一种用于复杂基质烟草提取物产地溯源的方法
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于烟用香精香料检测鉴别技术领域,具体涉及一种用于复杂基质烟草提取物产地溯源的方法。
背景技术
烟草提取物(浸膏、精油、净油)是从烟草中提取的具有烟草特征香气的一类化合物,其产品质量受烟草品种、产地等多方面影响。以云南和津巴布韦烟草提取物为例,云南烟草提取物具有沁人心脾、令人愉悦的清甜香,津巴布韦烟草提取物具有甜润、醇和的自然香韵,两种产地的烟草提取物香气成分组成较为相似。为了便于卷烟企业开展自主调香工作,有必要研究有效的产地来源鉴别方法加强对天然香原料属性认知。
现有技术主要依赖于常规物理指标检测和感官评价,不能客观准确地对其产地、组分及感官功效等进行针对性研究。鉴于烟草提取物的种类繁多和成分复杂,样品的前处理方式的选择对于分析结果的准确性和重现性非常重要。固相支持液液萃取作为一种新型前处理方法,具有吸水性强、性质稳定、及不存在乳化现象的特点,能够提高分析结果的重现性和降低前处理过程的损失,已成功应用于医药、食品和环境等领域的样品前处理。此外,大量研究表明非靶向代谢组学技术在食品认证和溯源方面的已得到成功应用,其具有明显技术优势,这为使用物质组分对烟草提取物产地鉴别研究提供了参考。
目前,利用代谢组学数据与化学计量学方法相结合开展烟草提取物产地溯源的研究处于空白状态。因此,提供一种用于烟草提取物产地溯源的方法,为如何科学、快速、准确实现靶向补充、改善、替代某些烟叶风格,增强天然香原料属性认知和提升自主调香核心技术提供技术支撑。
发明内容
针对现有技术存在的不足,本发明的目的在于提供一种用于复杂基质烟草提取物产地溯源的方法,所述方法可以客观、科学、准确有效地为增强天然香原料属性认知和提升自主调香核心技术掌控提供参考。
为达到此发明目的,本发明采用以下技术方案:
第一方面,本发明提供一种用于复杂基质烟草提取物产地溯源的方法,所述方法包括:
收集不同产地代表性烟草提取物样品;采用溶剂萃取和固相支持液液萃取处理样品,得到挥发物组进样溶液;采用溶剂萃取和衍生处理样品,得到非挥发物组进样溶液;采用气相色谱-质谱联用方法,对挥发物组进样溶液和非挥发物组进样溶液进行检测,采集指纹图谱;根据指纹图谱对挥发物组分和非挥发物组分分析;对质谱数据进行预处理,构建溯源模型,使用产地溯源模型进行识别,研判目标烟草提取物的产地身份。
本发明中使用代谢标志物进行烟草提取物产地溯源的检测方法,所述检测方法使用的代谢标志物是基于非靶向代谢组学筛选获得的,具有更好的普适性。本发明采用“解卷积+标准谱库检索+保留指数”分析手段,结合GC/MS内标法,创新构建了烟草提取物中挥发性组分的定性定量分析方法,克服了现有技术的不足,MPP软件独特的递归非靶向特征提取算法,可以保证查找到更多的化合物,可实现自动化样品分类,结合保留指数能够提高定性的精确度,彻底改变了单纯基于MS的未知样品的定性分析带来的误判。
优选地,所述方法包括以下步骤:
(1)样品前处理:收集不同产地代表性烟草提取物,经溶剂萃取和固相支持液液萃取,对样品进行处理,得到挥发物组进样溶液;
收集不同产地代表性烟草提取物,采用溶剂萃取和衍生处理,对样品进行处理,得到非挥发物组进样溶液;
(2)指纹图谱采集:使用气相色谱-质谱联用仪,对挥发物组进样溶液和非挥发物组进样溶液进行质谱分析,采集指纹图谱;
(3)挥发物组分分析:对采集得到的样品图谱进行解卷积,有效提取样品离子信息,结合数据库和保留指数,对样品挥发物进行非靶向分析;
