一类具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物及其制备方法和应用
文献发布时间:2023-06-19 18:37:28
技术领域
本发明属于药物化学合成技术领域,具体涉及一种具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物及其制备方法和应用。
背景技术
杂环化合物数量特别多,广泛分布在在自然界中,在生物的发展、成长等过程中起重大的作用。十九世纪初期,杂环化合物的获得多为天然产物的提取,但是由此得到的化合物产量很低,纯度不高,而且价格昂贵;随着有机合成技术的发展,为了充分利用杂环类化合物,化学工作者开始进行了大规模人工合成杂环类化合物,使其得到了广泛的应用,其中包括生物医药、储能材料、生物模拟材料,高性能染料等。其中含氮氧杂环化合物作为重要的杂环化合物,由于其具有独特的结构从而具有广谱的生物活性及药理活性,例如含氮氧杂环类化合物具有杀菌、抗病毒、抗肿瘤等生物活性,可以作为很多药物的先导化合物骨架,在医药创制领域有着重要的作用。
异噁唑含有相邻的一个氧原子和一个氮原子,被广泛应用于有机合成,是一类非常重要的化合物。而且这类化合物具有较好的药理学特性,在抗炎、抗真菌、抗高血压、抗肿瘤等方面起了较大作用。例如,NVP-AUY922 VIII可抑制人内皮细胞的增殖、化学迁移和管状分化,并具有抗血管生成活性,从而降低肿瘤异种移植瘤中的微血管密度;VER-50589IX在人结肠癌(HCT-116)异种移植瘤中导致肿瘤体积和重量有统计学显著降低(~30%)(Liu,J.;Sun,Y.ACS.Med.Chem.Lett.,2014,5,517;Girisha,M.;Adhikari,A.IndianJ.Chem.-Sect.B.Org.Med.Chem.,2009,48,430;Artym,J.;
但是,具有高抗肿瘤活性的药物仍然极少。不仅仅是因为目前抗肿瘤药物少,合成困难;抗肿瘤药物的研发是一个耗时长、投入高、风险大的过程,一个药物从合成研发到批准上市需要大量的实验测定:单具有抗肿瘤活性还远远不够,需要保证其在具有高抗肿瘤活性的同时,对人体的副作用最小,同能够较好的被吸收等等条件。在满足这些条件的基础上又希望尽可能的能降低合成成本使更多的病人有能力治疗。因此我们迫切希望研发一种具有应用前景的抗肿瘤化合物。鉴于异噁唑类化合物具有非常广泛的应用价值,本发明对其三个位点进行修饰,设计、合成这一类具有新型结构的异噁唑类化合物,并进行了抗肿瘤活性测试。
发明内容
针对现有技术存在的不足,本发明的目的之一在于提供一种具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物。
本发明的目的之二在于提供一种具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物的制备方法,制备工艺简便,易于调节。
本发明的目的之三在于提供一种具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物的应用。
本发明的目的通过以下技术方案实现:
一类具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物,结构通式如下:
其中,所述R
优选的,所述碳原子数1-8的烷基为辛基。
优选的,所述的具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物的结构式如下:
优选的,所述具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物对MiaPaCa-2、BXPC-3、PANC-1、A549、MCF-7、HCT-116、HepG2、NCI-H460、B16中的至少一种具有抑制作用。
上述的具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物的制备方法,反应路线如下:
制备方法包括以下步骤:
(1)采用Route 1反应路线:
(1-1)惰性气氛下,将化合物1和化合物2在催化剂存在的条件下,在有机溶剂中反应,纯化得到中间体酮;
(1-2)所述中间体酮和甲氧基胺盐酸盐、吡啶、无水硫酸钠在有机溶剂中反应,纯化得到化合物3;
(2)当所述异噁唑类化合物的结构通式为式(I)时,采用Route 2反应路线:
