LsARF3蛋白或其编码基因在调控叶用莴苣的高温抽薹性能中的应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及植物基因工程技术领域,尤其涉及LsARF3蛋白或其编码基因在调控叶用莴苣的高温抽薹性能中的应用。
背景技术
叶用莴苣叶用莴苣(Lactuca sativa L.),俗称生菜,喜冷凉气候,是一种重要的世界性鲜食蔬菜。因其口感好和营养品质优良,深受广大消费者青睐,近几年栽培面积也在迅速扩大,已成为绿色蔬菜的重要种类之一。但是,当夏季温度超过30℃时,极易导致叶用莴苣“先期抽薹”,进而导致产量降低,严重影响了叶用莴苣的食用品质和商品价值。
现阶段关于叶用莴苣高温抽薹在栽培技术与生理层面的研究已经取得一定的成果,而在蛋白质与基因水平的研究十分匮乏。因此结合基因工程技术对研究叶用莴苣抽薹具有重要的生物学意义,对于后续制定科学的措施调控抽薹,解决产业发展中面临的提高产量和品质、保证周年生产的核心问题,具有重要的理论指导意义和应用前景。
发明内容
为了解决现有技术的技术问题,本发明提供一种LsARF3蛋白或其编码基因在调控叶用莴苣的高温抽薹性能中的应用。
第一方面,本发明提供LsARF3蛋白或其编码基因、或含有其编码基因的生物材料在调控叶用莴苣高温抽薹性能中的应用。
本发明进一步提供LsARF3蛋白或其编码基因、或含有其编码基因的生物材料在叶用莴苣耐高温抽薹种质资源改良中的应用。
进一步地,所述高温耐抽薹性能为:
在31~35℃处理后,叶用莴苣的抽薹性能。
进一步地,所述生物材料为表达盒、载体或转基因细胞。
进一步地,所述LsARF3蛋白包括如SEQ ID NO.1所示的氨基酸序列。
第二方面,本发明提供一种提高叶用莴苣的耐抽薹性能的方法,抑制叶用莴苣中LsARF3蛋白的表达;所述LsARF3蛋白包括如SEQ ID NO.1所示的氨基酸序列。
进一步地,所述抑制叶用莴苣中LsARF3蛋白的表达为:
通过转基因、基因敲除、RNA干扰、杂交、回交、自交或无性繁殖的方法,调控叶用莴苣中LsARF3基因的表达;
所述LsARF3基因包括如SEQ ID NO.2所示的核苷酸序列。
进一步地,所述转基因包括利用Ti质粒、植物病毒载体、直接DNA转化、显微注射、基因枪、电导或农杆菌介导的方法将包含所述LsARF3基因的重组表达载体导入植物,获得转基因植物株系;和/或,
所述基因敲除包括利用DNA同源重组技术、Cre/LoxP技术或CRISPR/Cas9技术将所述LsARF3基因敲除,获得转基因植物株系。
进一步地,本发明进一步提供一种gRNA,所述gRNA包括如SEQ ID NO.3或SEQ IDNO.4所示的核苷酸序列。所述gRNA可以应用于基因敲除中。
本发明具备如下有益效果:
本发明研究发现LsARF3蛋白对于叶用莴苣的高温抽薹性能有较大程度的影响,通过调节LsARF3基因的表达可以实现对叶用莴苣的高温抽薹性能的调控。本发明提供的LsARF3基因在叶用莴苣的高温抽薹性能中的应用,可以有效调节叶用莴苣在高温下的抽薹性能,对于探讨高温抽薹机制并制定科学管理措施,创制耐抽薹优良种质具有重大意义。
附图说明
为了更清楚地说明本发明或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
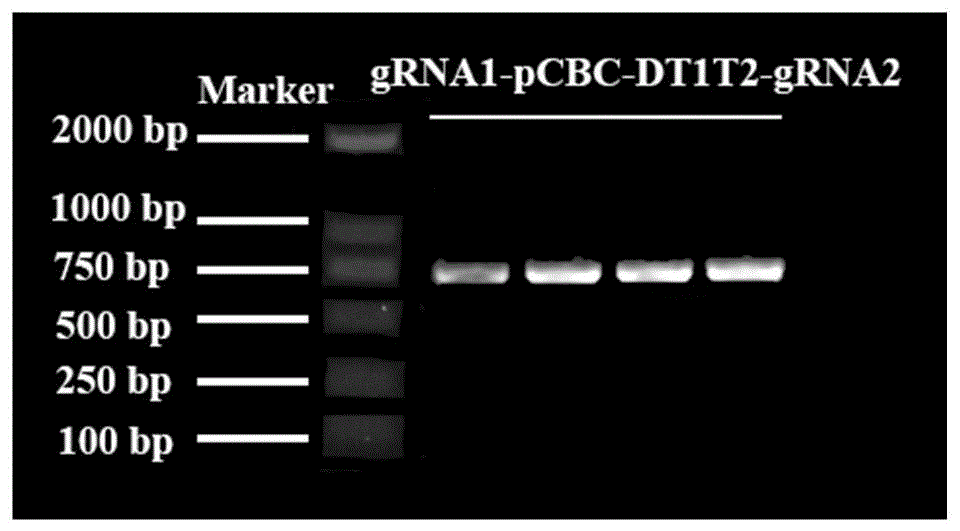
图1为本发明实施例1提供的电泳检测gRNA1-pCBC-DT1T2-g RNA2片段的结果,多条泳道为平行结果。
