髓鞘特异性磁共振造影剂及其制备方法和应用
文献发布时间:2024-04-18 19:52:40
技术领域
本发明属于医药技术领域,具体涉及髓鞘特异性磁共振造影剂及其制备方法和应用。
背景技术
脊椎动物神经元的轴突被一层由脂肪组织组成的细胞膜包围,称为髓鞘,髓鞘在保护轴突和促进冲动传递方面起作用。髓鞘的形成是脊椎动物神经系统最基本的生理过程之一。髓鞘病理学存在于许多神经系统疾病中,如多发性硬化(MS)、外伤性脊髓损伤(SCI)和各种白质营养不良。髓鞘损伤被认为是自身免疫性疾病的主要影响因素,如多发性硬化(MS),这是一种髓鞘功能退化的疾病。疾病可能会导致向肌肉传递信息的速度减慢,并最终失去对肌肉的控制。
磁共振成像(MRI)已成为诊断和监测MS及相关疾病进展的主要工具;然而,常规MRI信号强度的变化仅反映组织含水量的变化,不能区分脱髓鞘病变和其他炎症病变。它对髓鞘化程度和髓鞘再生的预后无指导作用。这只是宏观组织损伤总体变化的一个非特定测量。特殊的磁共振功能序列存在成像时间过长、微观分辨力差等原因,无法直观准确的反映髓鞘的变化。
髓鞘损伤治疗干预的重点是保护髓鞘完整性和促进髓鞘修复,这需要直接检测和量化体内髓鞘含量。然而,用于髓鞘可视化的组织学探针是不进入大脑的抗体或带电组织化学试剂。髓鞘特异性MR对比剂的研究仅基于Luxol Fast Blue(LFB),LFB是顺磁性铜核的组织学染色。然而,LFB具有低松弛性、低溶解度和有限的组织通透性。因此,需要一种有效的成像工具,将疾病进展与髓鞘化程度相关联,能够轻易进入大脑并选择性地结合到髓鞘化区域,并且具有高度松弛性。
为解决上述问题,本发明选用的二苯基取代的乙烯的衍生物为髓鞘特异性化合物,钆喷酸葡胺和钆特酸葡胺为磁共振造影剂基团,通过对中间的连接臂的优化筛选以达到优化药代动力学的目的,开发髓鞘特异性磁共振造影剂。
发明内容
本发明提供一种髓鞘特异性磁共振造影剂,其特征在于所述造影剂具有式I所示结构:
/>
其中,X为连接基团,选自-CO(CH
R为Gd螯合基团,选自
本发明的另一实施方案提供上述式I结构的髓鞘特异性磁共振造影剂,其特征在于选自如下化合物6a-d、12、18a-b、24a-d:
。
本发明的另一实施方案提供一种用于合成式I结构的髓鞘特异性磁共振造影剂的中间体II,其特征在于中间体II结构如下:
/>
其中,X为连接基团,选自-CO(CH
上述式II中间体选自如下化合物5a-d、11、17a-b、23a-d:
本发明的另一实施方案提供上述式I结构的化合物在制备髓鞘特异性磁共振造影剂中的应用。
本发明的另一实施方案提供上述式I结构的化合物在髓鞘损伤检测中的应用。
本发明所述“C1-C6的烷基”优选甲基、乙基、丙基、异丙基、环丙基等;“卤素”优选氟、氯、溴、碘。
附图说明
图1是化合物和6a-d,18a-b,24a-d,12和MeDAS的激发波长(Em 370±10nm)发射波长(Ex 460±10nm);
图2是化合物6a-d,18a-b,24a-d,12,Gd-DOTA和Gd-DTPA的随浓度变化的T1WI图像;
图3是化合物6a-d,18a-b,24a-d,12,Gd-DOTA和Gd-DTPA的弛豫率;
图4是化合物18a、18b、Gd-DOTA和Gd-DTPA的金属转移性图;
图5是在大脑部位的化合物6a-d,18a-b,24a-d,12的染色图;
图6是在小脑部位的化合物6a-d,18a-b,24a-d,12的染色图;
图7是在脊髓部位的化合物6a-d,18a-b,24a-d,12的染色图;
图8是化合物6a在MCAO模型组大脑的缺血同侧和缺血对侧的染色图,并与商用LFB进行对比;