A、将化合物3溶于有机溶剂中,在催化剂和1,2-萘醌存在的条件下与化合物4反应,纯化得到异噁唑类化合物式(I);
当所述异噁唑类化合物的结构通式为式(II)时,采用Route 3反应路线:
B1、将化合物3溶于有机溶剂中,在室温条件下与碘单质反应纯化得到中间体5;
B2、将中间体5和化合物6溶于有机溶剂中,在催化剂存在条件下,在惰性气体保护下反应,纯化得到式(II);
当所述异噁唑类化合物的结构通式为式(III)时,采用Route 4反应路线:
C、将化合物3溶于有机溶剂中,在催化剂和氧化剂存在的条件下,加入1-碘-2-(乙烯氧基)苯、碳酸钾和四丁基溴化铵反应纯化得到式(III)。
当所述异噁唑类化合物的结构式为式(IV)时,采用Route 5反应路线:
D、将化合物3和烯丙基溴溶于有机溶剂中,在催化剂存在的条件下,加入四丁基溴化铵,在室温下反应;纯化得到式(IV);
当R
E1、采用Route 2-5合成R
E2、将化合物7和三甲基硅基乙炔加入到有机溶剂中,在催化剂存在的条件下,在惰性气体下反应,得到上了保护基团的中间体8;
E3、将中间体8加入到有机溶剂中,在碱催化的条件下反应,纯化得到R
E4、将化合物7与吗啉、叔丁醇钾和三叔丁基磷加入到有机溶剂中,在催化剂存在的条件下,在惰性气体下反应,纯化得到R
优选的,步骤(1-1)中所述催化剂为双三苯基膦二氯化钯和碘化亚铜;有机溶剂为三乙胺,所述惰性气氛为氮气;所述反应的温度为10-50℃,时间为12-24h;所述化合物1与化合物2的摩尔比为1:(1.2-1.5);
优选的,步骤(1-2)中所述有机溶剂为甲醇;所述反应的温度为室温,时间为12-24h。
优选的,步骤(A)所述有机溶剂为乙二醇二甲醚;所述催化剂为醋酸钯;所述反应的温度为室温,时间为8-12h;
优选的,步骤(B1)所述有机溶剂为二氯甲烷;
优选的,步骤(B2)所述有机溶剂为三乙胺和DMF的混合溶剂;所述催化剂为双三苯基膦二氯化钯和碘化亚铜;所述惰性气体为氮气;所述反应的温度为50-100℃。
优选的,步骤(C)所述有机溶剂为四氢呋喃;所述催化剂为醋酸钯;所述氧化剂为氯化铜;所述反应的温度为30-90℃;
优选的,步骤(D)所述有机溶剂为N,N-二甲基甲酰胺;所述催化剂为醋酸钯。
优选的,步骤(E2)所述有机溶剂为三乙胺;所述催化剂为双三苯基膦二氯化钯和碘化亚铜;所述惰性气体为氮气;所述反应的温度为80-130℃;
优选的,步骤(E3)所述有机溶剂为二氯甲烷和甲醇混合溶剂;所述碱催化剂为碳酸钾;所述反应的温度室温;
优选的,步骤(E4)所述有机溶剂为无水甲苯;所述催化剂为醋酸钯;所述惰性气体为氮气;所述反应的温度为50-100℃。
优选的,步骤(1)-(2)所述反应均在搅拌下进行;搅拌速率为400-600rpm。
上述的具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物在制备抗癌药物中的应用。
优选的,所述抗癌药物为抗胰腺癌细胞MiaPaCa-2、BXPC-3、PANC-1,抗乳腺癌细胞A549、MCF-7,抗结肠癌细胞HCT-116,抗肝癌细胞HepG2,抗肺癌细胞NCI-H460以及抗黑色素瘤B16中的至少一种药物。
本发明具有以下优点和有益效果:
1、本发明的具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物,对3种胰腺癌细胞系MiaPaCa-2、BXPC-3、PANC-1,2种乳腺癌细胞系A549、MCF-7,1种结肠癌细胞HCT-116,1种肝癌细胞HepG2,一种肺癌细胞NCI-H460以及1种黑色素瘤B16中的至少一种具有抑制作用。
2、本发明还提供一种具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物的制备方法,其合成路线简单,反应效率高,具有较高的工业应用前景。
附图说明
图1为本发明的D1至D23的分子结构式;
图2为本发明D1至D23的合成路线图;
图3为D18的氢谱图;
图4为D18的碳谱图。
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
本发明中所使用的原料,如无特殊说明,均为常规的市售产品;本发明中所使用的方法,如无特殊说明,均为本领域的常规方法。
本发明提供一种具有抗肿瘤活性的3,4,5-三取代异噁唑类化合物,其分子结构式如图1所示,其合成路线如图2所示。
各个化合物的制备具体包括如下步骤:
实施例1
化合物3的合成