图2为本发明实施例1提供的电泳检测pKSE401载体线性化的结果,多条泳道为平行结果。
图3为本发明实施例1提供的电泳检测大肠杆菌菌落PCR产物的结果,多条泳道为平行结果。
图4为本发明实施例1提供的以引物CRISPR-DNA-F/R扩增阳性苗时的PCR产物的电泳结果。
图5为本发明实施例1提供的lsarf3纯合突变系编辑类型示意图;图中分别为Wildtype(WT),lsarf3-4、lsarf3-6、lsarf3-9;其中,ls arf3-4的Target 2后第7个碱基发生突变的,lsarf3-6在Target 1的前82个碱基插入一个碱基T,lsarf3-9为嵌合体,发生了3个碱基突变,C-G、A-G和A-T。
图6为本发明实施例1提供的高温32、70天的WT和lsarf3纯合突变体植株表型照片,标尺=6cm。
图7为本发明实施例1提供的高温32天WT和lsarf3纯合突变植株花芽分化状态;图中,1代表WT,2代表lsarf3-4,3代表lsarf3-6,4代表lsarf3-9,标尺=200μm。
图8为本发明实施例1提供的高温下野生型和突变系植株的抽薹、现蕾、开花时间的统计结果(*为p<0.05,**为p<0.01)。
图9为本发明实施例2提供的电泳检测加LsARF3同源臂片段的结果,多条泳道为平行结果。
图10为本发明实施例2提供的电泳检测PRI101载体线性化的结果,1为PRI101酶切结果,2为PRI101质粒,多条泳道为平行结果。
图11为本发明实施例2提供的电泳检测大肠杆菌菌落PCR产物的结果,多条泳道为平行结果。
图12为本发明实施例2提供的以引物G418-F/G418-R扩增阳性苗时的PCR产物的电泳结果,多条泳道为平行结果。
图13为本发明实施例2提供的正常生长条件下60天、108天的WT和过量表达植株表型照片;图中分别为WT,OE-1、OE-7、OE-6,标尺=6cm。
图14为本发明实施例2提供的正常生长60天时WT和过量表达植株中LsARF3基因的相对表达量(*为p<0.05,**为p<0.01)。
图15为本发明实施例2提供的正常生长条件下60天WT和过量表达植株花芽分化状态;图中,1代表WT,2代表OE-1,3代表OE-7,4代表OE-6,标尺=200μm。
图16为本发明实施例2提供的正常生长条件下野生型和过量表达植株的抽薹、现蕾、开花时间的统计结果(*为p<0.05,**为p<0.01)。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明中的附图,对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1CRISPR/Cas9-LsARF3载体构建
1、LsARF3基因靶向位点(gRNA)设计
本发明根据CRISPR/Cas9系统原间隔序列临近基序识别(PAM)原理及基因敲除靶位点guide RNAs(gRNAs)设计原则,使用CRISPR-gRNA设计网站(http://crispr.hzau.edu.cn/CRISPR2/)及NCBI-Blast潜在脱靶位点检测,挑选两条最适宜的gRNA。
2、设计四条引物
根据两条gRNA序列设计四条引物,最后引物序列如下:
DT1-BsF(SEQ ID NO.5):
5’-GCTAGAGTCGAAGTAGTGATTGCTCGAACAGCTACAATCCAGCGG-3’;
DT1-F0(SEQ ID NO.6):5’-CTCGAACAGCTACAATCCAGCGGGTTTTAGAGCTAGAAATAGC-3’;
