血浆中阿扑吗啡的定量测定方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于药物检测领域,特别是血浆中阿扑吗啡的定量测定方法。
背景技术
阿扑吗啡是一种白色至淡灰色粉末的化学品,可用于抢救误食毒物及难以洗胃的患者;或用于治疗吸入石油蒸馏液(如煤油、汽油、煤焦油、燃料油或清洁液等)的患者,以防止严重的吸入性肺炎;也可用于治疗帕金森病,以及诊断和治疗帕金森病患者由于长期接受左旋多巴治疗而出现的严重的症状波动。
阿扑吗啡在舌下给药后快速吸收,给药后10min血浆中可测出阿扑吗啡,40-60min达血药峰浓度,2-4mg剂量的Cmax平均值为0.7ng/mL,个体间的变异在40%~70%。阿扑吗啡的药物剂量与Cmax和AUC 0→∞呈正相关,有明显的首过效应,舌下给药的生物利用度为16%~18%,而内服的生物利用度仅为1%~2%,因而吞服后几乎无效。阿扑吗啡通过舌下途径治疗勃起障碍时,Cmax仍较低,约4-7ng/mL的消除半衰期很短,在24-30min之间。低药理学剂量、血浆中获得的低浓度、较短的消除半衰期已经越来越需要进行药代动力学研究,特别是开发更方便的方法,是目前存在的具有挑战性的分析问题。
阿扑吗啡绝大部分转化成代谢物,和要的代谢为葡萄醛化结合物和硫酸结全物,及N-脱甲基化生成的去甲基阿扑吗啡葡萄醛化结合的含量极少,去甲基阿扑吗啡无任何药理作用。
现有的技术文献中,阿扑吗啡的分析技术主要包括气相色谱检测、火焰电离检测、质谱检测,耗时样品制备和衍生,高效液相色谱(HPLC)结合紫外、荧光以及电化学检测。以上检测方法的检出限均≥0.5ng/mL的样品1mL。此外,阿扑吗啡还可以通过液质联用仪进行检测,虽然降低了检测限,但样品处理方法为固相萃取或液液萃取,只能检测少量样本,方法操作复杂,时间和原材料成本较高。
例如文献《液相色谱-串联质谱法测定人血浆中阿扑吗啡》(郭继芬等,分析测试学报,2004年9月,第23卷增刊,39、43页)中公开了采用液相色谱-串联质谱法测定人血浆中阿扑吗啡的方法,然而该文献中未使用稳定剂,明显不符合基本技术原理,因此其数据结果存在疑问;此外,所用的提取溶剂为乙醚,这种麻醉气体对操作环境和操作步骤有更严格的安全要求,例如需要在通风橱中进行等。
又例如文献《人血浆中纳美芬和阿扑吗啡测定方法的建立及应用》(李平,沈阳药科大学,2007年5月)中公开了人血浆中阿扑吗啡的测定方法,建立了灵敏、专属、快速的LC/MS/MS法,测定血浆中的阿扑吗啡,并用于低剂量制剂的临床药动学研究,然而该论文中在样品预处理时用到了有毒的稳定剂2-巯基乙醇,因此也对操作环境和操作步骤有更严格的安全要求。
发明内容
针对以上现有技术的不足,本发明提供了血浆中阿扑吗啡浓度的测定方法,具体通过以下技术实现,整个检测操作过程相对简便快捷,无毒环保,试剂容易购买,检测限低至0.02-50ng/mL。
血浆中阿扑吗啡的定量测定方法,包括以下步骤:
S1、制备阿扑吗啡的校正标样、质控样品;
S2、取步骤S1制备的校正标样、质控样品、待测样品各80μL,零浓度血浆样品取80μL含0.18%抗坏血酸的空白血浆分别加至EP管中,所述校正标样、质控样品、待测样品的EP管中各自加入20μL内标工作液和400μL甲基叔丁基醚(MTBE),涡旋混合;分别在4℃,10000g条件下离心5min;各自取300μL的上清液加至96孔板中,吹干,加入含有100μL复溶液进行复溶,涡旋混合5min,制成校正标样工作液、质控样品工作液、待测样品工作液、零浓度血浆样品工作液,待用;