图9是化合物18a在大鼠对照组和MCAO组浸泡前后的T1WI图像;
图10:化合物18a在大鼠对照组和MCAO组的侧脑室给药后,扫描T1WI和T1 mapping图像;
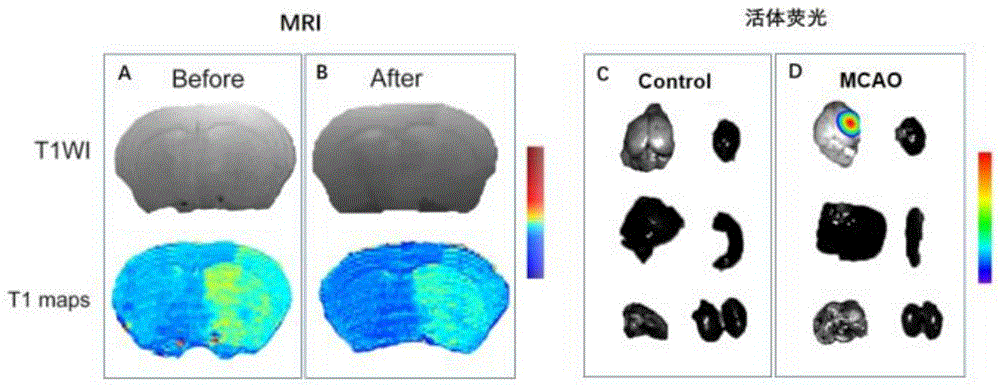
图11是A-B:化合物6a在MCAO小鼠尾静脉给药后的MRI扫描T1WI和T1mapping图像;C-D:MCAO对照组和MCAO尾静脉给药组的小动物活体荧光图像(从左到右,从上到下:脑、心、肝、脾、肺、肾)。
下面通过具体的制备例和实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,更非将本发明的保护范围局限于此。
具体实施方式
实施例1髓鞘特异性磁共振造影剂6a~6d的制备
本发明髓鞘特异性磁共振造影剂6a~6d的制备可以通过如下通式来进行:
概括地讲,本发明通过简单氨基酸连接的髓鞘特异性磁共振造影剂的制备包括以下步骤:
Dmedas在缩合剂催化下与Boc保护的不同碳链长度的氨基酸发生酰化反应,得到的产物在三氟乙酸条件下脱除Boc保护基,得到修饰的Dmedas.
不同碳链长度的氨基酸修饰的Dmedas在缩合剂的催化下,与单羧基暴露的DTPA发生酰化反应后,在三氟乙酸的条件下脱除叔丁基保护基暴露出四个羧基,四个羧基中的三个与三价钆离子发生螯合,经分离纯化即得化合物6a~6d。
具体的合成方法如下:
在100mL锥形瓶中依次加入Boc-甘氨酸(1.1g,6.3mmol),HBTU(3.2g,8.4mmol),DIPEA(3.66ml,21mmol)溶于DMF中,室温下搅拌反应0.5h,加入1(1g,4.2mmol),室温下搅拌反应过夜。TLC检测反应完成后,向反应液加水淬灭,乙酸乙酯萃取,饱和食盐水洗,无水硫酸镁干燥,硅胶柱层析(PE:EA=30:1-2:1),收集到产品棕红色固体678mg,收率46%。即化合物2a。MS(ESI):m/z:396.22[M+H]
1
13
2b~2d的合成方法同2a,只是将原料中的Boc-甘氨酸依次更换成4-Boc-氨基丁酸,5-Boc-氨基戊酸,6-Boc-氨基己酸。
2b,棕红色固体,收率49%,MS(ESI):m/z:424.25[M+H]
2c,棕红色固体,收率47%,MS(ESI):m/z:437.26,[M+H]
13
2d,棕红色固体,收率51%,MS(ESI):m/z:452.28,[M+H]
在50mL锥形瓶中依次加入2a(678mg,1.78mmol)溶于DCM中,冰浴下加入TFA,室温反应过夜。TLC检测反应完成后,将反应液蒸干,加入20ml二氯甲烷后再次蒸干,反复三遍,即得产品为棕色固体506mg。即化合物3a,收率98%,。MS(ESI):m/z:296.1688[M+H]
3b~3d的合成方法同3a,只是将原料中的2a分别更换为2b,2c,2d。
3b,棕色固体,收率为98%,MS(ESI):m/z:324.20[M+H]
3c,棕色固体,收率为97%,MS(ESI):m/z:338.21[M+H]
3d,棕色固体,收率为98%,MS(ESI):m/z:352.23[M+H]
在50mL锥形瓶中依次加入DTPA-3tBu(1.59g,2.57mmol),HATU(1.3g,3.43mmol),DIPEA(1.5ml,8.58mmol)溶于DMF中,室温下搅拌反应0.5h,加入3a(506mg,1.72mmol),室温反应过夜。TLC检测反应完成后,向反应液加水淬灭,EA萃取,饱和食盐水洗,无水硫酸镁干燥,硅胶柱层析(DCM—DCM:MeOH=20:1),再经分离,收集到产品棕红色油状液体100mg,收率6.7%。即化合物4a。MS(ESI):m/z:895.54[M+H]
