用于深度学习的高效体素化
文献发布时间:2024-04-18 19:58:21
优先权申请
本申请要求2022年3月24日提交的名称为“Efficient Voxelization For DeepLearning”的美国非临时专利申请号17/703,958(代理人案卷号ILLM 1048-2/IP-2143-US)的优先权,该美国非临时专利申请要求2021年4月16日提交的名称为“EfficientVoxelization For Deep Learning”的美国临时专利申请号63/175,767(代理人案卷号ILLM 1048-1/IP-2143-PRV)的优先权。
本申请还要求2022年3月24日提交的名称为“Multi-channel ProteinVoxelization To Predict Variant Pathogenicity Using Deep Convolutional NeuralNetworks”的美国非临时专利申请号17/703,935(代理人案卷号ILLM 1047-2/IP-2142-US)的优先权,该美国非临时专利申请要求2021年4月15日提交的名称为“Multi-channelProtein Voxelization To Predict Variant Pathogenicity Using DeepConvolutional Neural Networks”的美国临时专利申请号63/175,495(代理人案卷号ILLM1047-1/IP-2142-PRV)的优先权或权益。
据此优先权申请以引用方式并入以用于所有目的。
相关专利申请
本申请与同时提交的名称为“Multi-channel Protein Voxelization ToPredict Variant Pathogenicity Using Deep Convolutional Neural Networks”的PCT专利申请(代理人案卷号ILLM 1047-3/IP-2142-PCT)相关。该相关申请据此以引用方式并入本文用于所有目的。
技术领域
本发明所公开的技术涉及人工智能类型计算机和数字数据处理系统以及对应数据处理方法和用于仿真智能的产品(即,基于知识的系统、推断系统和知识采集系统);并且包括用于不确定性推断的系统(例如,模糊逻辑系统)、自适应系统、机器学习系统和人工神经网络。具体地,本发明所公开的技术涉及使用深度卷积神经网络来分析多通道体素化数据。
文献并入
以下文献以引用方式并入,即如同在本文完整示出一样,以用于所有目的:
Sundaram,L.等人,Predicting the clinical impact of human mutation withdeep neural networks.Nat.Genet.50,1161-1170(2018);
Jaganathan,K.等人,Predicting splicing from primary sequence with deeplearning.Cell 176,535-548(2019);
2017年10月16日提交的名称为“TRAINING A DEEP PATHOGENICITY CLASSIFIERUSING LARGE-SCALE BENIGN TRAINING DATA”的美国临时专利申请号62/573,144(代理人案卷号ILLM 1000-1/IP-1611-PRV);
2017年10月16日提交的名称为“PATHOGENICITY CLASSIFIER BASED ON DEEPCONVOLUTIONAL NEURAL NETWORKS(CNNs)”的美国临时专利申请号62/573,149(代理人案卷号ILLM 1000-2/IP-1612-PRV);
2017年10月16日提交的名称为“DEEP SEMI-SUPERVISED LEARNING THATGENERATES LARGE-SCALE PATHOGENIC TRAINING DATA”的美国临时专利申请号62/573,153(代理人案卷号ILLM 1000-3/IP-1613-PRV);
2017年11月7日提交的名称为“PATHOGENICITY CLASSIFICATION OF GENOMICDATA USING DEEP CONVOLUTIONAL NEURAL NETWORKS(CNNs)”的美国临时专利申请号62/582,898(代理人案卷号ILLM 1000-4/IP-1618-PRV);