S3、分别取80μL含0.18%抗坏血酸的空白血浆样品和80μL含0.3%抗坏血酸的甲醇溶液加至EP管中,并分别加入20μL含0.3%抗坏血酸的甲醇溶液和400μL的甲基叔丁基醚,涡旋混合,分别在4℃,10000g条件下离心5min;各自取300μL的上清液加至96孔板中,吹干,加入100μL复溶液复溶,涡旋混合5min,制成空白血浆样品工作液和空白试剂样品工作液,待用;
S4、以步骤S3制备的空白血浆样品工作液、空白试剂样品工作液和步骤S2制备的零浓度血浆样品工作液对液相色谱-质谱系统进行测试;
S5、以步骤S2制备的校正标样工作液、质控样品工作液和待测样品工作液分别进行液相色谱-质谱分析,建立阿扑吗啡标准曲线,计算得到待测样品工作液中阿扑吗啡的含量。
通过采用本发明提供的上述方法,对分析物和内标的色谱图采集和色谱峰积分由分析软件Analyst 1.6.3(AB Sciex)进行处理,在优化积分参数后,对目标色谱峰自动积分,不允许对色谱峰进行单独积分或手动积分。
分析物浓度计算方法为:用软件Analyst 1.6.3分别获得分析物与内标工作液中的甲苯磺丁脲的色谱峰面积,采用加权(W=1/x
优选地,步骤S1中,阿扑吗啡的校正标样和质控样品的制备方法为:精密称取一定量的阿扑吗啡,重量经校正因子计算后,加入含有质量分数为0.3%的抗坏血酸的甲醇溶液,配制成浓度为1mg/mL的溶液;再用含有质量分数为0.3%的抗坏血酸且体积比为1:1的甲醇水溶液依次稀释成浓度为1000ng/mL、800ng/mL、500ng/mL、160ng/mL、40ng/mL、10ng/mL、2ng/mL、1ng/mL、0.4ng/mL的校正标样储备液,以及浓度为600ng/mL、60ng/mL、3ng/mL的质控样品储备液;最后各取校正标样储备液和质控样品储备液10μL,加入190μL的含有质量分数为0.18%的抗坏血酸的空白血浆样品,得到校正标样和质控样品;
步骤S2的所述零浓度样品是由80μL含有质量分数为0.18%抗坏血酸的空白血浆样品、20μL内标工作液和400μL甲基叔丁基醚涡旋混合而成。
采用上述方法制得的校正标样的阿扑吗啡浓度为0.02ng/mL、0.05ng/mL、0.1ng/mL、0.5ng/mL、2ng/mL、8ng/mL、25ng/mL、40ng/mL、50ng/mL;制得的质控样品的阿扑吗啡浓度为0.15ng/mL、3ng/mL、30ng/mL。
优选地,步骤S2中,所述内标工作液是浓度为1000ng/mL甲苯磺丁脲溶液(采用0.3%的抗坏血酸的甲醇溶液作为溶剂配制而成)。
优选地,步骤S4和S5中的液相色谱的条件为:色谱柱Venusil C18 Plus,2.1×50mm,5.0μm;流动相A为质量分数为0.1%甲酸水溶液,流动相B为乙腈;自动进样器清洗溶液为甲醇,柱温35℃,流速0.6mL/min,色谱柱平衡状态时的柱压为11.2MPa,自动进样器温度为4℃,进样体积为5μL,压脚提升量为50mm;自动进样器清洗模式为进样前后,清洗液洗针体积为200μL,自动进样器进样针清洗时浸泡时间为1s;
流动相的梯度为:
0.01min,A:75%、B:25%;
0.5min,A:10%、B:90%;
1min,A:10%、B:90%;
1.8min,A:75%、B:25%;
2min,A:95%、B:5%;3.0min停止。
