卡前列素大环内酯化,以形成式3b化合物:
(2)层析分离式3b的化合物的异构体;及
(3)水解式3b的化合物,以形成含有不超过1%总异构体的高纯度卡前列素。
与缓血酸胺形成卡前列素盐
为了自卡前列素形成卡前列素缓血酸胺,本发明方法进一步包含与缓血酸胺(tromethamine)形成盐,及卡前列素缓血酸胺的结晶。盐形成及结晶步骤可遵循CN102336693或WO 2017/093770中所揭示的方法。大体而言,卡前列素可首先溶解于适合的溶剂中,诸如丙酮、乙腈、甲醇、乙醇、异丙醇及其组合;接着可将含缓血酸胺的适合溶剂(诸如水、甲醇、乙醇、异丙醇及其组合)添加至溶液中;且可将混合物加热至约85℃持续约1小时,以形成均质溶液。随后,可使均质溶液冷却至约60℃,且白色固体缓慢地开始沈淀。可将反应混合物进一步冷却至室温,且接着过滤以获得卡前列素缓血酸胺的白色晶体。由此获得的白色晶体的熔点通常约95℃至105℃,如最初由Pfizer所揭示者(例如,由Pfizer提供的
在CN 102336693的实例1的情况下,在降低温度期间自100ml乙腈及0.5ml水的混合物沈淀约1.20g卡前列素缓血酸胺,且白色晶体的熔点经量测为103.97℃(参见图2)。本案发明人重复CN 102336693的实例1且发现所获得卡前列素缓血酸胺晶体的熔点经量测为103.41℃。
在WO 2017/093770的实例1g的情况下,自异丙醇及丙酮的混合物沈淀约593g卡前列素缓血酸胺。WO 2017/093770未揭示自实例1g获得的卡前列素缓血酸胺晶体的熔点。本案发明人重复WO 2017/093770的实例1g且发现所获得卡前列素缓血酸胺晶体的熔点经量测为97.49℃。
上述已知卡前列素缓血酸胺晶体的熔点范围介于约97℃至约104℃范围内,其实际上在如最初由Pfizer提供的范围(95℃至105℃)内。
卡前列素缓血酸胺的再结晶
卡前列素缓血酸胺上的再结晶可对减少15(R)-异构体的含量具有影响。卡前列素缓血酸胺的再结晶方法已揭示于CN 102336693及WO 2017/093770中。本案发明人重复CN102336693所揭示的使用水与丙酮的方法,且发现再结晶的卡前列素缓血酸胺具有如所量测的103.85℃的熔点。本案发明人亦重复WO 2017/093770所揭示的使用异丙醇与丙酮的方法,且发现再结晶的卡前列素缓血酸胺具有如所量测的99.46℃的熔点。
出乎意料地发现,可藉由使用无水乙腈的特定溶剂的卡前列素缓血酸胺的再结晶方法来获得特定高熔点卡前列素缓血酸胺晶体。由此形成的高熔点卡前列素缓血酸胺晶体具有包含峰最大值为106.4±1.0℃的吸热峰的差示扫描热量测定(DSC)热谱图,所述峰最大值明显高于最初由Pfizer提供的熔点范围及先前技术中所揭示的上述卡前列素缓血酸胺晶体的熔点范围的上限。
制备高熔点卡前列素缓血酸胺晶体
本发明提供一种用于制备卡前列素缓血酸胺的高熔点结晶形式的方法,其包含以下步骤:
a.将卡前列素缓血酸胺添加至无水乙腈中以形成混合物,其中所述无水乙腈的量为每1g卡前列素缓血酸胺约80ml至250ml;
b.将所述混合物加热至范围介于约70℃至90℃的温度以得到均质溶液;
c.使所述均质溶液冷却以形成卡前列素缓血酸胺的所述结晶形式;及
d.视情况分离结晶产物。
在一些实施例中,可将混合物加热至约80℃以完全地溶解卡前列素缓血酸胺;接着可缓慢地冷却均质溶液,且白色固体缓慢地开始沈淀;且可将混合物进一步冷却至室温,且接着过滤并干燥,以获得卡前列素缓血酸胺的结晶形式。
在一些实施例中,步骤a中使用的无水乙腈的量范围介于每1g卡前列素缓血酸胺约80ml至250ml、约100mL至220ml或约150ml至200ml;无水乙腈的含水量(w/w)小于约0.05%、小于约0.01%、小于约0.005%或小于约0.001%;且所述方法优选在不添加水的情况下进行。由所述方法制备的卡前列素缓血酸胺的结晶形式具有包含峰最大值为106.4±1.0℃的吸热峰的差示扫描热量测定(DSC)热谱图,所述峰最大值高于卡前列素缓血酸胺的另一已知结晶形式。由于藉由本发明获得的卡前列素缓血酸胺晶体与所有其他已知卡前列素缓血酸胺晶体相比具有最高熔点,因此其为卡前列素缓血酸胺的最稳定结晶形式。