非挥发组分分析:对采集得到的样品图谱进行解卷积,有效提取样品离子信息,结合数据库和保留指数,对样品非挥发物进行非靶向分析;
(4)质谱数据预处理:对挥发物组分和非挥发物组分进行过滤、对齐和归一化,获取样品挥发物组分和非挥发物组分的数据矩阵;
(5)溯源模型构建:对不同产地代表性烟草提取物样品的数据进行分组,采用正交偏最小二乘—判别分析方法进行多元统计分析,构建不同产地烟草提取物样品产地溯源的模型;
(6)目标产地研判:对测试样品进行步骤(1)-(4)的处理,使用步骤(5)建立的产地溯源模型进行识别,研判目标烟草提取物的产地身份。
优选地,步骤(1)中,所述挥发物组进样溶液采用包括如下步骤的方法制备得到:
收集不同产地代表性烟草提取物样品,将样品、内标、盐水和乙醇混合得到稀释液,涡旋后进行固相支持液液萃取处理,收集淋洗液,浓缩,将浓缩后的淋洗液作为挥发物组进样溶液。
优选地,所述样品、内标、盐水和和乙醇的质量体积比为(0.1-0.3g):(50-100μL):(0.8-1.2mL):(0.8-1.2mL),其中,“0.1-0.3g”例如可以是0.1g、0.2g或0.3g等;“50-100μL”例如可以是50μL、80μL或100μL等;“0.8-1.2mL”例如可以是0.8mL、1mL或1.2mL等;“0.8-1.2mL”例如可以是0.8mL、1mL或1.2mL等。
优选地,所述内标为正十七碳烷的乙醇溶液,所述正十七碳烷的乙醇溶液的浓度为500-1000mg/L,例如可以是500mg/L、800mg/L或1000mg/L等。
优选地,所述盐水为无机盐与水的混合溶液,所述盐水的质量浓度为5-30%,例如可以是5%、10%、15%、20%、25%或30%等。
优选地,所述无机盐选自NaCl、MgSO
优选地,所述涡旋的转速1500-2000r/min,例如可以是1500r/min、1800r/min或2000r/min等,所述涡旋的时间为15-20min,例如可以是15min、18min或20min等。
优选地,所述固相支持液液萃取中采用的萃取柱为ProElut LLE+硅藻土固相萃取柱。
优选地,所述固相支持液液萃取中使用10-20柱体积的二氯甲烷进行萃取淋洗。
本发明中,将淋洗液浓缩20-30倍后作为挥发物组进样溶液。
优选地,步骤(1)中,所述非挥发物组进样溶液采用包括如下步骤的方法制备得到:
收集不同产地代表性烟草提取物样品,将样品、内标和甲醇水溶液混合,得到稀释液并涡旋振荡,取过滤后的稀释液采用氮吹吹干,加入甲氧氨盐酸盐的吡啶溶液,涡旋后温育,再加入衍生化试剂,涡旋后温育,得到非挥发物组进样溶液。
优选地,所述样品、内标和甲醇溶液的质量体积比为(0.1-0.2g):(0.2-0.3mL):(4-6mL);其中,“0.1-0.2g”例如可以是0.1g、0.15g或0.2g等;“0.2-0.3mL”例如可以是0.2mL、0.25mL或0.3mL等;“4-6mL”例如可以是4mL、5mL或6mL等。
优选地,所述内标为1-4丁二醇的甲醇溶液,所述1-4丁二醇的甲醇溶液的浓度为800-1000mg/L,例如可以是800mg/L、900mg/L或1000mg/L等。
优选地,所述甲醇水溶液的体积浓度为60-70%,例如可以是60%、65%或70%等。
优选地,所述涡旋振荡的转速1500-2000r/min,例如可以是1500r/min、1800r/min或2000r/min等,所述涡旋振荡的时间为10-30min,例如可以是10min、20min或30min等。
