一种度鲁特韦的高效绿色制备方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于药物合成技术领域,具体涉及一种度鲁特韦的高效绿色制备方法。
背景技术
人类免疫缺陷病(human immunodeficiency virus,HIV)于1981年在美国首次发现,是一种感染人类免疫系统细胞的慢病毒(lentivirus),属逆转录病毒的一类。该病毒破坏人体的免疫能力,导致免疫系统失去抵抗力,从而导致各种疾病及癌症得以在人体内生存,最终导致艾滋病(获得性免疫缺陷综合征),至今无有效根治疗法。高效抗逆转录病毒疗法(Highly Active Antiretroviral Therapy,HAART)是艾滋病的常规治疗方法,该方法采用一个基础药物与两个核苷类逆转录酶抑制剂联合应用。整合酶抑制剂通过抑制HIV逆转录生成的双链DNA整合至宿主染色体发挥抗HIV活性,可作为基础药物使用。
度鲁特韦(Dolutegraver)是一种安全高效且具有较高基因屏障和良好药代特性的整合酶抑制剂,是由美国葛兰素史克旗下公司所研发,于2013年由FDA批准上市。度鲁特韦通过与整合酶活性部位结合来阻断逆转录病毒脱氧核糖核酸(DNA)整合的链转移步骤。该药物每日仅服用一次,对于治疗首次感染HIV-1患者,疗效与每日服用两次的雷特格韦的作用相当,并且安全性高,具有强效的抗耐药属性,与现有的HIV整合酶抑制剂雷特格韦(raltegravir)、埃替格韦(slvitegravir)相比,该药物的安全性提高。葛兰素史克制药公司和日本盐野义制药公司日前表示,此前对该药物的三期临床试验中,在接受度鲁特韦(dolutegravir)药物以及其他两种老版艾滋病药物治疗48周之后,患者体内88%的病毒被成功抑制,而服用吉利德科技技术公司的依发韦仑(atripla)药物之后,患者体内81%的病毒被抑制,由此可以看出,葛兰素史克制药公司的度鲁特韦(dolutegravir)药物略胜一筹。度鲁特韦作为FDA优先审评药物,2020年销售额达26亿美元,因此开发一种高效绿色的度鲁特韦制备方法很有意义。
发明内容
本发明解决的技术问题是提供了一种操作简单易行、原料廉价易得且反应效率高的度鲁特韦制备方法。
本发明为解决上述技术问题采用如下技术方案,一种度鲁特韦的高效绿色制备方法,其特征在于具体制备过程包括以下步骤:
(1)、在反应瓶中,预先加入一定量的四氢呋喃和一定量氢化钠然后在10℃下缓慢滴加一定量的甲醇与4-氯乙酰乙酸甲酯,使内温控制在20℃以下,反应结束后加入2N盐酸溶液,保持内温在10℃以下,加完至反应液pH=5-6;浓缩反应液,浅黄色油状物在60℃的条件下用油泵高真空蒸馏后得到无色液体;
(2)、把一定量的4-甲氧基乙酰乙酸甲酯加入到反应瓶中,在氮气保护下,反应温度降至15~25℃,缓慢滴加一定量的DMF-DMA,滴加完毕搅拌反应至TLC监控原料反应完全,降低反应温度至10~15℃,缓慢滴加氨基乙醛缩二甲醇,滴加完毕,搅拌一段时间后加入一定溶剂,过滤反应液,滤饼烘干后得到(Z)-甲基-2-(((2,2-二甲氧基乙基)胺基)亚甲基)-4-甲氧基-3-氧桥丁酸;