准确称取酰氯类化合物(10mmol)于100mL圆底烧瓶中,加入双三苯基膦二氯化钯(0.4mol%)和碘化亚铜(2mol%),再加入端炔类化合物(12mmol)后加入50mL三乙胺,在圆底烧瓶口塞上连有氮气球的三通阀,抽真空且通入氮气(反复三次),将瓶内空气置换为氮气环境,然后放到温度设置为30℃的油浴锅中搅拌24h至完全反应。反应结束后加入100mL饱和氯化铵水溶液进行水洗,水层用乙酸乙酯萃取3次,合并有机相并使用无水硫酸钠干燥,过滤,减压浓缩除去溶剂,得到粗产物,再通过柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比100:1的混合溶剂,得到产物中间体酮。随后将所得中间产物酮溶于甲醇溶液中,在搅拌的条件下,加入甲氧基胺盐酸盐(1.2equiv)、吡啶(3.5mmol/1.5mL)以及无水硫酸钠(2equiv),室温搅拌至完全反应,加入50mL饱和氯化钠溶液进行水洗,水层用乙酸乙酯萃取3次,合并有机相并使用无水硫酸钠干燥,过滤,减压浓缩除去溶剂,得到粗产物,再通过柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比100:1的混合溶剂,提纯得到化合物3。
实施例2
化合物5的合成
取化合物3(0.25mmol)加入到试管中,然后加入碘单质(1.25mmol),二氯甲烷(2.5mL),在室温条件下反应6h,反应结束后用10mL饱和硫代硫酸钠水溶液洗涤,然后用10mL二氯甲烷萃取,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到中间体5。
实施例3
产物D1的合成
取(Z)-1,3-二苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),2-乙烯基萘(0.24mmol),加入醋酸钯(10mol%),室温条件下反应8h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D1,产率为42%。
IR(KBr)=3459,3054,3008,2924,1601,1515,1447,1409,1263,1173,1138,1026,963,754,694,583,475(cm
HRMS(ESI)Calcd for C
实施例4
产物D2的合成
取(Z)-1-(3,4-二甲氧基苯基)-3-苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),2-乙烯基萘(0.24mmol),加入醋酸钯(10mol%),室温条件下反应8h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D2,产率为37%。
IR(KBr)=3055,3004,2928,2838,1602,1523,1482,1426,1260,1175,1137,1026,966,863,812,748,695,477(cm
HRMS(ESI)Calcd for C
实施例5
产物D3的合成
取(Z)-1,3-二苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),2-乙烯基吡啶(0.24mmol),加入醋酸钯(10mol%),室温条件下反应10h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D3,产率为21%。
IR(KBr)=3589,2923,2855,1754,1642,1585,1456,1268,1208,1093,754,699,484(cm
HRMS(ESI)Calcd for C
实施例6
产物D4的合成
取(1E,3Z)-1,5-二苯基戊-1-烯-4-炔-3-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),2-乙烯基吡啶(0.24mmol),加入醋酸钯(10mol%),室温条件下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比10:1的混合溶剂,提纯得到产物D4,产率为18%。
IR(KBr)=3056,3004,2924,2853,1422,1268,967,754,697,475(cm
HRMS(ESI)Calcd for C
实施例7
产物D5的合成