此外,本发明的高熔点卡前列素缓血酸胺晶体具有展现在以下2θ反射角处的特征峰的X射线粉末绕射(XRPD)图:6.9±0.2°、10.3±0.2°、18.8±0.2°及21.9±0.2°,其显然不同于CN 102336693中所揭示的晶体的2θ反射角:6.6±0.2°、9.9±0.2°、18.5±0.2及21.6±0.2°。表示高熔点卡前列素缓血酸胺晶体为卡前列素缓血酸胺的新颖结晶形式。
在本发明的一个实施例中,卡前列素缓血酸胺晶体具有展现在以下2θ反射角处的特征峰的XRPD图:6.9±0.2°、10.3±0.2°、18.8±0.2°及21.9±0.2°。在一优选实施例中,XRPD图进一步包含在以下2θ反射角处的特征峰:9.1±0.2°、9.5±0.2°、11.0±0.2°及20.5±0.2°。更佳地,卡前列素缓血酸胺晶体的XRPD图与图1一致。卡前列素缓血酸胺晶体的特定资料展示于表1中。
表1
在一个实施例中,本发明提供具有实质上如图1中所展示的XRPD图的卡前列素缓血酸胺的结晶形式。
在一个实施例中,本发明提供五个不同批次(a)至(e)的卡前列素缓血酸胺晶体的XRPD图,如图3中所展示。四个主要经分离特征峰的特定数据清晰地标记且展示于表2中。特征峰的平均位置分别位于约6.9°、10.3°、18.8°及21.9°处。
表2
与由CN102336693所揭示的6.6±0.2°、9.9±0.2°、18.5±0.2°及21.6±0.2°相比,本发明的卡前列素缓血酸胺晶体的XRPD特征峰位置位于更高的2θ(2-theta)角(6.9±0.2°、10.3±0.2°、18.8±0.2°及21.9±0.2°)处。由CN 102336693所揭示的卡前列素缓血酸胺的6.621°处的特征峰表示
在一个实施例中,本发明提供具有DSC热谱图的卡前列素缓血酸胺晶体,所述DSC热谱图包含峰起始温度为大约103.9℃且峰最大值为大约106.4±1.0℃的吸热峰。在一优选实施例中,本发明提供具有实质上如图2中所展示的DSC热谱图的卡前列素缓血酸胺的结晶形式。
在一个实施例中,本发明提供五个不同批次(a)至(e)的卡前列素缓血酸胺晶体的DSC热谱图,如图4中所展示。单一吸热峰的峰最大值的特定数据清晰地标记且展示于表3中。此等吸热峰的平均峰最大值为约106.4℃。
表3
本发明的卡前列素缓血酸胺晶体的约106.4℃的DSC峰最大值温度远超出95℃至105℃的卡前列素缓血酸胺的所揭示熔点(如最初由Pfizer所提供)及CN 102336693中所揭示的103.97℃的峰最大值温度,从而指示与先前技术相比,本发明的卡前列素缓血酸胺晶体为具有更高热稳定性的新颖晶体。熟习此项技术者熟知包含最高熔点的晶体为热力学中的最稳定结晶形式;因此,与先前技术相比,本发明的新颖卡前列素缓血酸胺晶体为最稳定的一者。
美国药典(The United States Pharmacopeia)建议卡前列素缓血酸胺应储存于冰箱(-20℃)中。相反,本发明证明,即使在20℃下经历6个月,在5℃下经历18个月及在80℃下经历3天,卡前列素缓血酸胺的高熔点结晶形式亦为极稳定的。因此,本发明的卡前列素缓血酸胺晶体的稳定性得到显著改良。
尽管在所述范围内的最小值及最大值前均加上词语「约」,但除非另外明确指示,否则在本申请案中指定的多个定量值中的数值的使用陈述为近似值。以此方式,所述数值的微弱变化可用于实现与所述数值实质上相同的结果。此外,本揭示案的范围意欲为连续的范围,包含在所述最小值与最大值之间的每一数值以及可藉由此类数值所形成的任何范围。亦揭示于本文中的为可藉由将所陈述数值划分成任何其他所陈述数值所形成的任何及全部比率(及任何此模拟率的范围)。相应地,熟习此项技术者将了解许多所述等比率、范围及比率的范围可明显的来源于本文中所提出的数值,且在全部实例中所述等比率、范围及比率的范围代表本发明的多个实施例。