2018年10月15日提交的名称为“DEEP LEARNING-BASED TECHNIQUES FORTRAINING DEEP CONVOLUTIONAL NEURAL NETWORKS”的美国非临时专利申请号16/160,903(代理人案卷号ILLM 1000-5/IP-1611-US);
2018年10月15日提交的名称为“DEEP CONVOLUTIONAL NEURAL NETWORKS FORVARIANT CLASSIFICATION”的美国非临时专利申请号16/160,986(代理人案卷号ILLM1000-6/IP-1612-US);
2018年10月15日提交的名称为“SEMI-SUPERVISED LEARNING FOR TRAINING ANENSEMBLE OF DEEP CONVOLUTIONAL NEURALNETWORKS”的美国非临时专利申请号16/160,968(代理人案卷号ILLM 1000-7/IP-1613-US);以及
2019年5月8日提交的名称为“DEEP LEARNING-BASED TECHNIQUES FOR PRE-TRAINING DEEP CONVOLUTIONAL NEURAL NETWORKS”的美国非临时专利申请号16/407,149(代理人案卷号ILLM 1010-1/IP-1734-US)。
背景技术
本部分中讨论的主题不应仅因为在本部分中有提及就被认为是现有技术。类似地,在本部分中提及的或与作为背景技术提供的主题相关联的问题不应被认为先前在现有技术中已被认识到。本部分中的主题仅表示不同的方法,这些方法本身也可对应于受权利要求书保护的技术的具体实施。
基因组学在广义上也称为功能基因组学,其目的是通过使用基因组规模的测定(诸如基因组测序、转录组谱分析和蛋白质组学)来表征生物体的每种基因组元件的功能。基因组学作为数据驱动的科学出现-其通过从基因组规模数据的探索中发现新特性而不是通过测试预先设想的模型和假设来运作。基因组学的应用包括发现基因型与表型之间的关联、发现用于患者分层的生物标志物、预测基因功能,以及绘制有生化活性的基因组区域(诸如转录增强子)的图表。
基因组学数据太大太复杂,以至于不能仅通过可视化研究成对相关来挖掘。相反,需要分析工具来支持发现未预料到的关系,以导出新的假设和模型,并进行预测。机器学习算法与假设和领域专业知识被硬编码的一些算法不同,被设计成自动检测数据中的模式。因此,机器学习算法适合于数据驱动的科学,尤其适合于基因组学。然而,机器学习算法的性能可能强烈依赖于如何表示数据,也就是说,如何计算每个变量(也称为特征)。例如,为了从荧光显微镜图像中将肿瘤分类为恶性或良性,预处理算法可以检测细胞、识别细胞类型,以及生成针对每种细胞类型的细胞计数列表。
机器学习模型可以将估计的细胞计数(是手工特征的实例)作为输入特征来对肿瘤进行分类。核心问题是分类性能严重依赖于这些特征的质量和相关性。例如,相关视觉特征(诸如细胞形态、细胞间的距离或器官内的定位)在细胞计数中没有被捕捉到,对数据的这种不完整表示可能降低分类准确度。
深度学习(机器学习的分支学科)通过将特征的计算嵌入到机器学习模型本身中以产生端对端模型来解决这个问题。该成果已经通过开发深度神经网络来实现,这些深度神经网络是包括连续基本运算的机器学习模型,其中连续基本运算通过取在先运算的结果作为输入来计算越来越复杂的特征。深度神经网络能够通过发现高复杂度的相关特征(诸如上述实例中的细胞形态和细胞的空间组织)来提高预测准确性。通过数据爆炸、算法的进步以及计算能力的显著增加,特别是通过使用图形处理单元(GPU),已经能够实现深度神经网络的构建和训练。
监督学习的目标是获得将特征取作输入并返回对所谓目标变量的预测的模型。监督学习问题的一个示例是预测内含子是否被剪接掉RNA上的(目标)给定特征,诸如典型剪接位点序列是否存在、剪接分支点的位置或内含子长度。训练机器学习模型是指学习其参数,这通常涉及使关于训练数据的损失函数最小化,目的是对不可见数据进行准确预测。
对于计算生物学中的许多监督学习问题,输入数据可以表示为具有多个列或特征的表格,每个列或特征包含潜在可用于做出预测的数值数据或分类数据。一些输入数据自然地表示为表格中的特征(诸如温度或时间),而其他输入数据需要首先使用被称为特征提取的过程来变换(诸如将脱氧核糖核酸(DNA)序列变换为k-mer计数),以符合表格表示。对于内含子剪接预测问题,典型剪接位点序列是否存在、剪接分支点的位置和内含子长度可以是以表格格式收集的预处理特征。表格数据是多种多样监督机器学习模型的标准,范围从简单的线性模型(诸如逻辑回归)到更灵活的非线性模型(诸如神经网络),以及许多其他模型。