优选地,步骤S4和S5中的质谱条件为:离子源为电喷离子源,离子化模式为正离子模式,监测模式为多反应监测,电喷雾电压为5000V,离子喷雾温度为450℃。
优选地,步骤S3的复溶液为含有质量分数为0.3%的抗坏血酸且体积比为1:1的甲醇水溶液。
与现有技术相比,本发明的有益之处在于:本发明提供的上述阿扑吗啡的检测方法,利用在阿扑吗啡纯溶液中和血浆中加抗坏血酸做稳定剂(抗坏血酸价格亲民且对实验人员几乎无危害),采用液液萃取:甲基叔丁基醚进行提取,成本低,且用时短,提取后10-15min即可吹干,线性相关系数r均大于0.99,线性关系良好,此方法经济、快速且可靠。
附图说明
图1为实施例1的方法中,定量下限(0.02ng/mL,编号CAL-0.02)校正标样工作液的HPLC-MS/MS色谱图(正离子模式);
图2为实施例1的方法中,定量上限(50ng/mL,编号CAL-50)校正标样工作液的HPLC-MS/MS色谱图(正离子模式);
图3为实施例1的方法中,待测样品2h-D2工作液的HPLC-MS/MS色谱图(正离子模式);
图4为实施例1的方法中,零浓度样品工作液的HPLC-MS/MS色谱图(正离子模式);
图5为实施例1的方法中,空白血浆样品工作液的HPLC-MS/MS色谱图(正离子模式);
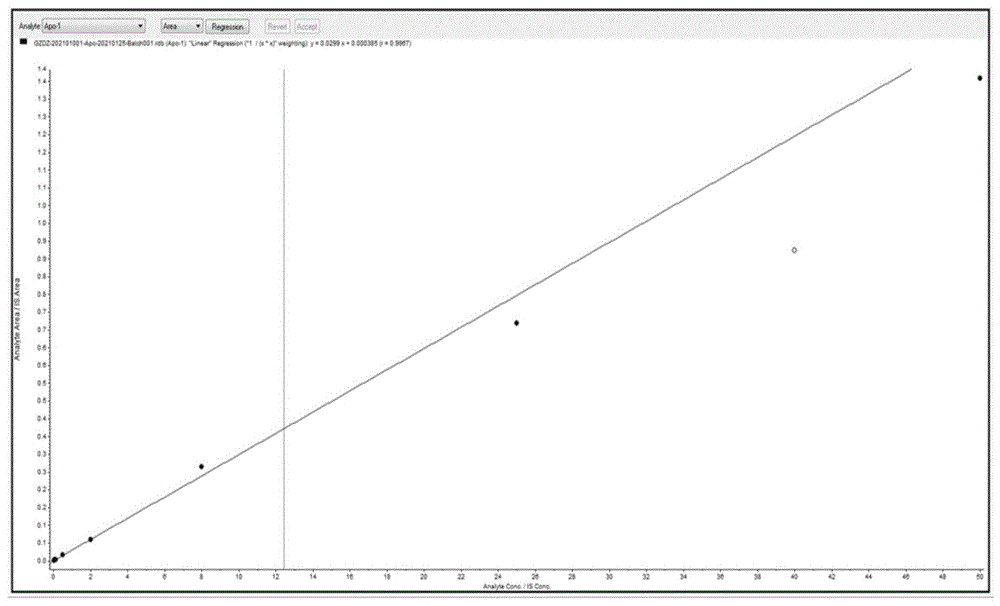
图6为实施例1的方法中,校正标样工作液的标准曲线图;
图1-5由上至下分别为总离子流图、内标色谱和阿扑吗啡的色谱图。
具体实施方式
下面将对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动条件下所获得的所有其它实施例,都属于本发明保护的范围。
实施例1
1、仪器和试剂
(1)试剂
本实施例采用的血浆中阿扑吗啡浓度的测定方法中,采用的血浆为湖北奥菲生物科技有限公司生产的含有抗凝剂EDTA-K
流动相A:用移液器移取1.00mL甲酸加入到装有1L蒸馏水的蓝盖瓶中,超声备用。储存条件:室温;有效期:一周。
含有质量分数为0.3%抗坏血酸的甲醇溶液:称量约0.3g抗坏血酸,加入甲醇至100mL,充分溶解,必要时超声;暂存条件:4℃;有效期:限当天使用;用于配制储备液和工作溶液。
含有质量分数为0.3%抗坏血酸且体积比为1:1的甲醇水溶液:分别量取250mL甲醇和250mL蒸馏水于同一蓝盖瓶中,混合均匀,称量约0.3g抗坏血酸,加入配置好的甲醇:水=1:1(V:V)溶液至100mL,充分溶解,必要时超声;暂存条件:4℃;有效期:限当天使用。
含6%的抗坏血酸溶液(稳定剂):称量约6g的抗坏血酸,加入蒸馏水至100mL,充分溶液,必要时超声;暂存条件:4℃;有效期:限当天使用。
含0.18%抗坏血酸的空白血浆样品:转移970μL猪空白血浆至EP管中,加入30μL含6%的抗坏血酸溶液,涡旋混合;暂存条件:4℃;有效期:限当天使用。
待测血浆(含有抗凝剂EDTA-K
(2)仪器
采用的各类仪器中,分析天平AS 60/220.R2十万分之一电子天平,采购自RADWAG公司;涡旋混合仪MX-S,采购自大龙兴创实验仪器(北京)有限公司;TGL-16.5M高速冷冻离心机,采购自上海卢湘仪离心机仪器有限公司;移液器,0.5-10μL,2-20μL,20-200μL,100-1000μL,采购自德国Brand公司;色谱柱为Venusil C18 Plus,2.1×50mm,5.0μm,Agela公司;高效液相色谱泵LC-20AD,采购自Shimadzu公司;自动进样器SIL-20AC,采购自Shimadzu公司;柱温箱CTO-20A,采购自Shimadzu公司;质谱仪API 4000,采购自AB Sciex公司;
