一种大规模生产唑吡坦的合成方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于化学合成技术领域。更具体地,涉及一种大规模生产唑吡坦的合成方法。
背景技术
唑吡坦是首先面市的新一代非苯二氮卓类镇静催眠药物,属于咪唑并吡啶类药物。通过选择性地与中枢神经系统的苯二氮卓受体——GABAA受体的一部分,以增加GABA的传递,产生药理作用。
现有技术公开了多条唑吡坦的合成路线及方法,如美国专利申请US4794185A公开了以5-甲基-2-吡啶胺和对甲基溴代苯乙酰为起始原料合成唑吡坦的方法,是目前商业化合成唑吡坦的主要路线之一;但是该路线反应步骤较多,生产周期较长,产率较低,且硼氢化钠法还原加入到酸性介质中会产生氢气迅速放出,具有一定的生产风险。为了提高中间体4的收率,中国专利申请CN113264932A对还原反应步骤进行了优化和调整,采用钯碳加氢的方式还原得到化合物4,提高中间产品(化合物4)收率,但该方法中氢化反应属于国家十八种危险性管控反应中的一种,生产时具有一定的危险性,并且,钯碳催化剂的价格昂贵,且该路线反应步骤仍然较多,生产周期仍很长,总体产率较低。
为了缩短反应路线,减少反应步骤,中国专利申请CN100408577C公开了采用N,N-二甲基乙醛酰胺半水合物固体与化合物1缩合反应得到化合物2,再经过三溴化磷还原、游离得到唑吡坦的合成路线与方法,该路线反应步骤较短,也避开了危险性反应,但该反应所使用的原料N,N-二甲基乙醛酰胺半水合物几乎无商业化销售途径,较难大量获取;此外,该方法仅在实验室进行了小试实验,投料量极小,数据不能支撑唑吡坦的商业化大规模生产,且并未公开使用该方法得到的唑吡坦的纯度数据;另一方面,通常仅采用三溴化磷还原时,化合物的转化率不高,还容易造成原料反应不完全而与产物分离困难,后处理纯化操作繁琐。
因此,迫切需要提供一种转化率高、产率高、经济、高效的商业化合成唑吡坦的方法。
发明内容
本发明要解决的技术问题是克服现有唑吡坦合成方法产率低、纯度不高杂质多、步骤繁琐等缺陷和不足,提供一种产率高、纯度高、经济高效的唑吡坦合成方法。
本发明上述目的通过以下技术方案实现:
一种大规模生产唑吡坦的合成方法,合成路线如下:
具体包括以下步骤:
将化合物2溶于有机溶剂中,加入路易斯酸催化剂,降温至-40~0℃,滴加三溴化磷溶液,滴加完毕后,升温至回流,反应完全,降温并将反应液加入碳酸钠溶液中淬灭反应,后处理,即得。
进一步地,所述路易斯酸催化剂选自氯化铝、氯化铁、氯化锌、三氟化硼、三氧化硫、冰乙酸、磷酸、硫酸中的一种或多种。
更进一步地,所述化合物2与路易斯酸催化剂的摩尔比为1:(0.4~1.2)。
进一步地,所述化合物2与三溴化磷的摩尔比为1:(2~10)。优选地,所述三溴化磷的与化合物2的摩尔比为1:(2~6.7)。
优选地,所述加入路易斯酸催化剂后,降温至-20~0℃。