本文中所揭示和主张的所有化合物和/或方法可根据本发明在无不当实验的情况下制成和执行。虽然已根据优选实施例描述本发明的化合物和方法,但熟习此项技术者应清楚变化可在不背离本发明的概念、精神和范畴的情况下应用于本文所描述的组合物和/或方法中和步骤中或方法的步骤顺序中。对熟习此项技术者显而易见的所有此类类似取代和修改均视为在由随附权利要求书所定义的本发明的精神、范畴和概念内。
实例
X射线粉末绕射(XRPD)分析:在具有固定发散狭缝及1D LYNXEYE侦测器的BrukerD2 PHASER绕射仪上收集XRPD图。将样品(大约100mg)平坦置放于样品固持器上。使用功率为10mA及30kV的CuK
差示扫描热量测定(DSC)分析:在TA DISCOVERY DSC25仪器上收集DSC热谱图。将样品称取至具有卷边封闭的铝盖的铝盘中。在氮气流(大约50ml/min)下以10℃/min的扫描速率自25℃至150℃分析所制备的样品。在量测的前藉由铟(In)来校准熔融温度及熔化热。藉由本发明的DSC量测期间吸热峰的峰最大值来测定所有样品的熔点。
实例1
(8aR,9R,10R,11aS,Z)-10-((三级丁基二甲基硅基)氧基)-9-((S,E)-3-((三级丁基二甲基-硅基)氧基)辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛(oxecin)-2(3H)-酮
在环境温度下在氮气下,将1.4kg的7-((1R,2R,3R,5S)-3-((三级丁基二甲基硅基)氧基)-2-((S,E)-3-((三级丁基二甲基硅基)氧基)辛-1-烯-1-基)-5-羟基环戊基)庚-5(Z)-烯酸(其包括藉由在HPLC分析中侦测其去保护产物所测定的约6.5%的5,6-反式异构体)及475g的吡啶溶解于6.5L的二氯甲烷中。接着,将843g苯甲酰氯添加至混合物中且搅拌一个小时。藉由TLC检查反应混合物以确认反应完成。将混合物用6L的饱和碳酸氢钠水溶液淬灭且搅拌10分钟。静置溶液以分离成两个相,且收集有机层。进一步蒸发掉有机层。将经浓缩残余物用6L的甲苯稀释,且分别用0.1N氢氯酸水溶液及盐水洗涤。收集且蒸发掉有机层以获得粗硅基保护的1,9-内酯化合物。使用己烷及乙酸乙酯的混合物作为梯度溶离剂藉由硅胶层析进一步纯化粗化合物。所获得的硅基保护的1,9-内酯的产率为1.0kg(74%)。
将0.1g产物进一步用水解反应及去保护反应处理以获得粗产物。HPLC分析展示,对于粗化合物未侦测到5,6-反式异构体。
实例2
(8aR,9R,10R,11aS,Z)-9-((S,E)-3-((三级丁基二甲基硅基)氧基)辛-1-烯-1-基)-10-((四-氢-2H-哌喃-2-基)氧基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮
在环境温度下在氮气下,将2.0g的7-((1R,2R,3R,5S)-2-((S,E)-3-((三级丁基二甲基硅基)氧基)辛-1-烯-1-基)-5-羟基-3-((四氢-2H-哌喃-2-基)氧基)环戊基)庚-5(Z)-烯酸(其包括藉由在HPLC分析中侦测其去保护化合物所测定的1.8%的5,6-反式异构体)及0.72g的吡啶溶解于20ml的二氯甲烷中。接着,将1.27g苯甲酰氯添加至混合物中且搅拌30分钟。藉由TLC检查反应混合物以确认反应完成。将混合物用15ml的饱和碳酸氢钠水溶液淬灭且搅拌10分钟。静置溶液以分离成两个相,且收集有机层。蒸发掉有机层,且获得粗乙醚保护的1,9-内酯化合物。使用己烷及乙酸乙酯的混合物作为梯度溶离剂藉由硅胶层析来纯化粗化合物。所获得的乙醚保护的1,9-内酯的产率为1.35g(70%)。