2、质谱条件为:
离子源:Electrospray Ionization(ESI);
离子化模式(Ionization mode):正离子模式(Positive);
监测模式(Mode):多反应监测(MRM);
电喷雾电压(Ion Spray Voltage):5500V;
离子喷雾温度(Turbo Ion Spray Temp):550℃;
气帘气种类(Curtain Gas Type):Nitrogen Setting:10;
雾化气种类(Nebulizing Gas,Gas1):Nitrogen Setting:40;
辅助气种类(Auxiliary Gas,Gas 2):Nitrogen Setting:55;
数据采集时间(Acquisition Time):2.40mins;
具体条件如下表1所示:
表1分析物和内标的质谱条件
3、色谱条件为
色谱柱:Venusil C18 Plus,2.1×50mm,5.0μm,Agela公司;
流动相A(Mobile PhaseA:MPA):含0.1%甲酸的水溶液;
流动相B(Mobile PhaseB:MPB):乙腈;
自动进样器清洗溶液(Rinse Port Wash Solution):甲醇;
柱温箱温度(Column Temperature):35℃;
流速(Flow Rate):0.600mL/min;
色谱柱平衡状态时的大致柱压(Initial Back Pressure):11.2MPa(Typical);
自动进样器温度:4℃;
进样体积(Injection Volume):5.00μL(Typical);
压脚提升量(Needle Stroke):50mm;
自动进样器清洗模式(Rinse Mode):Before and after aspiration(进样前后);
清洗液洗针体积(Rinse Volume):200μL;
自动进样器进样针清洗时浸泡时间(Rinse Dip Time):1s;
色谱流动相洗脱梯度如下表2所示。
表2色谱流动相洗脱梯度
4、本实施例的测定方法的具体步骤为:
S1、制备阿扑吗啡的校正标样、质控样品、零浓度血浆样品;
阿扑吗啡的校正标样和质控样品的制备方法为:精密称取一定量的阿扑吗啡,重量经校正因子计算后,加入含有质量分数为0.3%的抗坏血酸的甲醇溶液,配制成浓度为1mg/mL的溶液;再用含有质量分数为0.3%的抗坏血酸且体积比为1:1的甲醇水溶液依次稀释成浓度为1000ng/mL、800ng/mL、500ng/mL、160ng/mL、40ng/mL、10ng/mL、2ng/mL、1ng/mL、0.4ng/mL的校正标样储备液,以及浓度为600ng/mL、60ng/mL、3ng/mL的质控样品储备液;最后各取校正标样储备液和质控样品储备液10μL,加入190μL的含有质量分数为0.18%的抗坏血酸的空白血浆样品,得到校正标样和质控样品;
校正标样的阿扑吗啡浓度为0.02ng/mL(定量下限,编号CAL-0.02)、0.05ng/mL(编号CAL-0.05)、0.1ng/mL(编号CAL-0.1)、0.5ng/mL(编号CAL-0.5)、2ng/mL(编号CAL-2)、8ng/mL(编号CAL-8)、25ng/mL(编号CAL-25)、40ng/mL(定量上限,编号CAL-40)、50ng/mL(编号CAL-50);制得的质控样品的阿扑吗啡浓度为0.15ng/mL(编号QC-0.15)、3ng/mL(编号QC-3)、30ng/mL(编号QC-30);