优选地,所述过滤采用孔径为0.22-0.45μm滤膜,例如可以是0.22μm或0.45μm。
优选地,所述稀释液与甲氧氨盐酸盐的吡啶溶液的体积比为100:(80-100),例如可以是100:80、100:90或100:100等。
优选地,所述甲氧氨盐酸盐的吡啶溶液的浓度为20-30mg/mL,例如可以是20mg/mL、25mg/mL或30mg/mL等。
优选地,所述衍生化试剂的使用量为80-120μL,例如可以是80μL、100μL或120μL等。
优选地,所述衍生化试剂为纯度不低于99%的N,O-双(三甲基硅基)三氟乙酰胺。
优选地,所述涡旋的时间为0.5-1min,例如可以是0.5min或1min等,所述温育的时间为30-90min,例如可以是30min、60min或90min等,所述温育的温度为35-37℃,例如可以是35℃、36℃或37℃等。
优选地,步骤(2)中,所述采集指纹图谱采用包括如下步骤的方法进行:
使用气相色谱-质谱联用仪,对挥发物组进样溶液和非挥发物组进样溶液进行质谱分析,采集指纹图谱的色谱柱选用HP-5MS毛细管柱(30m×250μm×0.25μm)或等效色谱柱。
优选地,对所述挥发物组进样溶液进行检测的气相色谱条件和质谱条件:
气相色谱条件:载气:氦气(≥99.999%);进样口温度:250±10℃;柱流量:1±0.2mL/min,恒流模式;进样量:1±0.1μL;分流比:5:1;程序升温:50℃保持2min,以3℃/min的升温速度升至100℃保持5min,以2℃/min的升温速度升至250℃,再继续以10℃/min的升温速度升至300±5℃,保持10min;
质谱条件:电离方式:EI;离子源温度:230℃;电子能量:70eV;四极杆温度:150℃;扫描模式:全扫描;质量扫描范围:35-550amu;溶剂延迟:5±1min。
优选地,对所述非挥发物组进样溶液进行检测的气相色谱条件和质谱条件:
色谱条件:载气:氦气(≥99.999%);进样口温度:290±10℃;柱流量:1±0.2mL/min,恒流模式;进样量:1±0.1μL;分流比:20:1;程序升温:初始温度70℃,保持2min,以5℃/min的升温速率升温至310±5℃,保持10min;
质谱条件:电离方式:EI;传输线温度:310℃;电离能量:70eV;离子源温度:230℃;四极杆温度:150℃;溶剂延迟时间:11min;全扫描监测模式,扫描范围为50-500amu;溶剂延迟:4min。
优选地,步骤(3)中,所述对挥发物组分和非挥发物组分分析采用包括如下步骤的方法进行:
采用组分在质谱库NIST17和FLAVOR2中匹配度≥85%的方法进行定性分析,同时计算保留指数,并参照webbook.nist.gov、www.flavornet.org以及标准谱库中记载的相关化合物保留指数进行比对,将绝对值相差20以内的确定为同一化合物;
采用内标法定量,通过内标物的峰面积和样品溶液中各组分的峰面积比值,按公式计算各个组分的相对质量浓度:
式中:C
优选地,步骤(4)中,所述质谱数据预处理采用包括如下步骤的方法进行:
利用Agilent MassHunter Unknowns Analysis软件解卷积处理质谱原始数据,导出转换为cef格式文件,再导入到Agilent MassHunter Mass Profiler(MPP)软件中进行峰识别、峰对齐、过滤(在所有样品中出现50%以上)、归一化等操作有效提取样品离子信息,获取样品挥发物组和非挥发物组数据矩阵。