1
4b~4d的合成方法同4a,只是将原料中的3a分别更换为3b,3c,3d。
4b.棕红色油状液体,收率7%,MS(ESI):m/z:923.57[M+H]
4c.棕红色油状液体,收率9%,MS(ESI):m/z:937.59[M+H]
4d.棕红色油状液体,收率7%,MS(ESI):m/z:951.60[M+H]
在50mL锥形瓶中依次加入4a(100mg,0.114mmol)溶于DCM中,加入TFA,室温反应过夜。TLC检测反应完成后,向反应液蒸干,甲醇带两遍,即得5a 73mg,为无色油状液体,收率98%。MS(ESI):m/z:669.29[M-H]
13
5b~5d的合成方法同5a,只是将原料中的4a分别更换为4b,4c,4d。
5b,无色油状液体,收率为98%,MS(ESI):m/z:697.32[M-H]
5c,无色油状液体,收率为99%,MS(ESI):m/z:711.34[M-H]
5d,无色油状液体,收率为98%,MS(ESI):m/z:725.35[M-H]
在50mL锥形瓶中依次加入5a(19mg,0.023mmol)溶于MeOH/H
6b~6d的合成方法同6a,只是将原料中的5a分别更换为5b,5c和5d。
6b,白色粉末状固体,收率50%。HRMS:m/z[M+H]
6c,白色粉末状固体,收率52%。HRMS:m/z[M+H]
6d,白色粉末状固体,收率50%。HRMS:m/z[M+H]
实施例2髓鞘特异性磁共振造影剂12的制备
概括地讲,本发明通过三氮唑连接的髓鞘特异性磁共振造影剂的制备包括以下步骤:
Dmedas与4-叠氮基-1-丁酸在缩合剂的催化下发生酰化反应得到经丁酸修饰得DMedas。
丙炔胺在缩合剂的催化下与单羧基暴露的DTPA发生酰化反应得到经丙炔基修饰的DTPA。
经丁酸修饰得DMedas与经丙炔基修饰的DTPA在碘化亚铜的催化下经click反应后,在三氟乙酸的条件下脱除叔丁基保护基暴露出四个羧基,四个羧基中的三个与三价钆离子发生螯合,经分离纯化即得12。
具体的合成方法如下:
在50mL锥形瓶中依次加入7(2g,3.24mmol),丙炔胺(150uL,2.16mmol),PyBOP(2.25g,4.32mmol),DIPEA(1.88ml,10.8mmol)和DMF,室温下搅拌反应过夜。TLC检测反应完成后,向反应液水淬灭,EA萃取,饱和食盐水洗,无水硫酸镁干燥,浓缩得黄色油状液体2.16g,收率87%,未经纯化,直接用于下一步反应。即化合物8。MS(ESI):m/z:655.42[M+H]
在50mL锥形瓶中依次加入1(1g,4.2mmol),4-叠氮基丁酸(815mg,6.3mmol),PyBOP(4.37g,8.4mmol),DIPEA(3.66ml,21mmol)溶于DMF中,室温下搅拌反应过夜。TLC检测反应完成后,向反应液水淬灭,EA萃取,饱和食盐水洗,无水硫酸镁干燥,硅胶柱层析(PE:EA=30:1-2:1),收集到产品700mg,收率50%。即化合物9为红色粉末状固体。MS(ESI):m/z:350.19[M+H]
在50mL锥形瓶中依次加入8(281mg,0.43mmol),9(75mg,0.215mmol),CuI(8mg,0.04mmol),DIPEA(174ul,1mmol)溶于DMF中,N