逻辑回归是二元分类器,即,预测二元目标变量的监督学习模型。具体地,逻辑回归通过使用S型函数(一类激活函数)计算映射到[0,1]区间的输入特征的加权和,来预测正类的概率。逻辑回归或使用不同激活函数的其他线性分类器的参数是加权和中的权重。当用输入特征的加权和不能很好地区分类别(例如,被剪接掉或未被剪接掉的内含子的类别)时,线性分类器失效。为了提高预测性能,可以通过以新的方式(例如,通过取幂或成对乘积)变换或组合现有特征来手动添加新的输入特征。
神经网络使用隐藏层来自动学习这些非线性特征变换。每个隐藏层可以被认为是多个线性模型,其输出由非线性激活函数变换,该非线性激活函数诸如S型函数或更流行的整流线性单位函数(ReLU)。这些层一起将输入特征组成相关的复模式,这有助于区分两个类的任务。
深度神经网络使用许多隐藏层,其中一层在每个神经元接收到来自前一层的所有神经元的输入时,被称为是全连接层。神经网络通常使用随机梯度下降来训练,其中随机梯度下降是适合于在非常大的数据集上训练模型的一种算法。使用现代深度学习框架实现神经网络使得能够使用不同的架构和数据集进行快速原型设计。全连接神经网络可以用于许多基因组学应用,包括从序列特征(诸如存在剪接因子的结合基序或序列保守性)预测针对给定序列剪接的外显子的百分比;将潜在致病遗传变体按重要性排序;以及使用诸如染色质标记、基因表达和进化保守性的特征预测给定基因组区域中的顺式调控元件。
为了进行有效的预测,必须考虑空间数据和纵向数据的局部依赖性。例如,打乱DNA序列或图像的像素会严重破坏信息模式。这些局部依赖性设置除表格数据之外的空间或纵向数据,对于表格数据,特征的排序是任意的。考虑将基因组区域分类为由特定转录因子结合或不由特定转录因子结合的问题,其中结合区域被定义为染色质免疫沉淀、随后是测序(ChIP-seq)数据中的高置信度结合事件。转录因子通过识别序列基序与DNA结合。基于序列导出特征的全连接层,诸如序列中的k-mer实例的数量或位置权重矩阵(PWM)匹配,可以用于该任务。由于k-mer或PWM实例频率对于在序列内将基序移位具有稳健性,所以此类模型可以很好地推广到具有位于不同位置的相同基序的序列。然而,它们却不能识别转录因子结合依赖于具有明确定义间隔的多个基序的组合的模式。此外,可能的k-mer数量随着k-mer长度呈指数增加,这对存储和过拟合两方面提出了挑战。
卷积层是全连接层的一种特殊形式,其中相同的全连接层被局部地(例如在6bp窗口中)应用于所有序列位置。该方法也可以被视为使用多个PWM来扫描序列,例如,针对转录因子GATA1和TAL1。通过在不同位置使用相同的模型参数,参数总数急剧减少,并且网络能够检测在训练期间未看到的位置处的基序。每个卷积层通过在每个位置处产生标量值来用几个滤波器对序列进行扫描,该标量值量化滤波器与序列之间的匹配度。如在全连接神经网络中那样,在每一层处应用非线性激活函数(通常为ReLU)。接下来,应用池化操作,其将激活聚集在整个位置轴上的连续仓中,通常取每个通道的最大激活或平均激活。池化减小了有效序列长度,并使信号变得粗糙。随后的卷积层组成前一层的输出,并且能够检测GATA1基序和TAL1基序是否存在于某个距离范围内。最后,这些卷积层的输出可以用作全连接神经网络的输入,以执行最终的预测任务。因此,不同类型的神经网络层(例如,全连接层和卷积层)可以在单个神经网络内组合。
卷积神经网络(CNN)仅在DNA序列基础上就能够预测各种分子表型。应用包括对转录因子结合位点进行分类,以及预测分子表型,诸如染色质特征、DNA接触图、DNA甲基化、基因表达、翻译效率、RBP结合与微小RNA(miRNA)目标。卷积神经网络除了从序列预测分子表型之外,还可以应用于传统上由手工生物信息学流水线解决的更多技术任务。例如,卷积神经网络可以预测向导RNA的特异性、对ChIP-seq进行去噪、提高Hi-C数据分辨率、从DNA序列预测来源实验室,以及检出遗传变体。卷积神经网络也已经用于对基因组中的长程依赖性进行建模。尽管相互作用的调控元件在未折叠的线性DNA序列上可能远离彼此定位,但这些元件在实际的3D染色质构象中通常彼此邻近。因此,虽然由线性DNA序列对分子表型建模是对染色质的粗略近似,但却可以通过允许长范围依赖性和允许模型隐含地学习3D组织的各方面(诸如启动子-增强子成环)来改进。这通过使用扩张的卷积来实现,其具有高达32kb的感受野。扩张的卷积还允许使用10kb的感受野从序列预测剪接位点,从而使得能够跨越与典型的人内含子一样长的距离来整合遗传序列(参见Jaganathan,K.等人,Predictingsplicing from primary sequence with deep learning.Cell 176,535-548(2019))。