优选地,步骤(5)中,所述溯源模型构建采用包括如下步骤的方法进行:
将样品依据产地进行分组,进一步构建有监督的正交OPLS-DA模型;当R
优选地,步骤(6)中,所述目标产地研判体的过程为:
将已构建的正交OPLS-DA分类模型进行训练,判断模型的准确度,当准确度>90时,说明该模型可进行下一步预测;
对测试样品进行步骤(1)-(4)的处理,使用步骤(5)建立的正交OPLS-DA分类模型产地溯源模型进行识别,研判目标烟草提取物的产地身份。
优选地,所述的复杂基质烟草提取物包括烟草浸膏、精油或净油等。
本发明所述的数值范围不仅包括上述列举的点值,还包括没有列举出的上述数值范围之间的任意的点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
相对于现有技术,本发明具有以下有益效果:
(1)本发明采用的无机盐水溶液,具有促使烟草提取物组分快速从水相转入有机相的优势,最大程度地能够提高烟草提取物中不易溶于有机试剂组分的萃取效率;
(2)本发明采用的固相液液萃取柱,具有吸水性强、性质稳定及不存在乳化现象的特点,能够提高分析结果的重现性和降低前处理过程的损失;
(3)本发明使用代谢标志物进行烟草提取物产地溯源,该检测方法使用的代谢标志物是基于非靶向代谢组学筛选获得的,具有更好的普适性;
(4)本发明采用“解卷积+标准谱库检索+保留指数”分析手段,结合GC/MS内标法,创新构建了烟草提取物中挥发性组分的定性定量分析方法,克服了现有技术的不足,MPP软件独特的递归非靶向特征提取算法,可以保证查找到更多的化合物,可实现自动化样品分类,结合保留指数能够提高定性的精确度,彻底避免了单纯基于MS的未知样品的定性分析带来的误判。
附图说明
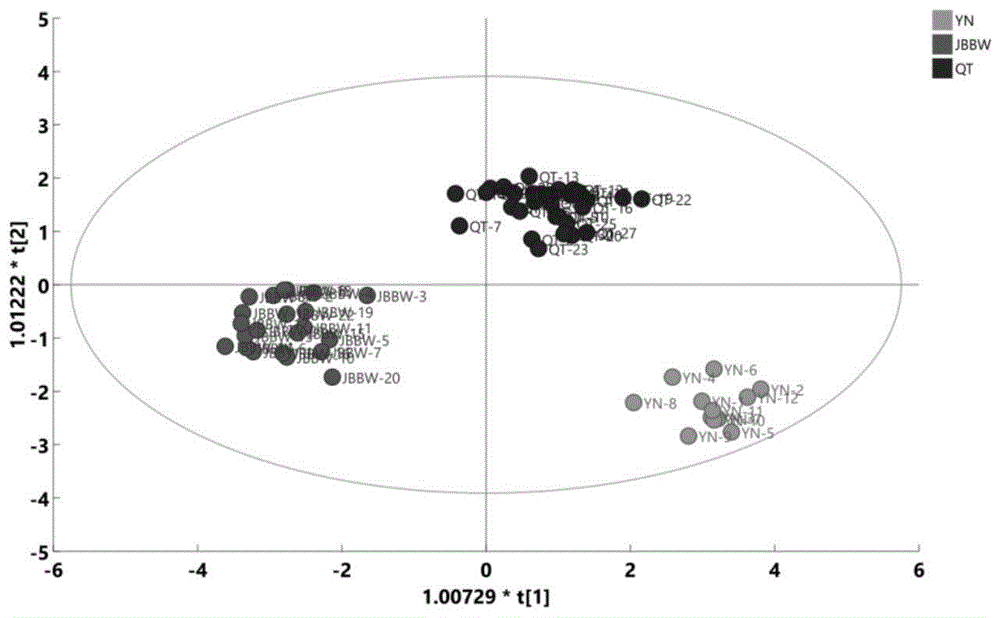
图1是不同烟草提取物的OPLS-DA得分图;
图2是置换检验图;
图3是因子载荷图及局部放大图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。
实施例1
一种用于复杂基质烟草提取物产地溯源的方法,具体步骤如下:
(1)样品前处理:收集不同产地代表性烟草提取物,经溶剂萃取和固相支持液液萃取(SLE),对样品进行处理,得到进样溶液。
选取64个国内外不同产地烟叶制备的烟草提取物样品(云南烟草提取物12个,津巴布韦烟草提取物22个和其他烟草提取物30个,分别简称YN、JBBW和QT,且QT样品主要包括河南、湖南、巴西和弗吉尼亚等国内外烟草提取物)作为研究对象。
①溶剂萃取:称取0.2g烟草提取物样品于离心管中,再准确移入50μL浓度为1000mg/L的正十七碳烷的乙醇溶液;然后加入盐水(25%氯化钠水溶液)和乙醇各1mL,使用涡旋混匀器于1500r/min条件下分散20min,得样品萃取液。
②固相液液萃取:采用ProElut LLE+硅藻土固相萃取柱进行活化,即用10mL甲醇去杂质,排干液体;然后将样品萃取液全部转入已活化过的固相萃取柱,同时用浓缩瓶接收洗脱液,待上样溶液全部流入小柱上筛板后,静置平衡约5min;随即用30mL二氯甲烷分3次对固相萃取柱进行淋洗,并收集所有淋洗液;最后在50℃、常压条件下,将淋洗液浓缩至1mL左右备用。