取(1E,3Z)-1,5-二苯基戊-1-烯-4-炔-3-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),4-甲氧基苯乙烯(0.24mmol),加入醋酸钯(10mol%),室温条件下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D5,产率为18%。
IR(KBr)=2924,2853,1753,1600,1509,1458,1267,1209,1097,1025,968,911,822,722,696,476(cm
HRMS(ESI)Calcd for C
实施例8
产物D6的合成
取(Z)-1,3-二苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),4-氯苯乙烯(0.24mmol),加入醋酸钯(10mol%),室温条件下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D6,产率为45%。
IR(KBr)=3056,2926,2855,1747,1594,1490,1448,1407,1267,1165,1088,1017,966,813,756,697,507(cm
HRMS(ESI)Calcd for C
实施例9
产物D7的合成
取(1E,3Z)-1,5-二苯基戊-1-烯-4-炔-3-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),4-氯苯乙烯(0.24mmol),加入醋酸钯(10mol%),室温条件下反应10h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D7,产率为50%。
IR(KBr)=2923,2852,1639,1580,1491,1418,1267,1087,753,691,467(cm
HRMS(ESI)Calcd for C
实施例10
产物D8的合成
取(Z)-1-(3,4-二甲氧基苯基)-3-苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2mL乙二醇二甲醚中,加入1,2-萘醌(0.3mmol),4-氯苯乙烯(0.24mmol),加入醋酸钯(10mol%),室温条件下反应10h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比5:1的混合溶剂,提纯得到产物D8,产率为47%。
IR(KBr)=3464,3060,3006,2928,2840,1598,1473,1426,1323,1259,1138,1024,967,863,813,748,695,505,462(cm
HRMS(ESI)Calcd for C
实施例11
产物D9的合成
取4-碘-3,5-二苯基异噁唑(0.5mmol)溶于5mL三乙胺和N,N-二甲基甲酰胺的混合溶液(1:1)中,在氮气环境下加入4-甲氧基苯乙炔(0.6mmol),加入双三苯基膦二氯化钯(1mol%),碘化亚铜(2mol%),80℃下反应12h至反应完全。将反应液用15mL饱和氯化铵或饱和氯化钠水溶液洗涤,然后用10mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D9,产率为20%。
IR(KBr)=2925,2850,1604,1509,1450,1265,1137,1029,832,754,696,574,481(cm
HRMS(ESI)Calcd for C
实施例12
产物D10的合成
取(E)-4-碘-5-苯基-3-苯乙烯基异噁唑(0.5mmol)溶于5mL三乙胺和N,N-二甲基甲酰胺的混合溶液(1:1)中,在氮气环境下加入4-甲氧基苯乙炔(0.6mmol),加入双三苯基膦二氯化钯(1mol%),碘化亚铜(2mol%),80℃下反应12h至反应完全。将反应液用15mL饱和氯化铵或饱和氯化钠水溶液洗涤,然后用10mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D10,产率为15%。
IR(KBr)=2926,2853,1756,1603,1507,1446,1285,1171,1074,967,919,833,755,694,478(cm
HRMS(ESI)Calcd for C
实施例13
产物D11的合成