(3)、在反应瓶中加入(Z)-甲基-2-(((2,2-二甲氧基乙基)胺基)亚甲基)-4-甲氧基-3-氧桥丁酸和无水甲醇,在室温条件搅拌下,加入草酸二甲酯和甲醇钠;然后反应体系缓慢升温至回流,保持反应回流搅拌至TLC监控原料反应完全;把反应体系冷却至5~10℃,保持该温度区间,在搅拌状态下缓慢滴加2N盐酸调节反应液pH为5~6,然后将反应液转入单口瓶中,在真空条件下蒸除溶剂甲醇;然后向浓缩物中加入乙酸乙酯,搅拌条件下使浓缩物溶解;然后反应温度降至10~15℃,加入2N盐酸调节反应液pH为3,搅拌下加入水洗涤将反应体系转入分液漏斗中,上层有机相分出,并快速加入饱和碳酸钠溶液洗涤有机相pH为8;分出有机层,真空浓缩得到产品1-(2,2-二甲氧基乙基)-1,4-二氢-3-甲氧基-4-氧代-2,5-吡啶二甲酸甲酯;
(4)、取1-(2,2-二甲氧基乙基)-1,4-二氢-3-甲氧基-4-氧代-2,5-吡啶二甲酸甲酯加入甲醇中,在氮气保护下,0℃条件下加入氢氧化锂,加完后反应至TLC显示原料反应完全保持反应温度0~5℃下缓慢滴加2N盐酸溶液调节反应液pH为6~7,反应体系转移至单口瓶,真空条件下蒸除溶剂甲醇;再加入乙酸乙酯,0℃下继续滴加2N盐酸溶液调节反应液pH为1~2,移至分液漏斗分层,分出上层有机相,有机相浓缩旋干后经甲醇重结晶提纯后得到1-(2,2-二甲氧基乙基)-1,4-二氢-3-甲氧基-4-氧代-2,5-吡啶二甲酸;
(5)、把1-(2,2-二甲氧基乙基)-1,4-二氢-3-甲氧基-4-氧代-2,5-吡啶二甲酸-2-甲酯加入到无水甲酸中,在氩气保护下,搅拌条件下加热至65℃;反应一段时间后浓缩旋蒸除去甲酸,加入乙腈,搅拌使溶解,加入R-3-氨基丁醇,体系升温至内温80~82℃搅拌反应至TLC检测原料反应完全;在45℃条件下浓缩蒸除溶剂,加入二氯甲烷,再加入2N盐酸调节反应液pH为1~2,分出下层有机相,上层水相用二氯甲烷萃取三次,合并有机相,然后在真空条件下浓缩得到粗品,最后在甲醇中重结晶纯化得到纯品;
(6)、在反应瓶中加入(4R,12aS)-3,4,6,8,12,12a-六氢-7-甲氧基-4-甲基-6,8-二氧代-2H-吡啶[1',2':4,5]吡嗪并[2,1-b][1,3]噁嗪-9-甲酸和N,N-二甲基甲酰胺,室温搅拌;加入HATU和DIPEA;滴加完后,通过滴液漏斗滴加2.4-二氟苄胺溶于N,N-二甲基甲酰胺的溶液,控制内温不超过35℃,约20min加完室温反应至TLC检测无原料,室温下加水洗涤,加入二氯甲烷萃取多次,合并有机相,减压蒸除溶剂后加入丙酮,洗涤后烘干得到类白色固体甲氧基化合物;
(7)、把一定量的甲氧基化合物加入到无水四氢呋喃中,在氮气保护下,室温搅拌均匀;然后加入溴化锂,然后缓慢升温至回流,保持该温度搅拌至原料消失,停止加热,冷却至25~30℃,加入水洗涤;40℃下减压旋蒸除去大部分四氢呋喃,加入二氯甲烷,室温搅拌下加入2N盐酸溶液调节反应液pH为2~4,萃取,分层,水相用二氯甲烷萃取三次,合并有机相,真空浓缩后加入甲醇,有大量固体析出,抽滤后烘干得到度鲁特韦。
本发明的技术优势:新设计的度鲁特韦合成路线进一步优化了原有的路线,把反应步骤直接缩短了三步,减少了中间体后处理损失,有效提高反应收率。
附图说明
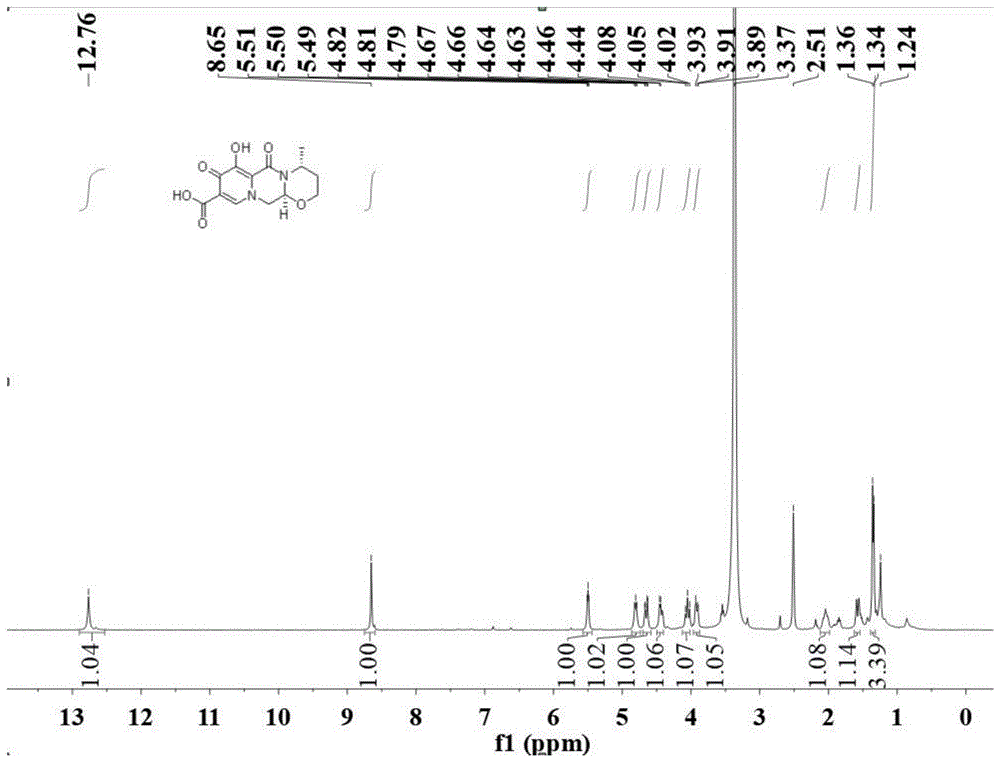
图1为实施例8中化合物8A的氢谱
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
实施例1