DT1-R0(SEQ ID NO.7):5’-GTGGGTGCTGGTATCGGAGGCGGCAATCTCTTAGTCGACTCTAC-3’;
DT1-BsR(SEQ ID NO.8):5’-CTTGCTATTTCTAGCTCTAAAACGTGGGTGCTGGTATCGGAGGCGG-3’。
3、添加同源臂
以pCBC-DT1T2载体质粒为模板,使用诺唯赞高保真酶
4、载体线性化
BsaI-HFv2限制性内切酶(NEB)线性化pKSE401载体,用1%琼脂糖凝胶电泳检测是否酶切完全(结果参见图2)。
5、同源重组
ClonExpressIIOne Step Cloning Kit(诺唯赞)同源重组PCR产物(gRNA1-pCBC-DT1T2-gRNA2片段)与线性化载(步骤4中获得的酶切产物)。反应程序为37℃30min,4℃冷却5min。反应体系如下表1。
表1重组反应体系(5μL)
6、大肠杆菌感受态转化
取5ul转化大肠杆菌感受态,Kan板筛选。
50μl冰上感受态加5μl步骤5中获得的同源重组产物,轻轻混匀,冰上放置30min;42℃水浴热激30s,快速转移到冰浴中放置2min。
以下工作在超净工作台中操作:
向离心管中加500μl空白的LB液体培养基,混匀后在37℃摇床200rpm摇1h。将活化的菌液吸取200μl涂布在含有Kan的LB固体培养基上,在37℃培养箱中,倒置平板过夜培养(具体步骤见全式金Trans1-T1 Phage Resistant Chemically Competent Cell说明书)。
7、检测阳性克隆
康为Taq酶(2xTaq MasterMix(Dye))与引物U626-IDF/U629-IDR,用于菌落PCR,反应程序为94℃,2min;35×(94℃,30s;59℃,30s;72℃,30s);72℃,2min。
反应体系如表2所示。用1%的琼脂糖凝胶检测PCR产物,条带大小约为750bp(结果参见图3)。将阳性的菌液送公司测序,测序引物为U626-IDF/U629-IDR。
菌落PCR及测序引物:
U626-IDF(SEQ ID NO.9):5’-TGTCCCAGGATTAGAATGATTAGGC-3’;
U629-IDR(SEQ ID NO.10):5’-AGCCCTCTTCTTTCGATCCATCAAC-3’。
表2菌落PCR反应体系(25μL)
8、农杆菌感受态转化
测序结果检测到2个gRNA的质粒可以进行农杆菌感受态的转化。
将50μl的GV3101感受态加入到5ng质粒中,依次于冰上静置5min、液氮5min、37℃水浴5min、冰浴5min。向离心管中加入700μl空白LB。在28℃摇床200r摇2-3h。6000rmp离心1min富集菌株,留100μl左右上清重悬菌株,将菌液涂布在含有Kan和Rif的固体LB培养基上。在28℃培养箱中倒置培养2-3天(具体步骤见唯地GV3101 Chemically Competent Cell说明书)。
9、叶用莴苣遗传转化
9.1培养基配方
MS液体培养基(1L):M519 4.43g+蔗糖30g,PH=5.8,121℃,20min灭菌;
MS固体培养基(1L):M519 4.43g+蔗糖30g+琼脂7g,PH=5.8,121℃,20min灭菌;
1/2MS固体培养基(1L):M519 2.215g+蔗糖30g+琼脂7g,PH=5.8,121℃,20min灭菌;
预培养/共培养培养基(1L):MS+0.6mg/L 6-BA+0.06mg/L NAA,PH=5.8;