取4-碘-3,5-二苯基异噁唑(0.5mmol)溶于5mL三乙胺和N,N-二甲基甲酰胺的混合溶液(1:1)中,在氮气环境下加入3,4-双甲氧基苯乙炔(0.6mmol),加入双三苯基膦二氯化钯(1mol%),碘化亚铜(2mol%),80℃下反应12h至反应完全。将反应液用15mL饱和氯化铵或饱和氯化钠水溶液洗涤,然后用10mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D11,产率为34%。
IR(KBr)=2925,2847,1580,1510,1450,1409,1322,1260,1130,1025,927,810,754,694,590,475(cm
HRMS(ESI)Calcd for C
实施例14
产物D12的合成
取(E)-4-碘-5-苯基-3-苯乙烯基异噁唑(0.5mmol)溶于5mL三乙胺和N,N-二甲基甲酰胺的混合溶液(1:1)中,在氮气环境下加入3,4-双甲氧基苯乙炔(0.6mmol),加入双三苯基膦二氯化钯(1mol%),碘化亚铜(2mol%),80℃下反应12h至反应完全。将反应液用15mL饱和氯化铵或饱和氯化钠水溶液洗涤,然后用10mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D12,产率为28%。
IR(KBr)=3005,2927,2849,1577,1510,1435,1267,1127,1025,846,755,681,479,429(cm
HRMS(ESI)Calcd for C
实施例15
产物D13的合成
取(E)-4-碘-5-苯基-3-苯乙烯基异噁唑(0.5mmol)溶于5mL三乙胺和N,N-二甲基甲酰胺的混合溶液(1:1)中,在氮气环境下加入4-乙炔基吡啶(0.6mmol),加入双三苯基膦二氯化钯(1mol%),碘化亚铜(2mol%),80℃下反应12h至反应完全。将反应液用15mL饱和氯化铵或饱和氯化钠水溶液洗涤,然后用10mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D13,产率为12%。
IR(KBr)=2924,2853,2215,1575,1493,1425,1268,963,813,754,685,609,500(cm
HRMS(ESI)Calcd for C
实施例16
产物D14的合成
取(Z)-1-(3,4-二甲氧基苯基)十一碳-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2.5mL四氢呋喃中,加入醋酸钯(15mol%),氯化铜(2equiv),1-碘-2-(乙烯氧基)苯(0.24mmol),碳酸钾(0.4mmol),四丁基溴化铵(0.24mmol),60℃下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比10:1的混合溶剂,提纯得到产物D14,产率为21%。
IR(KBr)=3325,2921,2781,2677,1701,1458,1256,1020,896,741,508(cm
HRMS(ESI)Calcd for C
实施例17
产物D15的合成
取(Z)-1,3-二苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2.5mL四氢呋喃中,加入醋酸钯(15mol%),氯化铜(2equiv),1-碘-2-(乙烯氧基)苯(0.24mmol),碳酸钾(0.4mmol),四丁基溴化铵(0.24mmol),60℃下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D15,产率为65%。
IR(KBr)=3853,3750,3652,3557,3328,3049,2904,2764,1708,1607,1459,1248,909,752,485(cm
HRMS(ESI)Calcd for C
实施例18
产物D16的合成
取(Z)-3-(4-溴苯基)-1-苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2.5mL四氢呋喃中,加入醋酸钯(15mol%),氯化铜(2equiv),1-碘-2-(乙烯氧基)苯(0.24mmol),碳酸钾(0.4mmol),四丁基溴化铵(0.24mmol),60℃下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D16,产率为43%。