更进一步地,所述有机溶剂选自四氢呋喃、醋酸异丙酯、1,4-二氧六环、1,2-二氯乙烷、乙腈、丙酮中的一种或多种。优选地,所述有机溶剂为四氢呋喃、1,4-二氧六环或1,2-二氯乙烷。
进一步地,所述化合物2与有机溶剂的质量比为1:(10~14)。优选地,所述化合物2与有机溶剂的质量比为1:(10~12)。
更进一步地,所述回流时间为0.5~4h。优选地,所述回流时间为0.5~2h。
进一步地,所述三溴化磷溶液为三溴化磷溶于有机溶剂中制成。
更进一步地,所述碳酸钠溶液的浓度为5~25%,温度为0~5℃;优选地,碳酸钠溶液的浓度为10~15%。
进一步地,所述后处理具体包括以下步骤:
将反应液减压浓缩至体系无明显液滴滴出,加入乙酸乙酯带走溶剂或杂质,再次减压浓缩至体系无明显液滴滴出;降温至20~30℃,用乙酸乙酯和水萃取1~3次,固液分离,固体用水洗涤、干燥,即得。
本发明具有以下有益效果:
本发明以三溴化磷和路易斯酸组成的复合催化剂为合成唑吡坦的催化体系,进一步优化控制反应条件,可商业化大规模生产得到纯度高达99.5%以上的唑吡坦,且收率较高;该方法步骤简单、操作简便、反应条件温和,并且生产周期短、成本较低、安全环保,非常适用于大规模产业化生产唑吡坦。
附图说明
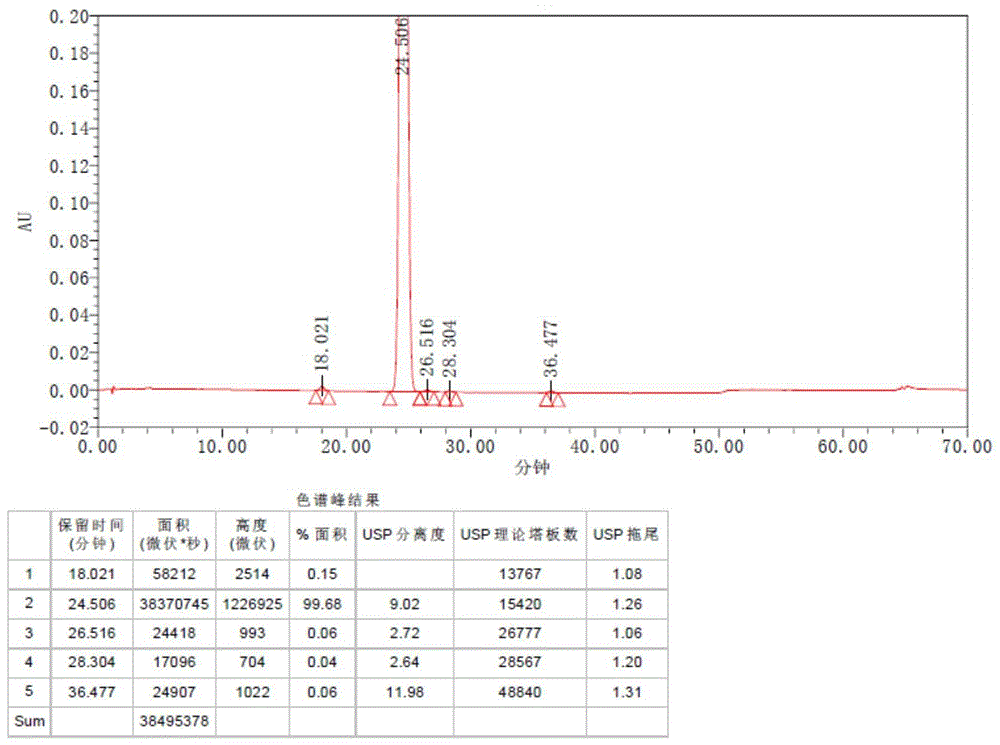
图1为本发明实施例3中批号为20201217X-2的检测色谱图。
图2为本发明实施例3、实施例6中批号为20210105X-1的检测色谱图。
图3为本发明实施例3中批号为20210119X-2的检测色谱图。
图4为本发明实施例4中批号为20210107X-1的检测色谱图。
图5为本发明实施例4中批号为20210122X-2的检测色谱图。
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
其中,本申请唑吡坦的合成方法反应路线如下:
其中,本发明中的化合物2参照申请人的中国专利申请CN113264932A所公开的方法合成得到。
实施例1唑吡坦大规模产业化生产
所述唑吡坦的合成步骤具体如下:
S1、称量物料,关闭罐底阀,向1000L反应罐中加入饮用水(400kg)、碳酸钠(72kg),开启搅拌,降温至0~5℃,得碳酸钠溶液,备用;
S2、称量物料,关闭罐底阀,向500L反应罐中加入化合物2(20kg)、四氢呋喃(160kg),开启搅拌,加入三氯化铝(3.3kg),降温,控制温度在0℃以下,缓慢滴加含三溴化磷(67kg)的四氢呋喃溶液;
S3、滴加完毕后,升温至回流,反应1小时后,降温至20℃以下,缓慢将反应液加入到步骤S1所得碳酸钠溶液中淬灭反应,淬灭结束后,测pH=7~9;
S4、减压浓缩至体系无明显液滴滴出,加入乙酸乙酯,减压浓缩至体系无明显液滴滴出;降温至25±5℃,加入乙酸乙酯(35kg)和饮用水(100kg),搅拌,将物料转移至离心机离心,固体用饮用水洗涤离心后,转移至真空干燥箱中干燥,得到唑吡坦15.988kg,收率84.1%,唑吡坦纯度为100.00%。
[M+H]308.17438;