不同类型的神经网络可以由它们的参数共享方案来表征。例如,全连接层不具有参数共享,而卷积层通过在其输入的每个位置处应用相同的滤波器来施加平移不变性。递归神经网络(RNN)是用于处理实现不同参数共享方案的顺序数据(诸如DNA序列或时间序列)的对卷积神经网络的替代方案。递归神经网络对每个序列元素应用相同的操作。该操作将前一个序列元素和新输入作为存储器的输入。该操作将存储器更新并任选地发出输出,该输出被传递到后续层或被直接用作模型预测结果。由于在每个序列元素处应用相同的模型,递归神经网络对于所处理的序列中的位置索引保持不变。例如,递归神经网络可以检测DNA序列中的开放阅读框,而不管在序列中的位置是怎样的。该任务需要识别特定系列的输入,诸如起始密码子之后是框内终止密码子。
递归神经网络优于卷积神经网络的主要优势在于,在理论上,它们能够经由存储器通过无限长的序列来携带信息。此外,递归神经网络可以自然地处理长度变化很大的序列,诸如mRNA序列。然而,在序列建模任务(例如音频合成和机器翻译)方面,与各种技巧(诸如扩张的卷积)组合的卷积神经网络可以达到与递归神经网络相当、甚至更好的性能。递归神经网络可以聚集卷积神经网络的输出,用于预测单细胞DNA甲基化状态、RBP结合、转录因子结合和DNA可及性。此外,由于递归神经网络应用顺序操作,所以不能轻易并行化,因此计算速度比卷积神经网络慢得多。
虽然每个人都有独特的遗传密码,但是人类遗传密码的大部分是所有人共有的。在一些情况下,人类遗传密码可以包括异常值,称为遗传变体,其在相对小群的人群的个体之中可能是共有的。例如,特定的人蛋白质可以包含特定的氨基酸序列,而该蛋白质的变体可以在其他方面相同的特定序列中有一个氨基酸不同。
遗传变体可以具有致病性,从而导致疾病。尽管大多数这样的遗传变体已经通过自然选择从基因组中耗尽,但是识别哪些遗传变体可能具有致病性的能力可以帮助研究人员集中于这些遗传变体以获得对相应疾病及其诊断、治疗或治愈的理解。对数百万个人类遗传变体的临床解释仍不清楚。一些最常见的致病性变体是改变蛋白质氨基酸的单核苷酸错义突变。然而,并非所有的错义突变都具有致病性。
可以直接从生物序列预测分子表型的模型可以用作计算机扰动工具来探测遗传变异与表型变异之间的关联,并且已经成为用于数量性状基因座识别和变体优先排序的新方法。这些方法非常重要,因为通过复杂表型的全基因组关联分析识别的大多数变体是非编码的,这使得估计它们对表型的作用和贡献具有挑战性。此外,连锁不平衡导致变体的块被共遗传,这在查明单个因果变体方面产生了困难。因此,可以用作评估此类变体的影响的探询工具的基于序列的深度学习模型提供了一种有前途的方法来发现复杂表型的潜在驱动因素。一个示例包括从两种变体在转录因子结合、染色质可及性或基因表达预测方面之间的差异间接预测非编码单核苷酸变体和短插入或缺失(indel)的影响。另一个示例包括根据序列或根据遗传变体对剪接的定量影响,来预测新剪接位点的产生。
应用用于预测变体效应的端对端深度学习方法,从蛋白质序列和序列保守性数据预测错义变体的致病性(参见Sundaram,L.等人,Predicting the clinical impact ofhuman mutation with deep neural networks.Nat.Genet.50,1161-1170(2018),本文中称为“PrimateAI”)。PrimateAI使用在已知具有致病性的变体上训练的深度神经网络,其中使用跨物种信息进行数据增强。特别地,PrimateAI使用野生型蛋白质和突变型蛋白质的序列来比较差异,并且使用受过训练的深度神经网络来决定突变的致病性。这种利用蛋白质序列进行致病性预测的方法是有前途的,因为其可以避免圆度问题和对先前知识的过度拟合。然而,与有效训练深度神经网络的数据数量充分相比,ClinVar中可用的临床数据数量相对较少。为了克服这种数据匮乏,PrimateAI使用常见的人类变体和灵长类动物变体作为良性数据,而将基于三核苷酸背景的模拟变体用作未标记数据。
当直接根据序列比对进行训练时,PrimateAI的性能优于现有方法。PrimateAI直接从由约120,000个人类样品组成的训练数据中学习重要的蛋白质结构域、保守氨基酸位置和序列依赖性。PrimateAI在区分候选发育障碍基因中的良性和致病性从头突变方面,以及在复制ClinVar中的先验知识方面,明显胜过其他变体致病性预测工具的性能。这些结果表明PrimateAI是变体分类工具的重要进步,可以减少临床报告对先验知识的依赖。