在50mL锥形瓶中依次加入10(40mg,0.0399mmol)溶于10ml DCM中,加入6ml TFA,室温下震荡反应5h.TLC检测反应完成后,反应液蒸干,甲醇带两遍,直接用于下一步反应.即化合物11,收率98%,为红色油状液体。MS(ESI):m/z:778.36[M-H]-。
在50mL锥形瓶中依次加入11(35mg,0.045mmol)溶于MeOH/H
实施例3髓鞘特异性磁共振造影剂18a,18b的制备
概括地讲,本发明通过取代的磺酰脒基连接的髓鞘特异性磁共振造影剂的制备包括以下步骤:
Dmedas在碱催化下与溴丙炔发生取代反应,得到炔基修饰的Dmedas.炔基修饰的Dmedas在碘化亚铜的催化下与对位取代的苯磺酰叠氮、单Boc保护的丙二胺发生三组分偶联生成带磺酰脒基的中间体,该中间体在三氟乙酸的条件下脱除Boc保护基后,与单羧基暴露的DOTA发生酰化反应后,在三氟乙酸的条件下脱除叔丁基保护基暴露出四个羧基,四个羧基中的三个与三价钆离子发生螯合,经分离纯化即得18a~18b。
具体的合成方法如下:
在50mL锥形瓶中依次加入1(2g,8.4mmol)溶于DMF后,加入3-溴丙炔(1.5g,12.6mmol)和DIPEA(4.4mL,25mmol),室温下搅拌反应5~6h。TLC检测反应完成后,水淬灭,EA萃取,饱和食盐水洗,无水硫酸镁干燥,硅胶柱层析:PE:EA=10:1~5:1得13粉红色固体377mg,收率37%。MS(ESI):m/z:277.1624[M+H]
1
在50mL锥形瓶中依次加入N-叔丁氧羰基-1,3-丙二胺(94mg,0.54mmol)溶于DCM中,加入三乙胺,氮气保护下室温下搅拌反应10min,加入13(150mg,0.54mmol),4-氟苯磺酰基叠氮(128mg,0.65mmol),CuI(10.2mg,0.054mmol),搅拌反应过夜。TLC检测反应完成后,向反应中加入适量水和DCM稀释,搅拌反应30min,DCM萃取,饱和食盐水洗,无水硫酸镁干燥,浓缩,硅胶柱层析:PE:EA=30:1~1:1,得固体240mg,为绿色油状液体,收率46%,即化合物14a。MS(ESI):m/z:625.29[M+H]
1
14b的合成方法同14a,只是将原料中的4-氟苯磺酰叠氮更换成对甲苯磺酰叠氮,14b为绿色油状液体,收率49%。MS(ESI):m/z:620.31[M+H]
在50mL锥形瓶中依次加入14a(240mg,0.39mmol)溶于7ml DCM后,加入3mL TFA,室温下搅拌反应过夜。TLC(DCM:MeOH=10:1)。TLC检测反应完成后,将反应液蒸干,加入三乙胺调pH碱性,浓缩蒸干,得棕红色油状液体,即化合物15a,收率95%.MS(ESI):m/z:524.24[M+H]
15b的合成方法同15a,只是将原料中的14a更换成14b,15b为绿色油状液体,收率96%。MS(ESI):m/z:520.26[M+H]
在50mL锥形瓶中依次加入15a(182mg,0.351mmol),DOTA-3tBu(301mg,0.526mmol),HATU(266mg,0.701mmol),DIPEA(305uL,1.75mmol)溶于5ml DMF后,室温下搅拌反应过夜。TLC检测反应完成后,水淬灭,DCM萃取,饱和食盐水洗,无水硫酸镁干燥,过滤,浓缩蒸干,硅胶柱层析:DCM:MeOH=10:1,刮大板,收集到产品74.5mg为淡黄色油状液体,即化合物16a,收率20%。MS(ESI):m/z:1078.60[M+H]
1
16b的合成方法同16a,只是将原料中的15a更换成15b,16b为淡黄色油状液体,收率23%。MS(ESI):m/z:1074.62[M+H]