分化培养培养基(1L):MS+0.06mg/L NAA+0.6mg/L 6-BA+Tim+Kan,PH=5.8;
生根培养培养基(1L):1/2MT(北京酷来搏)2.32g+1mg/L IBA+0.1mg/L IAA+活性炭,pH=5.8。
9.2遗传转化步骤
本试验中农杆菌介导的叶用莴苣遗传转化体系以子叶为外植体,培养温度为25±3℃,光周期为16/8h,光照强度12000lux。
具体步骤如下:
(1)无菌实生苗的培养
用50%的84消毒液消毒15min,无菌水冲洗4次,每次1min;将消毒过的种子摆放在铺有1层无菌滤纸(无菌水浸湿)的灭菌皿中培养,放入光照培养室。1-2d后种子萌发,取4-7天龄的两片子叶作为实验材料。
(2)预培养
剪下子叶切口在叶柄处(无生长点即可),放在铺有滤纸的预培养培养基(MS+0.5mg/L KT+0.05mg/L NAA)上。预培养1天(暗培养)
(3)侵染
侵染前菌液划线28℃培养,待菌板长出单菌落,挑取单菌落于MS侵染液中,28℃摇3小时左右,待菌液浑浊后,测OD值在0.1-0.3。把切好的子叶放入灭菌的皿或瓶中,倒入准备好的菌液,轻轻摇晃,侵染7分钟。将经过侵染的子叶外植体一片取出放到滤纸上,吸干表面的多余侵染液。然后转至共培养培养基(MS+0.5mg/L KT+0.05mg/L NAA)上。封口膜密封后,暗培养培养30小时左右(大于等于30小时)。
(4)分化培养
转移到分化培养基MS+0.5mg/L KT+0.05mg/L NAA+300mg/L Tim+50mg/L Kan每皿放20个子叶,两到三周后换新培养基中,是否有苗分化出来。
(5)切芽生根
预先准备无菌的生根培养基:1/2MS+0.05mg/L NAA+300mg/L特美汀,灭菌的镊子、剪刀、刀以及滤纸。待愈伤组织长出无菌苗之后,取出放到滤纸上,小心切下具有生长点的无菌芽。
(6)移栽和驯化
再生苗长出根系并足够健壮后,小心取出,在水中洗净培养基,移栽至营养土中,浇透水,对组培苗进行保湿处理,适应一周左右开始一点点揭开保湿膜至完全揭除,在温室中继续生长。
10、阳性苗鉴定及纯合突变体获得
在生菜基因组DNA序列(LOC111884676)上,两条gRNA前后200bp左右设计引物CRISPR-DNA-F/R,扩增阳性苗靶点序列(结果参见图4),扩增酶为诺唯赞高保真酶,反应程序为:95℃,3min;35×(95℃,15s;60℃,15s;72℃,60s);72℃,5min。反应体系如表3。PCR产物送测序,鉴定编辑类型。编辑类型主要有未编辑、纯合、杂合和嵌合四种。其中,未编辑与纯合编辑类型可直接通过PCR产物测序结果判定,杂合与嵌合类型PCR产物测序结果为双峰,需将PCR产物连接到Blunt-Zero克隆载体后,至少每个样品送10个单克隆进行测序后判定。最终得到3个株系的F
鉴定引物:
Cas9-F(SEQ ID NO.11):5’-GAACAGCGACAAGCTCATCG-3’;
Cas9-R(SEQ ID NO.12):5’-GAGCTGAGACAGGTCGATGC-3’;
CRISPR-DNA-F(SEQ ID NO.13):5’-CTCGAACAGCTACAATCCAGCGG-3’;
CRISPR-DNA-R(SEQ ID NO.14):5’-GTGGGTGCTGGTATCGGAGGCGG-3’。
表3 DNA序列扩增PCR反应体系(50μL)
11、高温下叶用莴苣lsarf3纯合突变体抽薹表型鉴定