IR(KBr)=3324,2885,2783,2680,1700,1467,1267,1015,902,831,747,668,486(cm
HRMS(ESI)Calcd for C
实施例19
产物D17的合成
取产物D16(0.2mmol)加入封管中,氮气氛围下,加入三甲基硅基乙炔(0.24mmol),加入双三苯基膦二氯化钯(5mol%),碘化亚铜(5mol%),在40℃下反应12h至完全。将反应液用10mL饱和氯化钠溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物;将所得粗产物溶于4mL二氯甲烷和甲醇的混合溶液中,在室温条件下加入碳酸钾颗粒(0.6mmol)反应12h至完全,反应液用饱和氯化钠溶液洗涤,然后用二氯甲烷萃取,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D17,产率为35%。
IR(KBr)=3461,3010,2912,2791,1596,1407,1266,1153,1021,753,538,473(cm
HRMS(ESI)Calcd for C
实施例20
产物D18的合成
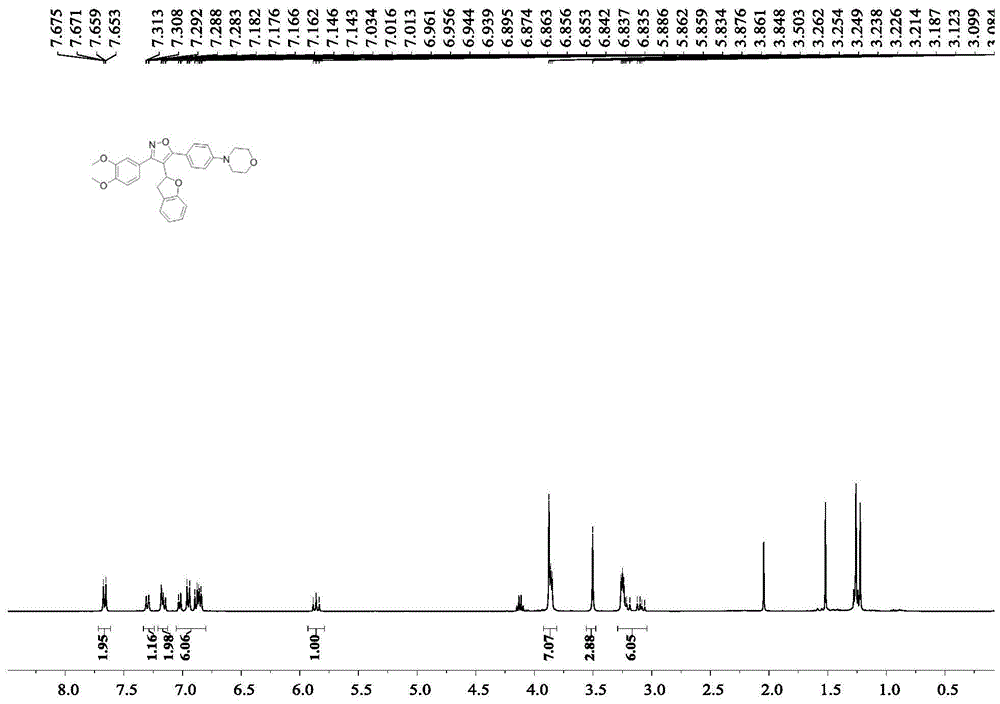
取产物D16(0.2mmol)加入封管中,加入吗啉(0.24mmol)、叔丁醇钾(0.3mmol)和三叔丁基磷(0.3mmol),加入醋酸钯(20mol%),在甲苯溶剂中120℃下氮气保护中反应12h至完全。将反应液用饱和氯化钠溶液洗涤,然后用乙酸乙酯萃取,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D18,产率为42%。(氢谱图如图3,碳谱图如图4)
IR(KBr)=3462,2884,2789,1595,1408,1319,1252,1020,824,747,511(cm
HRMS(ESI)Calcd for C
实施例21
产物D19的合成
取(Z)-1,3-二苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于2mL二甲基亚砜中,加入冰醋酸(0.24mmol),三氟乙酸钯(10mol%),氯化铜(2equiv),在氮气氛围下60℃下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比50:1的混合溶剂,提纯得到产物D19,产率为80%。
IR(KBr)=3675,3323,3028,2894,2779,1731,1612,1437,1280,925,838,690(cm
HRMS(ESI)Calcd for C
实施例22
产物D20的合成
取(Z)-1,3-二苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于N,N-二甲基甲酰胺中(2mL),加入四丁基溴化铵(0.3mmol),烯丙基溴(0.24mmol),醋酸钯(10mol%),室温条件下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比100:1的混合溶剂,提纯得到产物D20,产率为82%。
IR(KBr)=3666,3322,3059,2886,2780,2675,1697,1425,1300,925,697,490(cm
HRMS(ESI)Calcd for C
实施例23
产物D21的合成
取(Z)-3-(4-溴苯基)-1-苯基丙-2-炔-1-酮-O-甲基肟(0.2mmol)溶于N,N-二甲基甲酰胺中(2mL),加入四丁基溴化铵(0.3mmol),烯丙基溴(0.24mmol),醋酸钯(10mol%),室温条件下反应12h至完全。将反应液用10mL饱和氯化钠水溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比100:1的混合溶剂,提纯得到产物D21,产率为70%。
IR(KBr)=3461,2949,2833,2718,1597,1360,1266,1070,763(cm
HRMS(ESI)Calcd for C
实施例24
产物D22的合成