S2、取步骤S1制备的校正标样、质控样品、待测样品各80μL,零浓度血浆样品取80μL含0.18%抗坏血酸的空白血浆分别加至EP管中,所述校正标样、质控样品、待测样品和零浓度血浆样品的EP管中各自加入20μL内标工作液和400μL甲基叔丁基醚,涡旋混合;分别在4℃,10000g条件下离心5min;各自取300μL的上清液加至96孔板中吹干,加入100μL 0.3%抗坏血酸且甲醇和水的体积比为1:1的甲醇水溶液进行复溶,涡旋混合5min,制成校正标样工作液、质控样品工作液、待测样品工作液、零浓度血浆样品工作液,待用;
零浓度样品是由80μL含有质量分数为0.18%抗坏血酸的空白血浆样品、20μL内标工作液和400μL甲基叔丁基醚涡旋混合而成。
S3、分别取80μL含0.18%抗坏血酸的空白血浆样品和80μL含0.3%抗坏血酸的甲醇溶液加至EP管中,并分别加入20μL含0.3%抗坏血酸的甲醇溶液和400μL的甲基叔丁基醚,涡旋混合,分别在4℃,10000g条件下离心5min;各自取300μL的上清液加至96孔板中,吹干,加入100μL 0.3%抗坏血酸且甲醇和水的体积比为1:1的甲醇水溶液进行复溶,涡旋混合5min,制成空白血浆样品工作液和空白试剂样品工作液,待用;
S4、以步骤S3制备的空白血浆样品工作液、空白试剂样品工作液和步骤S2制备的零浓度血浆样品工作液对液相色谱-质谱系统进行测试;
零浓度样品工作液的HPLC-MS/MS色谱图如图3所示,空白血浆样品工作液的HPLC-MS/MS色谱图如图4所示;
S5、以步骤S2制备的校正标样工作液、质控样品工作液和待测样品工作液分别进行液相色谱-质谱分析,建立阿扑吗啡标准曲线,计算得到待测样品工作液中阿扑吗啡的含量。
校正标样工作液的定量下限(0.02ng/mL,编号CAL-0.02)的HPLC-MS/MS色谱图如图1所示,校正标样工作液的定量上限(50ng/mL,编号CAL-50)的HPLC-MS/MS色谱图如图2所示,待测样品2h-D2工作液的HPLC-MS/MS色谱图如图3所示;零浓度样品工作液的HPLC-MS/MS色谱图如图4所示,空白血浆样品工作液的HPLC-MS/MS色谱图如图5所示。
最终绘制的校正标样工作液的标准曲线图如图6所示,标准曲线方程y=0.0299x+0.000385(r=0.9967)。
下表3为校正标样工作液和质控样品工作液的回算浓度(ng/mL)及准确度偏差(%)汇总表;接受标准为接受标准:每条标准曲线至少75%并且至少6个校正标样的准确度偏差不超过±20%,质控样品至少2/3并且每个浓度至少50%质控样品的准确度偏差不超过±20%。
表3校正标样工作液和质控样品工作液的回算浓度(ng/mL)及准确度偏差(%)汇总表
经检测,待测样品2h-D2工作液中阿扑吗啡浓度0.23ng/mL。由上表3、4和附图1-6可以得出,采用本发明的方法,此方法利用在阿扑吗啡纯溶液中和血浆中加抗坏血酸做稳定剂(抗坏血酸价格亲民且对实验人员几乎无危害),采用液液萃取:甲基叔丁基醚进行提取,成本低,且用时短,提取后10-15min即可吹干,线性相关系数r均大于0.99,线性关系良好,此方法经济、快速且可靠。
- 一种血浆中丹酚酸B、阿司匹林、水杨酸的联合定量测定方法
- 一种血浆中特异性IgM抗体的定量测定方法