在50mL锥形瓶中依次加入16a(124mg,0.156mmol)溶于10mLTFA中,室温下搅拌反应3h。TLC检测反应完成后,反应液蒸干,即得化合物17a,为绿色油状液体,收率95%。MS(ESI):m/z:908.42[M-H]
17b的合成方法同17a,只是将原料中的16a更换成16b,17b为淡黄色油状液体,收率95%。MS(ESI):m/z:904.44[M-H]
在50mL锥形瓶中依次加入17a(20mg,0.022mmol),溶于去离子水后,调pH 5,GdCl
18b的合成方法同18a,只是将原料中的17a更换成17b,18b为白色粉末状固体,收率50%,HRMS:m/z[M+H]
实施例4髓鞘特异性磁共振造影剂24a-24d的制备
概括地讲,本发明通过取代的磺酰脒基连接的髓鞘特异性磁共振造影剂的制备包括以下步骤:
Dmedas在碱催化下与不同碳链长度的炔酸发生缩合反应,得到炔基修饰的Dmedas.炔基修饰的Dmedas在碘化亚铜的催化下与对位取代的苯磺酰叠氮、单Boc保护的丙二胺发生三组分偶联生成带取代的磺酰脒基的中间体,该中间体在三氟乙酸的条件下脱除Boc保护基后,与单羧基暴露的DTPA发生酰化反应后,在三氟乙酸的条件下脱除叔丁基保护基暴露出四个羧基,四个羧基中的三个与三价钆离子发生螯合,经分离纯化即得24a-24d
本发明髓鞘特异性磁共振造影剂24a-24d的制备可以通过如下通式来进行:
具体的合成方法如下:
在10mL锥形瓶中依次将5-己炔酸(706mg,6.3mmol),PyBOP(4.4g,8.4mmol),DIPEA(3.66ml,21mmol)溶于DMF中,室温下搅拌反应0.5h,加入1(1g,4.2mmol),室温下搅拌反应过夜。TLC检测反应完成后,向反应液加水淬灭,乙酸乙酯萃取,饱和食盐水洗,无水硫酸镁干燥,硅胶柱层析(PE:EA=30:1-2:1),收集到产品641mg,为黄色粉末状固体,收率46%。即化合物19a。MS(ESI):m/z:332.1899[M+H]
19b的合成方法与19a相同,只是将原料中的5-己炔酸更换成6-庚炔酸。19b为黄色粉末状固体,收率48%,MS(ESI):m/z:346.18[M+H]
在50mL锥形瓶中依次加入N-叔丁氧羰基-1,3-丙二胺(52mg,0.3mmol)溶于DCM中,加入三乙胺,氮气保护下室温下搅拌反应10min,加入19a(100mg,0.3mmol),4-氟苯磺酰基叠氮(73mg,0.36mmol),CuI(6mg,0.003mmol),搅拌反应过夜。TLC检测反应完成后,向反应中加入适量水和DCM稀释,搅拌反应30min,DCM萃取,饱和食盐水洗,无水硫酸镁干燥,浓缩,硅胶柱层析:PE:EA=30:1~1:1,得红色油状液体130mg,收率61%。即化合物20a.MS(ESI):m/z:680.32[M+H]
20b,20c和20d的合成方法与20a相同,只是分别将原料中19a依次更换成19b,19c,19d,4-氟苯磺酰基叠氮更换成对甲苯磺酰叠氮,4-氟苯磺酰基叠氮和对甲苯磺酰叠氮。
20b,红色油状液体,收率64%。MS(ESI):m/z:676.34[M+H]
20c,红色油状液体,收率65%。MS(ESI):m/z:694.33[M+H]
20d,红色油状液体,收率64%。MS(ESI):m/z:690.36[M+H]
在50mL锥形瓶中依次加入20a(130mg,0.19mmol)溶于7ml DCM后,加入3mL TFA,室温下搅拌反应过夜。TLC(DCM:MeOH=10:1)。TLC检测反应完成后,将反应液蒸干,加入三乙胺调pH碱性,浓缩蒸干,得棕红色油状液体,收率96%,即化合物21a。MS(ESI):m/z:580.26[M+H]