取产物D21(0.2mmol)加入封管中,氮气氛围下,加入三甲基硅基乙炔(0.24mmol),加入双三苯基膦二氯化钯(5mol%),碘化亚铜(5mol%),在40℃下反应12h至完全。将反应液用10mL饱和氯化钠溶液洗涤,然后用5mL乙酸乙酯萃取三次,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物;将所得粗产物溶于4mL二氯甲烷和甲醇的混合溶液中,在室温条件下加入碳酸钾颗粒(0.6mmol)反应12h至完全,反应液用饱和氯化钠溶液洗涤,然后用二氯甲烷萃取,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D22,产率为43%。
IR(KBr)=3734,3645,3326,2890,2780,1699,1470,1285,1022,925,863,748(cm
HRMS(ESI)Calcd for C
实施例25
产物D23的合成
取产物D21(0.2mmol)加入封管中,加入吗啉(0.24mmol)、叔丁醇钾(0.3mmol)和三叔丁基磷(0.3mmol),加入醋酸钯(20mol%),在甲苯溶剂中120℃下氮气保护中反应12h至完全。将反应液用饱和氯化钠溶液洗涤,然后用乙酸乙酯萃取,合并有机层,有机层用无水硫酸钠干燥,过滤和减压浓缩得到粗产物,再经柱层析分离纯化,所用的柱层析洗脱液为石油醚:乙酸乙酯体积比20:1的混合溶剂,提纯得到产物D23,产率为46%。
IR(KBr)=3455,2926,1596,1510,1425,1322,1250,1136,1029,925,830,748(cm
HRMS(ESI)Calcd for C
实施例26
产物D1到D23的抗肿瘤活性测试
实施例3到实施例25制备得到的产物D1到D23进行抗肿瘤活性测试,具体步骤如下所示:
收集对数生长期细胞,计数,用完全培养基重悬细胞,调整细胞浓度至合适浓度(MiaPaCa-2,3000cell/well;BXPC-3,3000cell/well;PANC-1,3000cell/well;A549,3000cell/well;MCF-7,3000cell/well;HCT-116,3000cell/well;HepG2,3000cell/well;NCI-H460,3000cell/well;B16,3000cell/well),接种至96孔板,每孔加90μL细胞悬液,在37℃,5%CO
待细胞孵育过夜后,依次加入10μL对应10个梯度浓度的工作液,溶剂对照孔加10μL含1%DMSO完全培养基,置于37℃,5%CO
加药72小时后,按照CCK8操作说明,用完全培养基配成含10%的检测液,将9株贴壁细胞MiaPaCa-2、;BXPC-3、PANC-1、A549、MCF-7、HCT-116、HepG2、NCI-H460、B16上清吸除,每孔加入100μL配置好的CCK8检测液,半放置培养箱1-4h,再进行检测。
采用CCK-8法对多种癌细胞进行生长抑制活性筛选,同时以星形孢菌素作为阳性对照药。表1显示的是产物D1到D23分别在10μmol·L
表1
由表1数据可以看出产物D13对HepG2有一定的抑制作用。
产物D13对HepG2的IC
用非线性回归模型绘制S型剂量-抑制率曲线并计算IC
表2 CTG法检测产物D13对HepG2的IC
从表2数据可以看出异噁唑衍生物D13对肝癌细胞HepG2的IC
由表1数据可以看出产物D18对NCI-H460、HCT-116均有一定的抑制作用。
产物D18对NCI-H460、HCT-116的IC
用非线性回归模型绘制S型剂量-抑制率曲线并计算IC
表3 CTG法检测产物D18对H460、HCT-116的IC
从表3数据可以看出异噁唑衍生物D18对肺癌细胞NCI-H460的IC
由表1数据可以看出产物D23对HepG2、MCF-7均有一定的抑制作用。
产物D23对HepG2、MCF-7的IC
用非线性回归模型绘制S型剂量-抑制率曲线并计算IC
表4 CTG法检测产物D23对HepG2、MCF-7的IC
从表4数据可以看出异噁唑衍生物D23对肝癌细胞HepG2的IC
以上所述是本发明的优选实施方式而已,当然不能以此来限定本发明之权利范围,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和变动,这些改进和变动也视为本发明的保护范围。
- 一类具有抗肿瘤活性的脱氢枞酸芳胺基苯并咪唑衍生物及其制备方法和应用
- 三环异恶唑类化合物及其制备方法与应用
- 3,5-二取代的和3,4,5-三取代2-异噁唑啉和异噁唑,其制备方法及其作为药物的用途
- 3,4,5-三甲氧基苯基取代含色酮结构的螺吲唑-异噁唑衍生物及制备方法与应用