在带有搅拌的反应瓶中氮气保护下,把含量为60%氢化钠3g加入四氢呋喃30mL,在10℃左右缓慢滴加甲醇2g与4-氯乙酰乙酸甲酯5g的混合溶液,边搅拌边滴加使反应温度不超过20℃,滴加完后升至室温下搅拌反应2.5h,然后降温至0℃,缓慢加入2N盐酸溶液,保持内温在10℃以下,加完至反应液pH=5-6;真空浓缩反应液,蒸除反应液中的四氢呋喃,加入乙酸乙酯50mL搅拌萃取反应液2次,合并所有有机相,用水30mL洗涤两次,然后真空浓缩得到浅黄色油状物,浅黄色油状物在60℃的条件下用油泵高真空蒸馏后得到3.3g无色液体化合物2,即为4-甲氧基乙酰乙酸甲酯。
实施例2
把4-甲氧基乙酰乙酸甲酯150g(1.0mol)加入到反应瓶中,在氮气保护下,反应温度降至15~25℃,缓慢滴加DMF-DMA(120g,1.0mol),滴加完毕搅拌反应1.0h,TLC监控原料反应完全,降低反应温度至10~15℃,缓慢滴加氨基乙醛缩二甲醇(115g,1.1mol),滴加完毕,搅拌60min后加入甲基叔丁基醚100mL,搅拌20min,使固体分散悬浮,过滤反应液,滤饼烘干后得到固体191g,即为(Z)-基-2-(((2,2-二甲氧基乙基)胺基)亚甲基)-4-甲氧基-3-氧桥丁酸。
实施例3
把4-甲氧基乙酰乙酸甲酯150g(1.0mol)和乙醚100mL加入到反应瓶中,在氮气保护下,反应温度降至15~25℃,缓慢滴加DMF-DMA(120g,1.0mol),滴加完毕搅拌反应5.0h,TLC监控原料反应完全,降低反应温度至10~15℃,缓慢滴加氨基乙醛缩二甲醇(115g,1.1mol),滴加完毕,搅拌1.5h,可见反应体系中有大量固体出现,过滤反应液,滤饼烘干后得到固体172g,即为(Z)-基-2-(((2,2-二甲氧基乙基)胺基)亚甲基)-4-甲氧基-3-氧桥丁酸。
实施例4
在反应瓶中加入(Z)-甲基-2-(((2,2-二甲氧基乙基)胺基)亚甲基)-4-甲氧基-3-氧桥丁酸(化合物4)260g和无水甲醇2000mL,在室温条件和氩气保护下搅拌至完全溶解;保持室温搅拌,加入草酸二甲酯480g,搅拌10min;控制内温在室温条件下,快速加入甲醇钠125g,可见反应体系升温;然后反应体系缓慢升温至回流,可见溶液逐渐呈深红棕色,保持反应回流搅拌4h,TLC监控原料反应完全;把反应体系冷却至10℃,在搅拌状态下缓慢滴加2N盐酸调节反应液pH为5~6,可见溶液颜色逐渐由深棕红色变为土黄色;然后将反应液转入单口瓶中,在真空条件下,内温小于40℃下蒸除溶剂甲醇;然后向浓缩物中加入乙酸乙酯2000mL,搅拌条件下使浓缩物溶解;然后反应温度降至10~15℃,加入2N盐酸调节反应液pH为3,搅拌下加入水200mL,可见溶液上层有机相部分基本澄清;将反应体系转入分液漏斗中,上层有机相分出,并快速加入饱和碳酸钠溶液洗涤有机相pH为8;下层水相加入乙酸乙酯200mL萃取一次,震摇(若乳化加入酸水少量即可澄清)再次分层,有机相分出,加入饱和碳酸钠洗涤有机相pH为8;水相再次用同样办法反萃取三次,每次若发现有轻微乳化现象,可加入少量的2N盐酸溶液破除乳化;这三次的有机相分出后均快速加入饱和碳酸钠溶液调节pH为8;最后合并所有得到的有机相,加入水200mL,搅拌30.0min,分出有机层,真空浓缩得到产品(化合物5)269g。
实施例5
在反应瓶中,取(化合物5)32g(0.1mol)加入甲醇150mL中,在氮气保护下,0℃条件下加入无水LiOH(7.2g,0.3mol),加完后反应1.0h,TLC显示原料反应完全保持反应温度0~5℃下缓慢滴加2N盐酸溶液调节反应液pH为6~7,反应体系转移至单口瓶,真空条件下蒸除溶剂甲醇;再加入乙酸乙酯200mL,0℃下继续滴加2N盐酸溶液调节反应液pH为1~2,转移至分液漏斗分层,分出上层有机相,下层水相用乙酸乙酯50mL萃取3次,合并有机相,用饱和食盐水20mL洗涤一次,有机相浓缩旋干后经甲醇重结晶提纯后得到精品18.1g。