蛋白质生物学的核心是理解结构元件如何产生观察到的功能。蛋白质结构数据过剩使得能够开发计算方法来系统地导出支配结构-功能关系的规则。然而,这些方法的性能在很大程度上取决于对蛋白质结构表示的选择。
蛋白质位点是蛋白质结构内的微环境,通过其结构或功能作用来区分。位点可以由三维(3D)位置和该位置周围的其中存在结构或功能的局部邻域来定义。合理蛋白质工程的核心是理解氨基酸的结构排列如何在蛋白质位点内产生功能特征。确定蛋白质内各个氨基酸的结构和功能作用提供了有助于工程化和改变蛋白质功能的信息。识别功能或结构上重要的氨基酸允许集中的工程努力,诸如用于改变靶蛋白功能特性的定点诱变。替代性地,这种知识可以有助于避免会破坏期望功能的工程设计。
由于已经确定结构比序列保守得多,所以蛋白质结构数据增加提供了使用数据驱动的方法系统地研究支配结构-功能关系的潜在模式的机会。任何蛋白质计算分析的基本方面都是如何表示蛋白质结构信息。机器学习方法的性能通常更多地取决于对数据表示的选择,而不是所采用的机器学习算法。良好的表示高效地捕获最关键的信息,而差的表示产生没有底层图案的噪声分布。
蛋白质结构过剩和最近深度学习算法的成功提供了开发用于自动提取蛋白质结构的任务特异性表示的工具的机会。因此,有机会使用3D蛋白质结构的多通道体素化表示作为深度神经网络的输入来预测变体的致病性。
附图说明
本专利或专利申请文件包含至少一幅彩色附图。具有彩色附图的本专利或本专利申请公布的副本将在提出请求并支付必要费用后由专利局提供。
彩色附图也可通过补充内容选项卡成对地获得。在附图中,在所有不同视图中,类似的参考符号通常是指类似的部件。另外,附图未必按比例绘制,而是重点说明所公开的技术的原理。在以下描述中,参考以下附图描述了本发明所公开的技术的各种实施方式,其中:
图1是展示根据本发明所公开技术的各种具体实施的用于确定变体致病性的系统的过程的流程图。
图2示意性地展示了根据本发明所公开技术的一个具体实施,蛋白质的示例参考氨基酸序列和该蛋白质的替代性氨基酸序列。
图3展示了根据本发明所公开技术的一个具体实施,图2的参考氨基酸序列中氨基酸原子的氨基酸式分类。
图4展示了根据本发明所公开技术的一个具体实施,图3中在氨基酸基础上分类的α-碳原子的3D原子坐标的氨基酸式归属。
图5示意性地展示了根据本发明所公开技术的一个具体实施的确定体素式距离值的过程。
图6示出了根据本发明所公开技术的一个具体实施的21个氨基酸式距离通道的实例。
图7是根据本发明所公开技术的一个具体实施的距离通道张量的示意图。
图8示出了根据本发明所公开技术的一个具体实施,来自图2的参考氨基酸和替代性氨基酸的独热编码。
图9是根据本发明所公开技术的一个具体实施,体素化独热编码的参考氨基酸和体素化独热编码的变体/替代性氨基酸的示意图。
图10示意性地展示了根据本发明所公开技术的一个具体实施的连结过程,该连结过程按体素方式将图7的距离通道张量与参考等位基因张量连结。
图11示意性地展示了根据本发明所公开技术的一个具体实施的连结过程,该连结过程按体素方式将图7的距离通道张量、图10的参考等位基因张量和替代性等位基因张量连结。
图12是展示根据本发明所公开技术的一个具体实施,用于确定和分配相对于体素最接近的原子的泛氨基酸保守频率(体素化)的系统的过程的流程图。
图13展示了根据本发明所公开技术的一个具体实施的体素到最接近氨基酸的映射。
图14示出了根据本发明所公开技术的一个具体实施的跨99个物种的参考氨基酸序列的示例多序列比对。
图15示出了根据本发明所公开技术的一个具体实施的确定特定体素的泛氨基酸保守频率序列的实例。
图16示出了根据本发明所公开技术的一个具体实施,使用图15中描述的位置频率逻辑针对相应体素确定的相应泛氨基酸保守频率。
图17展示了根据本发明所公开技术的一个具体实施的体素化每体素进化谱。
图18描绘了根据本发明所公开技术的一个具体实施的进化谱张量的实例。
图19是展示根据本发明所公开技术的一个具体实施,用于确定和分配相对于体素最接近的原子的每氨基酸保守频率(体素化)的系统的过程的流程图。
图20示出了根据本发明所公开技术的一个具体实施,与距离通道张量连结的体素化注释通道的各种实例。
图21展示了根据本发明所公开技术的一个具体实施的输入通道的不同组合与排列,其可以作为用于确定目标变体的致病性的致病性分类器的输入提供。
图22示出了根据本发明所公开技术的各种具体实施的计算本发明所公开的距离通道的不同方法。
图23示出了根据本发明所公开技术的各种具体实施的进化通道的不同实例。
图24示出了根据本发明所公开技术的各种具体实施的注释通道的不同实例。