③衍生化萃取:称取0.1g样品,精确至0.0001g,置于10mL离心管中。分别准确加入0.2mL浓度为1000mg/L的1-4丁二醇内标溶液和4mL甲醇/水(6:4)稀释溶剂,2000r/min涡旋振荡10min。稀释液过0.22μm有机相滤膜,取100μL稀释液用氮吹吹干。然后加入80μL 20mg/mL的甲氧氨盐酸盐的吡啶溶液,涡旋1min后在37℃下温育90min,再加入100μL BSTFA,涡旋30s后在37℃下温育30min。最后将衍生后的溶液转入微量进样装置进行GC-MS分析。
(2)指纹图谱采集:使用气相色谱-质谱联用仪,对不同产地来源烟草提取物样本进行质谱分析,采集指纹图谱。
挥发组分:
采集指纹图谱的色谱柱选用HP-5MS毛细管柱(30m×250μm×0.25μm);
色谱条件:载气:高纯氦气(≥99.999%);进样口温度:250℃;柱流量:1mL/min(恒流模式);进样量:1μL;分流比:5:1;程序升温:50℃保持2min,以3℃/min的升温速度升至100℃保持5min,以2℃/min的升温速度升至250℃,再继续以10℃/min的升温速度升至300℃,保持10min。
质谱条件:电离方式:EI;离子源温度:230℃;电子能量:70eV;四极杆温度:150℃;扫描模式:全扫描;质量扫描范围:35-550amu;溶剂延迟:5min。
难挥发组分:
色谱条件:载气:高纯氦气(≥99.999%);进样口温度:290℃;柱流量:1mL/min(恒流模式);进样量:1μL;分流比:20:1;程序升温:初始温度70℃,保持2min,以5℃/min的速率升温至310℃,保持10min。
质谱条件:电离方式:EI;传输线温度:310℃;电离能量:70eV;离子源温度:230℃;四极杆温度:150℃;溶剂延迟时间:11min;全扫描监测模式,扫描范围为50-500amu;溶剂延迟:4min。
(3)挥发物和难挥发物组分分析:
采用组分在质谱库NIST17和FLAVOR2中匹配度≥85%的方法进行定性分析,同时计算保留指数,并与文献资料相关化合物保留指数进行比对,将绝对值相差20以内的确定为同一化合物。
采用内标法定量,通过内标物的峰面积和样品溶液中各组分的峰面积比值,按公式计算各个组分的相对质量浓度:
式中:C
(4)质谱数据预处理:
利用Agilent MassHunter Unknowns Analysis B10.1软件解卷积处理质谱原始数据,导出转换为cef格式文件,再导入到Agilent MassHunter Mass Profiler(MPP)软件中进行峰识别、峰对齐、过滤(在所有样品中出现50%以上)、归一化等操作,有效提取样品离子信息,获取高质量的样品挥发物组数据矩阵。
表1为不同烟草提取物样品挥发性和难挥发性成分测定结果,表1中包括3组烟草提取物的挥发性、难挥发性成分数据结果。
表1
/>
/>
/>
/>
注:①字母A到M依次按照醇类、酯类、醛类、酮类、酸类、烷烃类、烯烃类、酚类、胺类、醚类、其他类、烟碱和糖类进行编号;②YN、JBBW和QT分别表示云南、津巴布韦和其他烟草提取物,表中数据为各自组样品的平均质量浓度;③RI为保留指数鉴定,MS为质谱鉴定。
(5)溯源模型构建
采用正交OPLS-DA对YN、JBBW和QT样品进行分析,以考察YN、JBBW和QT样品之间的差异特征挥发性成分。由模型得分情况(图1)可知,基本上所有样本均在95%置信区间内,极个别样品处于置信区间附近,JBBW样品主要分布在左侧象限,QT和YN样品分别位于右侧象限上下两端,显示不同组样品能够得到有效区分。使用有监督的正交OPLS-DA模型时容易产生过拟合现象,因此,又采用200次响应的置换检验验证分析模型是否有过拟合现象。如图2所示,置换检验得到的R