实施例6
在反应瓶中,把1-(2,2-二甲氧基乙基)-1,4-二氢-3-甲氧基-4-氧代-2,5-吡啶二甲酸-2-甲酯(化合物6,30g,0.1mol)加入到无水甲酸150mL中,在氩气保护下,搅拌条件下加热至70℃;反应约3h后,真空浓缩旋蒸除去甲酸,加入乙腈200mL,搅拌使溶解,加入R-3-氨基丁醇(12.5g,0.14mol),体系升温至内温80℃搅拌反应3h,TLC检测原料反应完全;真空浓缩蒸后加入二氯甲烷200mL,搅拌下加入水100mL,再加入2N盐酸调节反应液pH为1~2,搅拌10min后分出下层有机相,上层水相用二氯甲烷100mL萃取三次,合并有机相,用饱和食盐水100mL洗涤3次;然后在真空条件下浓缩得到粗品,最后在甲醇中重结晶纯化得到纯品24.7g
实施例7
在反应瓶中加入化合物8(6g,1.0eq)和N,N-二甲基甲酰胺100mL,室温搅拌;加入HATU(1.0eq),搅拌20min后,保持室温状态,逐滴入DIPEA(2.0eq);滴加完后,通过滴液漏斗滴加2.4-二氟苄胺(1.1eq)溶于N,N-二甲基甲酰胺的溶液30mL,控制内温不超过30℃反应1~2h,TLC检测无原料。室温下加水50mL,加入二氯甲烷100mL萃取多次,分层,水层再用二氯甲烷50mL萃取2次,合并有机相,减压蒸除溶剂后加入丙酮50mL,洗涤后烘干得到类白色固体化合物9(7.3g)
实施例8
在反应瓶中,把化合物8(0.20g,0.65mmol)加入到无水四氢呋喃10mL中,在氮气保护下,加入无水溴化锂(0.13mg,1.5mmol),然后缓慢升温至回流,TLC检测至反应原料消失,产物生成,停止加热,冷却至室温,体系旋蒸除去大部分四氢呋喃旋干后,加入少量水,室温搅拌下加入2N盐酸溶液调节反应液pH为2~3,加入二氯甲烷50mL,萃取、分层,水相用二氯甲烷萃取三次,合并有机相,浓缩烘干得到产品8A:0.17g;将化合物8A(0.17g,1.0eq)和2.4-二氟苄胺(91mg,1.1eq)溶到N,N-二甲基甲酰10mL中,加入HATU(0.22g,1.0eq)和DIPEA(0.22g,3.0eq),体系在室温下反应1~2h,TLC监控反应原料消失,产物生成停止搅拌,体系加入适量二次水,体系析出大量固体抽滤,滤饼用少量异丙醇淋洗烘干后得到类白色产物10(0.22g)。
实施例9
在反应瓶中,把化合物9(4g,0.01mol)加入到无水四氢呋喃40mL中,在氮气保护下,室温搅拌均匀;然后快速加入无水溴化锂(2.6g,0.03mol),然后缓慢升温至回流,TLC检测反应原料反应完全,停止加热,冷却至25~30℃,加入水50mL,搅拌20min;真空旋蒸除去大部分四氢呋喃,加入二氯甲烷50mL,室温搅拌下加入2N盐酸溶液调节反应液pH为2~4,萃取,分层,水相用二氯甲烷40mL萃取三次,合并有机相,真空浓缩后加入甲醇20mL,有大量固体析出,抽滤后烘干得到固体3.3g。
以上实施例仅为说明本发明的技术思想,不能以此限定本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案基础上所做的任何改动,均落入本发明保护范围之内。
- 一种改进的度鲁特韦制备工艺
- 一种度鲁特韦中间体的合成方法
- 一种度鲁特韦关键中间体的制备方法
- 一种度鲁特韦母核中间体的制备方法