图25示出了根据本发明所公开技术的各种具体实施的结构置信度通道的不同实例。
图26示出了根据本发明所公开技术的一个具体实施的致病性分类器的示例处理架构。
图27示出了根据本发明所公开技术的一个具体实施的致病性分类器的示例处理架构。
图28、图29、图30和图31使用PrimateAI作为基准模型来证明本发明所公开的PrimateAI 3D相对于PrimateAI的分类优势。
图32A和图32B示出了根据本发明所公开技术的各种具体实施的本发明所公开的高效体素化过程。
图33描绘了根据本发明所公开技术的一个具体实施,原子如何与包含原子的体素相关联。
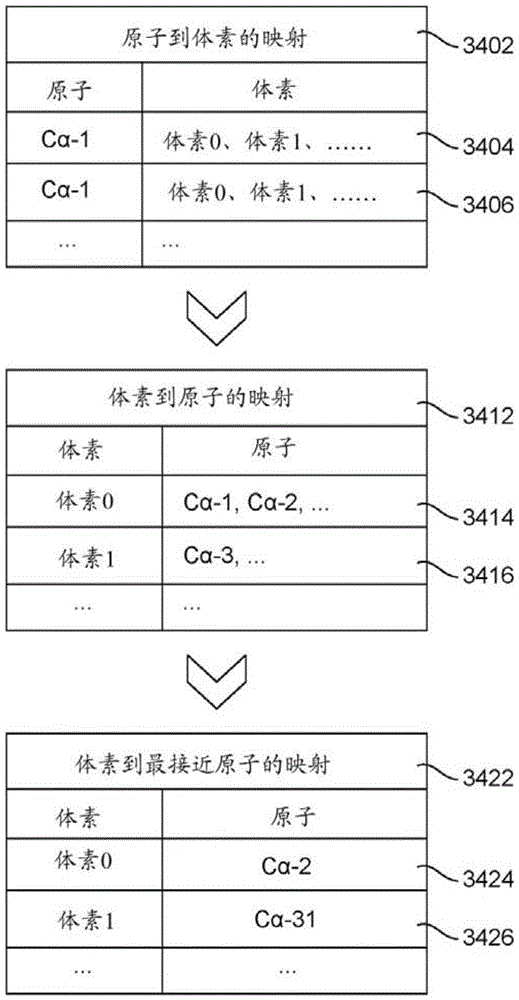
图34示出了根据本发明所公开技术的一个具体实施,从原子到体素映射生成体素到原子映射以在逐个体素的基础上识别最接近原子。
图35A和图35B展示了本发明所公开的高效体素化如何具有为O(#原子)的运行时复杂度,对比在不使用本发明所公开的高效体素化的情况下为O(#原子*#体素)的运行时复杂度。
图36是可以用于实现本发明所公开技术的示例计算机系统。
具体实施方式
呈现以下讨论以使得本领域的任何技术人员能够实现和使用所公开的技术,并且在特定应用及其要求的上下文中提供以下讨论。对所公开的具体实施的各种修改对于本领域的技术人员而言将是显而易见的,并且在不脱离所公开的技术的实质和范围的情况下,本文所定义的一般原理可应用于其他具体实施和应用。因此,所公开的技术并非旨在限于所示的具体实施,而是要符合与本文所公开的原理和特征一致的最广范围。
当结合附图阅读时,将更好地理解各种具体实施的详细描述。就附图例示各种具体实施的功能块的图而言,功能块不一定指示硬件电路之间的划分。因此,例如,功能块(例如,模块、处理器或存储器)中的一者或多者可以在单件硬件(例如,通用信号处理器或随机存取存储器块、硬盘等)或多件硬件中实现。类似地,程序可以是独立程序,可作为子例程并入操作系统中,可以是已安装软件包中的功能等。应当理解,各种具体实施不限于附图中所示的布置和工具。
附图中被指定为模块的处理引擎和数据库可在硬件或软件中实现,并且不需要按如附图所示那样精确地划分成相同的块。这些模块中的一些模块还可在不同的处理器、计算机或服务器上实现,或者分布在多个不同的处理器、计算机或服务器之间。此外,应当理解,在不影响所实现的功能的情况下,可组合、同步操作或以与图中所示不同的序列操作模块中的一些。附图中的模块也可被认为是方法中的流程图步骤。模块也不一定需要将其所有代码连续地放置在存储器中;代码的一些部分可与代码的其他部分分离,来自其他模块或其他功能的代码设置在两者之间。
基于蛋白质结构的致病性测定
图1是展示用于确定变体致病性的系统的过程100的流程图。在步骤102处,系统的序列存取器104存取参考氨基酸序列和替代性氨基酸序列。在步骤112处,系统的3D结构生成器114生成参考氨基酸序列的3D蛋白质结构。在一些具体实施中,3D蛋白质结构是人蛋白质的同源模型。在一个具体实施中,所谓的SwissModel同源性建模流水线提供了预测的人蛋白质结构的公共知识库。在另一个具体实施中,所谓的HHpred同源性建模使用称为Modeller的工具从模板结构预测目标蛋白质的结构。
蛋白质由原子集合及其在3D空间中的坐标表示。氨基酸可以具有多种原子,诸如碳原子、氧(O)原子、氮(N)原子和氢(H)原子。这些原子可以进一步分类为侧链原子和主链原子。主链碳原子可以包括α-碳(C
在步骤122处,系统的坐标分类器124基于氨基酸对3D蛋白质结构的3D原子坐标进行分类。在一个具体实施中,氨基酸式分类涉及将3D原子坐标归属于21个氨基酸类别(包括终止氨基酸类别或缺口氨基酸类别)。在一个示例中,α-碳原子的氨基酸式分类可以分别列出在21个氨基酸类别中的每个类别下的α-碳原子。在另一个示例中,β-碳原子的氨基酸式分类可以分别列出在21个氨基酸类别中的每个类别下的β-碳原子。
在又一个示例中,氧原子的氨基酸式分类可以分别列出在21个氨基酸类别中的每个类别下的氧原子。在又一个示例中,氮原子的氨基酸式分类可以分别列出在21个氨基酸类别中的每个类别下的氮原子。在又一个示例中,氢原子的氨基酸式分类可以分别列出在21个氨基酸类别中的每个类别下的氢原子。
本领域的技术人员将会知道,在各种具体实施中,氨基酸式分类可以包括21个氨基酸类别的子集和不同原子元素的子集。
在步骤132处,系统的体素网格生成器134将体素网格实例化。体素网格可以具有任何分辨率,例如3×3×3、5×5×5、7×7×7等。体素网格中的体素可以具有任何尺寸,例如,每侧1埃