OPLS-DA因子载荷图可直观地反映出每一个变量在得分图上的贡献。对不同烟草提取物中主要挥发性和难挥发性成分进行分析,结果如图3所示:区别于JBBW和QT样品,A4(丝氨醇)、B4(γ-丁内酯)、D1(4-环戊烯-1,3-二酮)、D7(2,2,5-三甲基-3,4-己二酮)、D9(2,5-二甲基-3-己酮)、H2(菜籽多酚)、F13(1-碘十二烷)、G1(4-乙酰氧基-3-甲氧基苯乙烯)、K7(2-苄基硫基-4-甲氧基苯甲腈)等成分是凸显YN样品特征的物质组分;区别于YN和QT样品,B1(乳酸乙酯)、F19(3-甲基三十一烷)、A7[2-(2-羟基丙氧基)-1-丙醇]、A8(一缩二丙二醇)、B13(2,4-二叔丁基-5-羟基苯基戊酸酯)等成分是凸显JBBW样品特征的组分,以酯类和醇类较为突出;区别于YN和JBBW样品,H5(α-生育酚)、K1(2-羟基-5-甲基吡啶)、B3(1-羟基-2-丙基乙酸酯)、E2(乙酰丙酸)、D19(6,7-二甲氧基-1,4-二氢-2,3-喹啉二酮)、D6[5-(羟甲基)二氢呋喃-2(3H)-酮]和D2(2-吡咯烷酮)等成分是QT组烟草提取物的特征组分。
(6)目标产地研判
正交OPLS-DA模型不仅能够验证3组烟草提取物的分类结果,并且能够预测未知烟草提取物的类别。本正交OPLS-DA模型由每个实际烟草提取物构建,3组样品具有良好的分类。在预测之前,先对该模型进行训练,判断模型的准确度,结果如表2所示:准确度均为100%,识别率高达100%。说明该模型准确度较高,可用于下一步的样本预测。
为了证明该模型的预测能力,除了用于寻找特征差异物和建立分类模型的64个烟草提取物样品外,又分别使用1个云南烟草提取物和1个津巴布韦烟草提取物,导入正交OPLS-DA模型进行预测,所有不同组预测样本均没有出现分类错误,模型预测结果与实际样本产地结果均一致,准确度为100%。进一步表明构建的正交OPLS-DA模型具有良好的预测能力,能够对云南、津巴布韦和其他产地烟草提取物进行正确分类,可用于烟草提取物的产地溯源。表2为正交OPLS-DA模型预测烟草提取物的分类结果。
表2
本实施例所用到的云南烟草提取物和津巴布韦烟草提取物只是做一个例子进行说明,其他产地的烟草提取物产地溯源与上述实施例相同,在此不在一一列举。
实施例2
本实施例探究了不同的样品前处理步骤对建模结果的影响,本实施例中的建模方法参照实施例1进行,其中样品前处理步骤包括以下方式:
样品前处理方式1:
①溶剂萃取:称取0.2g烟草提取物样品于离心管中,再准确移入100μL浓度为500mg/L的正十七碳烷的乙醇溶液;然后加入盐水(25%氯化钠水溶液)和乙醇各1mL,使用涡旋混匀器于2000r/min条件下分散15min,得样品萃取液。
②固相液液萃取:采用ProElut LLE+硅藻土固相萃取柱进行活化,即用10mL甲醇去杂质,排干液体;然后将样品萃取液全部转入已活化过的固相萃取柱,同时用浓缩瓶接收洗脱液,待上样溶液全部流入小柱上筛板后,静置平衡约5min;随即用30mL二氯甲烷分3次对固相萃取柱进行淋洗,并收集所有淋洗液;最后在50℃、常压条件下,将淋洗液浓缩至1mL左右备用。