21b,21c和21d的合成方法与21a相同,只是将原料中的20a分别更换成20b,20c,和20d,
21b,棕红色油状液体,收率97%。MS(ESI):m/z:576.29[M+H]
21c,棕红色油状液体,收率96%。MS(ESI):m/z:594.28[M+H]
21d,棕红色油状液体,收率97%。MS(ESI):m/z:590.30[M+H]
在50mL锥形瓶中依次加入DTPA-3tBu(1.05g,1.71mmol),PyBOP(1.19g,2.28mmol),DIPEA(0.93ml,5.7mmol)溶于DMF中,室温下搅拌反应0.5h,加入21a(400mg,1.14mmol),室温反应过夜。TLC检测反应完成后,向反应液加水淬灭,EA萃取,饱和食盐水洗,无水硫酸镁干燥,硅胶柱层析(DCM—DCM:MeOH=20:1),再经分离,收集到产品496mg为红棕色油状液体,收率37%。即化合物22a。MS(ESI):m/z:1179.64[M+H]
22b,22c和22d的合成方法与22a相同,只是将原料中的21a依次更换成21b,21c,21d。
22b,为红棕色油状液体,收率39%,MS(ESI):m/z:1175.67[M+H]
22c,为红棕色油状液体,收率45%,MS(ESI):m/z:1193.66[M+H]
22d,为红棕色油状液体,收率为44%,MS(ESI):m/z:1189.68[M+H]
在50mL锥形瓶中依次加入22a(100mg,0.08mmol)溶于DCM中,加入TFA,室温反应过夜。TLC检测反应完成后,向反应液蒸干,甲醇带两遍,即得23a98为红棕色油状液体,收率98%。MS(ESI):m/z:953.39[M-H]
23b,23c和23d的合成方法与23a相同,只是将原料中的22a依次更换成22b,22c和22d,
23b,为红棕色油状液体,收率97%,MS(ESI):m/z:949.42[M-H]
23c,为红棕色油状液体,收率96%,MS(ESI):m/z:967.41[M-H]
23d,为红棕色油状液体,收率为97%,MS(ESI):m/z:963.43[M-H]
在50mL锥形瓶中依次加入23a(100mg,0.10mmol)溶于MeOH/H
24b,24c和24d的合成方法与24a相同,只是将原料中的23a分别更换成23b,23c,23d.
24b,粉红色粉末状固体,收率为47%。HRMS:m/z[M+H]
24c,粉红色粉末状固体,收率为53%。HRMS:m/z[M+H]
24d,粉红色粉末状固体,收率为55%。HRMS:m/z[M+H]
实施例5
1、荧光属性
将化合物6a-d,18a-b,24a-d,12和MeDAS配置成PBS溶液(2ml,10μM),使用荧光分光光度计确定激发波长和发射波长的峰值及范围(图1)。
2、磁共振弛豫率
将不同Gd浓度(0-0.8mM)的化合物6a-d,18a-b,24a-d,12,Gd-DTPA和Gd-DOTA分散液(在PBS中,PH=7.0)装入300μL的离心管中,使用7.0T MR成像扫描仪(Biospec 70/20,Bruker,德国)进行检测。扫描T1加权成像:参数TR/TE=693/6ms。T1反演恢复快速自旋回波序列(T1_map_RARE)进行。T1_map_RARE参数:TE=8.5ms,TR=60-5500ms。通过使用Bruker提供的ParaVision 6.0.1工具在每个样本中选择相同的感兴趣水平区域,通过1/T1弛豫时间(s-1)的曲线拟合得到化合物6a-d,18a-b,24a-d,12,Gd-DTPA和Gd-DOTA的T1弛豫度(r1)。