在步骤142处,系统的体素网格中心定位器144将体素网格中心定位在经历氨基酸水平上的目标变体的参考氨基酸处。在一个具体实施中,体素网格中心定位在经历目标变体的参考氨基酸的特定原子的原子坐标处,例如,经历目标变体的参考氨基酸的α-碳原子的3D原子坐标处。
距离通道
体素网格中的体素可以具有多个通道(或特征)。在一个具体实施中,体素网格中的体素具有多个距离通道(例如,分别针对21个氨基酸类别(包括终止或缺口氨基酸类别)的21个距离通道)。在步骤152处,系统的距离通道生成器154为体素网格中的体素生成氨基酸式距离通道。对于这21个氨基酸类别中的每个类别独立地生成距离通道。
例如,考虑丙氨酸(A)氨基酸类别。进一步考虑,例如,体素网格的尺寸为3×3×3,具有27个体素。然后,在一个具体实施中,丙氨酸距离通道分别包括体素网格中的27个体素的27个距离值。丙氨酸距离通道中的27个距离值是从体素网格中的27个体素的相应中心到丙氨酸氨基酸类别中的相应最接近原子测量的。
在一个示例中,丙氨酸氨基酸类别仅包括α-碳原子,因此最接近原子是分别最靠近体素网格中的27个体素的那些丙氨酸α-碳原子。在另一个示例中,丙氨酸氨基酸类别仅包括β-碳原子,因此最接近原子是分别最靠近体素网格中的27个体素的那些丙氨酸β-碳原子。
在又一个示例中,丙氨酸氨基酸类别仅包括氧原子,因此最接近原子是分别最靠近体素网格中的27个体素的那些丙氨酸氧原子。在又一个示例中,丙氨酸氨基酸类别仅包括氮原子,因此最接近原子是分别最靠近体素网格中的27个体素的那些丙氨酸氮原子。在又一个示例中,丙氨酸氨基酸类别仅包括氢原子,因此最接近原子是分别最靠近体素网格中的27个体素的那些丙氨酸氢原子。
类似于丙氨酸距离通道,距离通道生成器154针对剩余氨基酸类别中的每一者生成距离通道(即,体素式距离值的集合)。在其他具体实施中,距离通道生成器154仅针对21个氨基酸类别的子集生成距离通道。
在其他具体实施中,对最接近原子的选择不限于特定原子类型。即,在主题氨基酸类别内,选择与特定体素最接近的原子,而不考虑最接近原子的原子元素,并且计算特定体素的距离值以便包括在主题氨基酸类别的距离通道中。
在还有其他的具体实施中,在原子元素基础上生成距离通道。作为用于氨基酸类别的距离通道的替代或补充,可以为原子元素类别生成距离值,而不考虑原子所属的氨基酸。例如,考虑参考氨基酸序列中的氨基酸的原子跨越七个原子元素:碳、氧、氮、氢、钙、碘和硫。然后,体素网格中的体素被配置为具有七个距离通道,使得这七个距离通道中的每一个都具有二十七个体素式距离值,这些距离值指定了仅在对应的原子元素类别内到最接近原子的距离。在其他具体实施中,可以生成仅用于七个原子元素的子集的距离通道。在还有其他的具体实施中,可以将原子元素类别和距离通道生成进一步分层为相同原子元素的变型,例如,α-碳(C
在还有其他的具体实施中,可以在原子类型基础上生成距离通道,例如,仅用于侧链原子的距离通道和仅用于主链原子的距离通道。
可以在距体素中心的预定义最大扫描半径(例如,六埃
计算体素中心的3D坐标与原子的3D原子坐标之间的距离。另外,利用中心定位在相同位置处(例如,中心定位在经历目标变体的参考氨基酸的α-碳原子的3D原子坐标处)的体素网格生成距离通道。
这些距离可以是欧几里得距离。另外,这些距离可以由原子尺寸(或原子影响)来参数化(例如,通过使用所考虑原子的伦纳德-琼斯势和/或范德瓦尔斯原子半径)。另外,这些距离值可以通过最大扫描半径来归一化,或者通过主题氨基酸类别或主题原子元素类别或主题原子类型类别内的最接近原子的最大观察距离值来归一化。在一些具体实施中,基于体素和原子的极坐标来计算体素与原子之间的距离。极坐标由体素与原子之间的角度来参数化。在一个具体实施中,该角度信息用于生成体素的角度通道(即,独立于距离通道)。在一些具体实施中,最接近原子与邻近原子(例如,主链原子)之间的角度可以用作利用体素编码的特征。
参考等位基因通道和替代性等位基因通道
体素网格中的体素还可以具有参考等位基因通道和替代性等位基因通道。在步骤162处,系统的独热编码器164生成参考氨基酸序列中的参考氨基酸的参考独热编码和替代性氨基酸序列中的替代性氨基酸的替代性独热编码。参考氨基酸经历了目标变体。替代性氨基酸是目标变体。参考氨基酸和替代性氨基酸分别位于参考氨基酸序列和替代性氨基酸序列中的相同位置处。参考氨基酸序列和替代性氨基酸序列具有相同的位置式氨基酸组成,但有一个例外。该例外是在参考氨基酸序列中具有参考氨基酸且在替代性氨基酸序列中具有替代性氨基酸的位置。