③衍生化萃取:称取0.2g样品,精确至0.0001g,置于10mL离心管中。分别准确加入0.3mL浓度为800mg/L的1-4丁二醇内标溶液和5mL甲醇/水(6:4)稀释溶剂,1500r/min涡旋振荡30min。稀释液过0.22μm有机相滤膜,取100μL稀释液用氮吹吹干。然后加入100μL20mg/mL的甲氧氨盐酸盐的吡啶溶液,涡旋1min后在37℃下温育90min,再加入120μLBSTFA,涡旋30s后在37℃下温育30min。最后将衍生后的溶液转入微量进样装置进行GC-MS分析。
样品前处理方式2:
①溶剂萃取:称取0.2g烟草提取物样品于离心管中,再准确移入50μL浓度为1000mg/L的正十七碳烷的乙醇溶液;然后加入盐水(氯化钠水溶液)0.5mL和乙醇1.5mL,使用涡旋混匀器于1500r/min条件下分散20min,得样品萃取液。
②固相液液萃取和③衍生化萃取:参照实施例1。
样品前处理方式3:
①溶剂萃取:称取0.2g烟草提取物样品于离心管中,再准确移入50μL浓度为1000mg/L的正十七碳烷的乙醇溶液;然后加入盐水(氯化钠水溶液)1.5mL和乙醇0.5mL,使用涡旋混匀器于1500r/min条件下分散20min,得样品萃取液。
②固相液液萃取和③衍生化萃取:参照实施例1。
样品前处理方式4:
①溶剂萃取和②固相液液萃取参照实施例1。
③衍生化萃取:称取0.1g样品,精确至0.0001g,置于10mL离心管中。分别准确加入0.1mL浓度为1000mg/L的1-4丁二醇内标溶液和2mL甲醇/水(6:4)稀释溶剂,2000r/min涡旋振荡10min。稀释液过0.22μm有机相滤膜,取100μL稀释液用氮吹吹干。然后加入80μL 20mg/mL的甲氧氨盐酸盐的吡啶溶液,涡旋1min后在37℃下温育90min,再加入100μL BSTFA,涡旋30s后在37℃下温育30min。最后将衍生后的溶液转入微量进样装置进行GC-MS分析。
样品前处理方式5:
①溶剂萃取和②固相液液萃取参照实施例1。
③衍生化萃取:称取0.1g样品,精确至0.0001g,置于10mL离心管中。分别准确加入0.2mL浓度为1000mg/L的1-4丁二醇内标溶液和4mL甲醇/水(4:6)稀释溶剂,2000r/min涡旋振荡10min。稀释液过0.22μm有机相滤膜,取100μL稀释液用氮吹吹干。然后加入80μL 20mg/mL的甲氧氨盐酸盐的吡啶溶液,涡旋1min后在37℃下温育90min,再加入100μL BSTFA,涡旋30s后在37℃下温育30min。最后将衍生后的溶液转入微量进样装置进行GC-MS分析。
不同的处理方式所构建的模型预测效果如表3所示。
表3
通过实施例1和方式2和3的结果对比可知,在萃取过程中盐水和乙醇使用量和比例影响后续萃取和分散的效果,从而进一步影响后续的检测和建模。
通过实施例1和方式1的结果可知,当实验条件在权利保护范围之内时,所采集的数据所构建的模型参数R
通过实施例1和方式4和5的结果对比可知,当R
综上,本发明提供的方法使用的代谢标志物是基于非靶向代谢组学筛选获得的,具有更好的普适性和精确度。所述方法可以客观、科学、准确有效地为增强天然香原料属性认知,和提升自主调香核心技术掌控提供参考。
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
- 一种烟草核酸提取物的制备方法及其在烟草中的应用
- 一种基于多元素和稳定同位素的牦牛肉产地溯源方法
- 一种乌龙茶产地溯源方法
- 一种利用红外光谱溯源识别成品六堡茶产地的方法
- 一种农产品产地身份溯源装置、溯源系统及溯源方法
- 一种气溶胶生成基质、烟草提取物及其制备方法