于计算机温室中种植叶用莴苣野生型和上述获得的纯合突变植株,培养条件为:光照/黑暗:14h/10h;光强12000lux;相对湿度为65~75%。常温下,昼温/夜温:20±2℃/13±2℃。待幼苗长至六叶一心时,进行持续高温处理,昼温/夜温:33±2℃/25±2℃。高温处理lsarf3突变株系,植株表现出明显延迟抽薹现象(结果参见图6)。高温后32天植株茎尖石蜡切片结果显示,敲除突变体顶端生长锥突出明显,包被在叶原基中间,呈现半圆形,仍处在营养生长期,还未开始花芽分化;野生型植株顶端生长点扁平且圆缓已进入花芽分化始期(结果参见图7)。具体统计其抽薹、现蕾、开花时间,结果显示相较于野生型抽薹时间晚了20天左右,现蕾时间晚了32天,开花时间晚了48天(结果参见图8)。
实施例2OE-LsARF3载体构建
1、LsARF3基因克隆
PCR物序列如下:
LsARF3-F:5’-ATGATGTGTGGTTTAATCGATCTGAACACC-3’;
LsARF3-R:5’-TCACAATCCTTGTACACAACCATCATTTGATC-3’。
2、添加同源臂
以PRI101载体质粒为模板,使用诺唯赞高保真酶
3、载体线性化
SmaI-HFv2限制性内切酶(NEB)线性化PRI101载体,用1%琼脂糖凝胶电泳检测是否酶切完全(结果参见图10)。
4、同源重组
ClonExpress II One Step Cloning Kit(诺唯赞)同源重组PCR产物与线性化载体(步骤4中获得的酶切产物)。反应程序为37℃,30min,4℃,5min。反应体系如下表4。
表4重组反应体系(10μL)
6、大肠杆菌感受态转化
步骤如实施例1中步骤6
7、检测阳性克隆
康为Taq酶(2xTaq MasterMix(Dye))与引物G418-F/G418-R,用于菌落PCR,反应程序为94℃,2min;35×(94℃,30s;59℃,30s;72℃,30s);72℃,2min。反应体系如表5所示。用1%的琼脂糖凝胶检测PCR产物,条带大小约为363bp(结果参见图11)。将阳性的菌液送公司测序。测序引物为G418-F/G418-R。
菌落PCR及测序引物:
G418-R:5’-AAAAGCGGCCATTTTCCACC-3’;
G418-R:5’-AAAAGCGGCCATTTTCCACC-3’。
表5菌落PCR反应体系(25μL)
/>
8、农杆菌感受态转化
步骤如实施例1中步骤8
9、遗传转化
步骤如实施例1中步骤9
10、阳性苗鉴定
提取转基因苗叶片DNA,以DNA为模板,418-F/418-R为引物,扩增出现亮带的则为T
检测引物:
LsARF3-qF:5’-TGGTTGGAGTGCGTTCGTGAC-3’;
LsARF3-qR:5’-TGCTTGTGTCGCTCTTCGGATTC-3’。
11、植株表型结果鉴定
于计算机温室中种植叶用莴苣野生型和阳性过量表达植株,生长条件为光照/黑暗:14h/10h;光强12000lux;相对湿度为65~75%。常温下,昼温/夜温:20±2℃/13±2℃。同野生型植株相比,过量表达LsARF3转基因植株表现出明显的提前抽薹现象(结果参见图14)。表达量分别野生型植株表达量的4倍,8倍,16倍左右(结果参见图15)。花芽分化结果显示其在60天开始抽薹。统计其具体的抽薹、现蕾、开花时间,发现过量表达植株比野生型植株抽薹时间提前了约48天,现蕾时间提前了约33天,开花时间提前了约40天(结果参见图16)。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 株型相关蛋白OsSLA1及其编码基因在调控水稻叶倾角中的应用
- TaYgl蛋白及其编码基因在调控小麦叶色中的应用