确定了化合物6a-d,18a-b,24a-d,12,Gd-DTPA和Gd-DOTA的纵向质子弛豫时间(T1)与Gd
3、磁共振金属转移性
在t=0min时,将1ml 2.5mM Gd配合物加入磷酸盐缓冲液(pH=7.0,50mM)中,与10μl 250mM ZnCl
如图4所示,化合物18a、18b与Gd-DOTA是大环类造影剂,因此,在氯化锌的条件下,随着时间的改变,不会产生沉淀,因此,弛豫率不会下降。而线性造影剂化合物Gd-DTPA,会随着时间的增加,弛豫率下降,但仍然可以用于MNR造影。
4、染色
将小鼠富有髓鞘的各部分脑组织(大脑、小脑、脊髓)经多聚甲醛浸泡24h后,依次沉入10%、20%、30%的蔗糖梯度脱水,使用OCT包埋后制作成冰冻切片。使用化合物6a-d,18a-b,24a-d,12与其孵育30min,PBS清洗10min后置于共聚焦下进行成像。
如图5所示,在大脑富有髓鞘的胼胝体、纹状体等区域化合物均看到清晰的荧光,并且与无髓鞘区域分辨清晰。不仅在大脑部位、在小脑部位和脊髓部位依然适用。表现出我们的化合物具有良好的靶向髓鞘的能力。
5、缺血性脑卒中MCAO模型的髓鞘损伤染色
缺血性脑卒中的同侧,髓鞘损伤严重,可见化合物6a的荧光强度较低。而缺血性脑卒中对侧,髓鞘尚未受损,髓鞘结构清晰可见,荧光强度较强。对照商用LFB染色,发现类似情况,表明化合物6a可以分辨髓鞘损伤部位。
6、体外MRI浸泡实验
将对照组和MCAO组的大鼠和小鼠的脑组织切成2mm厚的切片,分别浸泡在化合物18a和化合物6a的PBS溶液中24h(5mM,1ml),然后用PBS溶液清洗七天,每天更换一次PBS溶液。在浸泡前后分别进行T1WI的扫描。
浸泡后的对照组较浸泡前,髓鞘的对比度清晰明显,且左右分布对称。浸泡后的MCAO组按箭头处的纹状体部位信号强度增加,表明该部位髓鞘受损。化合物18a、6a可以在磁共振的条件下清晰的识别出髓鞘受损部位。
7、体内MRI成像
侧脑室给药
将大鼠固定在大脑立体定位仪上,在头部皮肤做一个3cm纵行切口,然后用牙科钻在前囟(AP=±1.5mm,ML=0.5mm,DV=4.3mm)位置做一个圆形切口,两侧分别注入5μl,7mM化合物18a。
如图10所示,相比给药前,对照组给药后全脑T1WI图像信号减低,髓鞘部位信号更低,T1 mapping图像可见到分辨更加清晰的髓鞘部位。MCAO组T1WI可见损伤侧信号稍高,T1mapping可见损伤侧的纹状体部位弛豫时间并未降低。表明本发明化合物可在大鼠体内与髓鞘进行结合,并通过磁共振进行监测。
8、尾静脉给药
将化合物6a配置成40mg/kg的浓度,通过尾静脉打入小鼠体内,扫描完磁共振T1WI和T1mapping序列后,将小鼠处死进行小动物活体荧光成像(430nm,500nm)。
与给药前相比,尾静脉给药后小鼠TIWI信号降低,脑梗死损伤侧的信号增高,T1mapping可见未受伤侧髓鞘部位对比度更加明显。通过小动物活体荧光可见脑梗死一侧有荧光的存在,而其他心肝脾肺肾并未见到荧光。表明我们的化合物6a可通过尾静脉循环,通过脑梗死血脑屏障破坏一侧与颅内的髓鞘进行结合。并通过MRI和小动物活体荧光监测到。
- 靶向TSPO磁共振成像造影剂及其制备方法
- 可特异性清除髓鞘组织的转基因斑马鱼及其制备方法和应用
- 可特异性清除髓鞘组织的转基因斑马鱼及其制备方法和应用