将0.1g产物进一步用水解反应及去保护反应处理以获得粗产物。HPLC分析展示,对于粗化合物未侦测到5,6-反式异构体。
实例3
(8aR,9R,10R,11aS,Z)-9-((E)-3-侧氧基辛-1-烯-1-基)-10-((四氢-2H-哌喃-2-基)氧基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮
在环境温度下在氮气下,将1.5g的7-((1R,2R,3R,5S)-5-羟基-2-((E)-3-侧氧基辛-1-烯-1-基)-3-((四氢-2H-哌喃-2-基)氧基)环戊基)庚-5(Z)-烯酸及0.68g的吡啶溶解于15ml的二氯甲烷中。接着,将1.21g苯甲酰氯添加至混合物中且搅拌30分钟。藉由TLC检查反应混合物以确认反应完成。将混合物用10ml的饱和碳酸氢钠水溶液淬灭且搅拌10分钟。静置溶液以分离成两个相,且收集有机层。蒸发掉有机层,且获得粗1,9-内酯化合物。使用己烷及乙酸乙酯的混合物作为梯度溶离剂藉由硅胶层析来纯化粗化合物。所获得的1,9-内酯的产率为1.02g(71%)。
实例4
(8aR,9R,10R,11aS,Z)-10-羟基-9-((S,E)-3-羟基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮
在环境温度下,将480g(8aR,9R,10R,11aS,Z)-10-((三级丁基二甲基硅基)氧基)-9-((S,E)-3-((三级丁基二甲基-硅基)氧基)辛-1-烯-1-)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮(来自实例1)溶解于4.5L的四氢呋喃中。接着,将775g四丁基氟化铵三水合物添加至溶液中。在45℃下加热均质溶液且搅拌6小时。藉由TLC检查反应混合物以确认反应完成。在环境温度下冷却混合物且随后用8L的饱和碳酸氢钠水溶液淬灭。静置溶液以分离成两个相,且收集有机层。蒸发掉有机层以获得粗1,9-内酯二醇化合物。使用己烷及乙酸乙酯的混合物作为梯度溶离剂藉由硅胶层析来纯化粗化合物。接着,使二醇化合物在乙酸乙酯及己烷的混合溶剂中进一步结晶。二醇化合物的所获得晶体的产率为259g(91%)。
实例5
(8aR,9R,10R,11aS,Z)-10-羟基-9-((E)-3-侧氧基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮
在环境温度下,将300g的(8aR,9R,10R,11aS,Z)-10-羟基-9-((S,E)-3-羟基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环-戊[b]奥克辛-2(3H)-酮(来自实例4)溶解于3L的四氢呋喃中,且添加2,3-二氯-5,6-二-氰基-1,4-苯并-醌。在55℃下加热反应混合物且搅拌2小时。在反应完成后,在环境温度下冷却混合物且蒸发掉。接着,用二氯甲烷稀释深棕色残余物,且沈淀黄色固体。过滤出混合物,且将过滤物进一步浓缩以获得粗酮化合物。使用己烷及乙酸乙酯的混合物作为梯度溶离剂藉由硅胶层析来纯化粗化合物。所获得的15-酮1,9-内酯的产率为276g(93%)。
实例6
(8aR,9R,10R,11aS,Z)-10-羟基-9-((S,E)-3-羟基-3-甲基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮
方法A:在环境温度下在氮气下,将275g的(8aR,9R,10R,11aS,Z)-10-羟基-9-((E)-3-侧氧基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮(来自实例5)溶解于4.5L的四氢呋喃中且在-70℃下冷却。在-70℃下将1.0L的甲基锂(2M于醚中)缓慢添加至反应混合物且藉由TLC检查反应。在反应完成后,将混合物用饱和氯化铵水溶液淬灭且搅拌10分钟。接着,将1L的乙酸乙酯添加至反应混合物中且使其在环境温度下升温。静置混合物以分离成两个相,且收集有机层并经无水硫酸钠干燥。随后,滤出固体且蒸发掉过滤物。获得粗15-甲基混合物化合物且藉由HPLC测试。粗化合物的15-R/15-S的比率为约25/75。使用二氯甲烷及丙酮的混合物作为梯度溶离剂藉由硅胶层析进一步纯化粗化合物。所获得的纯15-甲基化合物的产率为163g(57%)。
方法B:在环境温度下在氮气下,将2g的(8aR,9R,10R,11aS,Z)-10-羟基-9-((E)-3-侧氧基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮(来自实例5)溶解于20ml的四氢呋喃中且在-70℃下冷却。接着,在-70℃下将21ml的甲基溴化镁(1M于THF中)缓慢添加至反应混合物且在0℃下升温。藉由TLC检查混合物以确认反应完成。取样反应混合物以检查产物的15-R/15-S的比率为约35/65。
实例7
(8aR,9R,10R,11aS,Z)-10-((三级丁基二甲基硅基)氧基)-9-((S,E)-3-羟基-3-甲基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮
在环境温度下,2.0g的(8aR,9R,10R,11aS,Z)-10-((三级丁基二甲基硅基)氧基)-9-((E)-3-侧氧基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮于20ml的四氢呋喃中且在-70℃下冷却。在-70℃下将3.5ml的甲基锂(2M于乙醚中)缓慢添加至反应混合物且藉由TLC检查反应。在反应完成后,将混合物用饱和氯化铵水溶液淬灭且搅拌10分钟。接着,将1ml的乙酸乙酯添加至反应混合物中且使其在环境温度下升温。相分离混合物,且经无水硫酸钠干燥有机层。随后,滤出固体,且蒸发掉过滤物以获得粗15-甲基混合物化合物。使用二氯甲烷及丙酮的混合物作为梯度溶离剂藉由硅胶层析来纯化粗化合物。所获得的纯15-甲基化合物的产率为1.05g(51%)。
MS(m/z,EI):C
实例8
(Z)-7-((1R,2R,3R,5S)-3,5-二羟基-2-((S,E)-3-羟基-3-甲基辛-1-烯-1-基)环戊基)庚-5-烯酸,卡前列素
将143g的(8aR,9R,10R,11aS,Z)-10-羟基-9-((S,E)-3-羟基-3-甲基辛-1-烯-1-基)-4,5,8,8a,9,10,11,11a-八氢环戊[b]奥克辛-2(3H)-酮(来自实例6)溶解于750ml的甲醇中,且添加1.5L的1N氢氧化锂水溶液。将反应混合物在60℃下加热2小时。在反应完成后,在室温下冷却混合物,且将溶液的pH值调整至约8.5。浓缩混合物以移除甲醇且藉由酸碱萃取进一步纯化残余物。所获得卡前列素的产率为129g。
MS(m/z,EI):C
实例9
卡前列素缓血酸胺盐形成
2-胺基-2-(羟基甲基)丙烷-1,3-二醇;(Z)-7-((1R,2R,3R,5S)-3,5-二羟基-2-((S,E)-3-羟基-3-甲基辛-1-烯-1-基)环戊基)庚-5-烯酸酯,卡前列素缓血酸胺
方法A:
在60℃下将114g的卡前列素溶解于1.2L的乙腈中,且将溶液加热至65℃。将含37.4g缓血酸胺的120ml水缓慢添加至溶液中。将反应混合物进一步加热至85℃以进行回流持续10分钟。使均质溶液冷却至60℃,且白色固体缓慢地开始沈淀。将混合物进一步冷却至室温且搅拌超过16小时。过滤反应混合物以获得卡前列素缓血酸胺的白色结晶形式(118g)。藉由DSC量测到晶体的熔点为103.41℃,如图5(a)中所展示。
MS(m/z,ESI):C
方法B:
藉由以下CN 102336693的实例3中所揭示的方法由丙酮/水溶液来制备卡前列素缓血酸胺晶体。本案发明人量测到晶体的熔点为103.22℃,如图5(b)中所展示。