实施例2唑吡坦大规模产业化生产
所述唑吡坦的合成步骤具体如下:
S1、称量物料,关闭罐底阀,向1000L反应罐中加入饮用水(300kg)、碳酸钠(54kg),开启搅拌,降温至0~5℃,得碳酸钠溶液,备用;
S2、称量物料,关闭罐底阀,向500L反应罐中加入化合物2(15kg)、四氢呋喃(120kg),开启搅拌,加入氯化锌(3.2kg),降温,控制温度在0℃以下,缓慢滴加含三溴化磷(50.25kg)的四氢呋喃溶液;
S3、滴加完毕后,升温至回流,反应1小时后,降温至20℃以下,缓慢将反应液加入到步骤S1所得碳酸钠溶液中淬灭反应,淬灭结束后,测pH=7~9;
S4、减压浓缩至体系无明显液滴滴出,加入乙酸乙酯复溶,减压浓缩至体系无明显液滴滴出;降温至25±5℃,加入乙酸乙酯(26kg)和饮用水(75kg),搅拌,将物料转移至离心机离心,固体用饮用水洗涤离心后,转移至真空干燥箱中干燥,得到唑吡坦11.863kg,收率83.2%,唑吡坦纯度为99.91%。
实施例3不同催化剂对反应的影响
参考实施例1的生产参数,采用表1所示不同催化体系制备得到唑吡坦。结果参见表1,其中,批号20201217X-2、20210105X-1、20210119X-2的色谱图参图1~3,纯度均在99.5%以上。
表1不同催化剂对反应的影响
注:杂质1~4为检测时发现的未知杂质。
由表可见,单独采用PBr
实施例4不同三溴化磷添加量对反应的影响
参考实施例1的生产参数,采用表2所示不同溴化磷添加量制备得到唑吡坦。结果参见表2,其中,批号20210107X-1的色谱图参图4。
表2不同三溴化磷添加量对反应的影响
注:杂质1~2为检测时发现的未知杂质。
由表可见,采用路易斯酸/PBr
实施例5不同三溴化磷滴加温度对反应的影响
参考实施例1的生产参数,采用表3所示不同三溴化磷滴加温度制备得到唑吡坦。结果参见表3。
表3不同三溴化磷滴加温度对反应的影响
由表可见,PBr
实施例6不同路易斯酸催化剂添加量对反应的影响
参考实施例1的生产参数,采用表4所示不同路易斯酸催化剂并进一步限定其添加量制备得到唑吡坦。结果参见表4,其中,批号为20210105X-1的色谱图参见图2。
表4不同路易斯酸催化剂添加量对反应的影响
由表可见,路易斯酸(磷酸)的添加量不同,对该反应的收率及纯度有一定的影响,优选地,路易斯酸的添加量为0.4~1.2eq.。
实施例7不同溶剂添加量对反应的影响
参考实施例1的生产参数,采用表5所示不同溶剂添加量制备得到唑吡坦。结果参见表5,其中,批号为20210122X-2的色谱图参见图5。
表5不同溶剂添加量对反应的影响
由表可见,溶剂的使用量对反应的进行及产物的纯度和收率有一定的影响,优选地,溶剂的添加量为10~14倍。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 一种他唑巴坦手性异构体的合成方法
- 一种利用光催化三组分反应合成唑吡坦的方法
- 一种电化学三组分合成唑吡坦的方法