方法C:
藉由以下CN 102336693的实例7中所揭示的方法由乙醚/水溶液来制备卡前列素缓血酸胺晶体。本案发明人量测到晶体的熔点为103.43℃,如图5(c)中所展示。
方法D:
藉由以下WO 2017/093770的实例1g中所揭示的方法由异丙醇/丙酮溶液来制备卡前列素缓血酸胺晶体。本案发明人量测到晶体的熔点为97.49℃,如图5(d)中所展示。
实例10
卡前列素缓血酸胺的再结晶
方法A:
在85℃下,将10.0g卡前列素缓血酸胺溶解于1L含有0.02%水的乙腈中以得到均质溶液,且接着将均质溶液冷却至60℃,且白色固体缓慢地开始沈淀。将混合物进一步冷却至室温。过滤及干燥所得沈淀晶体。在再结晶后,所获得的高纯度卡前列素缓血酸胺晶体的产率为9.0g。藉由DSC量测到晶体的熔点为106.40℃,如图6(a)中所展示。在HPLC分析后,15(R)-差向异构体的量为0.05%,且未侦测到5,6-反式异构体。
方法B:
藉由以下CN 102336693的实例2、4、6或8中所揭示的方法由丙酮/水溶液来制备卡前列素缓血酸胺晶体。本案发明人量测到晶体的熔点为103.85℃,如图6(b)中所展示。
方法C:
藉由以下WO2017093770的实例1h中所揭示的方法由异丙醇/丙酮溶液来制备卡前列素缓血酸胺晶体。本案发明人量测到晶体的熔点为99.46℃,如图6(c)中所展示。
实例11
低纯度卡前列素缓血酸胺的纯化
将3g市售卡前列素缓血酸胺(其包括2.5%的5,6-反式异构体及1.5%的15(R)-差向异构体)溶解于pH为3的酸性水中且用乙酸乙酯萃取以获得2g卡前列素。用1.73g的2,2'-二吡啶基二硫化物及2.06g的三苯基膦处理卡前列素于8ml无水无氧二甲苯中的溶液。在25℃下搅拌18小时后,用500ml二甲苯稀释混合物且在回流下加热4小时。进一步蒸发反应混合物以移除溶剂,且将残余物分配于低温碳酸氢钠水溶液与乙酸乙酯之间。将有机层收集且用盐水洗涤,并用无水硫酸钠干燥。过滤混合物且浓缩以获得粗1,9-内酯化合物。使用二氯甲烷及丙酮的混合物作为梯度溶离剂藉由硅胶层析来纯化粗化合物。所获得的15-甲基1,9-内酯的产率为1.67g。
1,9-内酯产物进一步藉由水解反应处理以获得包括2.5%的15(R)-差向异构体但在HPLC分析中无可侦测5,6-反式异构体的卡前列素。卡前列素缓血酸胺是根据实例9的方法由卡前列素及缓血酸胺形成,且根据实例10(方法A)及实例11的方法结晶及再结晶以获得高纯度卡前列素缓血酸胺。在HPLC分析后,15(R)-差向异构体的量为0.15%,且未侦测到5,6-反式异构体。
实例12
卡前列素缓血酸胺晶体的稳定性
表4及表5中所展示的稳定性数据展示,即使在20℃下处理6个月、在5℃下处理18个月及在80℃下处理3天,高熔点卡前列素缓血酸胺晶体亦为稳定的。高熔点卡前列素缓血酸胺晶体在室温下、甚至在更高温度下为极稳定的,使得可有效地避免降解杂质的形成。
表4
表5
另一方面,量测由实例10(方法A)制备的高熔点卡前列素缓血酸胺晶体及由实例10(方法B)制备的低熔点卡前列素缓血酸胺晶体的吸湿性。在25℃下将样品置放于具有99%RH的玻璃瓶中持续3小时,且藉由卡尔费歇尔滴定法(Karl Fischer titration)来量测样品的含水量,如表6中所展示。高熔点卡前列素缓血酸胺晶体展示与低熔点卡前列素缓血酸胺晶体相比的相对低速率的水吸收,从而指示高熔点卡前列素缓血酸胺晶体更稳定且可在高湿度条件下储存更长时间,因此高熔点卡前列素缓血酸胺晶体更有利于产物处置、储存及运送。
表6
虽然已参考说明性实例描述了本发明,但应理解,所属领域的技术人员可易于实现的任何修改或更改将属于本说明书和所附权利要求书的揭示内容的范围内。
- 制备苯并前列环素类似物的方法和中间物,以及由其制得的苯并前列环素类似物
- 用于减脂的比马前列素、比马前列素类似物、前列腺酰胺和前列腺素的持续释放