在步骤172处,系统的连结器174将氨基酸式距离通道与参考独热编码和替代性独热编码连结。在另一个具体实施中,连结器174将原子元素式距离通道与参考独热编码和替代性独热编码连结。在又一个具体实施中,连结器174将原子类型式距离通道与参考独热编码和替代性独热编码连结。
在步骤182处,系统的运行时逻辑184通过致病性分类器(致病性测定引擎)处理连结的氨基酸式距离通道/原子元素式距离通道/原子类型式距离通道与参考独热编码和替代性独热编码,以测定目标变体的致病性,该致病性进而被推断为在氨基酸水平上产生目标变体的基础核苷酸变体的致病性测定结果。使用良性变体和致病性变体的标记数据集,例如使用反向传播算法来训练致病性分类器。关于良性变体和致病性变体的标记数据集以及致病性分类器的示例架构和训练的附加细节可以在共同拥有的美国专利申请号16/160,903、16/160,986、16/160,968和16/407,149中找到。
图2示意性地展示了蛋白质200的参考氨基酸序列202和蛋白质200的替代性氨基酸序列212。蛋白质200包含N个氨基酸。蛋白质200中氨基酸的位置标记为1、2、3......N。在所展示的该实例中,位置16是经历由基础核苷酸变体引起的氨基酸变体214(突变)的位置。例如,对于参考氨基酸序列202,位置1具有参考氨基酸苯丙氨酸(F),位置16具有参考氨基酸甘氨酸(G)204,而位置N(例如,序列202的最后一个氨基酸)具有参考氨基酸亮氨酸(L)。尽管为了清楚起见并未示出,但是参考氨基酸序列202中的剩余位置以对于蛋白质200具有特异性的顺序包含各种氨基酸。替代性氨基酸序列212与参考氨基酸序列202相同,位置16处的变体214除外,其含有替代性氨基酸丙氨酸(A)214,而不是参考氨基酸甘氨酸(G)204。
图3展示了参考氨基酸序列202中的氨基酸原子的氨基酸式分类,在本文中也称为“原子分类300”。在列302中列出的20种天然氨基酸中,特定类型的氨基酸可以在蛋白质中重复出现。即,特定类型的氨基酸可以在蛋白质中出现不止一次。蛋白质还可以具有一些未确定的氨基酸,这些氨基酸按21个终止或缺口氨基酸类别分类。图3中的右侧列含有来自不同氨基酸的α-碳(C
具体地,图3示出了参考氨基酸序列202中氨基酸的α-碳(C
图4展示了基于图3中的原子分类300,参考氨基酸序列202的α-碳原子的3D原子坐标的氨基酸式归属。这在本文中称为“原子坐标分组聚合400”。在图4中,列表404至440以表格形式列出了被分组聚合到21个氨基酸类别中的每个类别下的α-碳原子的3D原子坐标。
在所展示的该具体实施中,图4中的分组聚合400遵循图3的分类300。例如,在图3中,丙氨酸氨基酸类别具有11个α-碳原子,因此,在图4中,丙氨酸氨基酸类别具有来自图3的对应11个α-碳原子的11个3D原子坐标。对于其他氨基酸类别,该分类至分组聚合逻辑也从图3流向图4。然而,该分类至分组聚合逻辑仅用于代表性目的,在其他具体实施中,本发明所公开的技术不需要执行分类300和分组聚合400来定位体素式最接近原子,而且可以执行较少的、额外的或不同的步骤。例如,在一些具体实施中,本发明所公开的技术可以通过使用排序和搜索算法来定位体素式最接近原子,该排序和搜索算法响应于被配置为接受查询参数的搜索查询而从一个或多个数据库返回体素式最接近原子,这些查询参数如排序准则(例如,氨基酸式、原子元素式、原子类型式)、预定义最大扫描半径,以及距离类型(例如,欧几里得、马氏、归一化、非归一化)。在本发明所公开技术的各种具体实施中,本领域的技术人员可以类似地使用来自当前或未来技术领域的多种排序和搜索算法来定位体素式最接近原子。
在图4中,3D原子坐标由笛卡尔坐标x、y、z表示,但是可以使用任何类型的坐标系,诸如球面或柱面坐标,并且要求保护的主题在此方面不受限制。在一些具体实施中,一个或多个数据库可以包括关于蛋白质中α-碳原子和其他氨基酸原子的3D原子坐标的信息。此类数据库可以通过特定的蛋白质进行搜索。
如上文所论述的,体素和体素网格是3D实体。然而,为清楚起见,附图描绘了并且说明书论述了二维(2D)格式的体素和体素网格。例如,27个体素的3×3×3体素网格在本文中被描绘和描述为具有9个2D像素的3×32D像素网格。本领域的技术人员将会知道,2D格式仅用于代表性目的,并且旨在覆盖3D对应物(即,2D像素表示3D体素,而2D像素网格表示3D体素网格)。另外,附图也不是按比例绘制的。例如,使用单个像素来描绘尺寸为2埃
体素式距离计算
图5示意性地展示了确定体素式距离值的过程,该过程在本文中也称为“体素式距离计算500”。在所展示的该实例中,仅针对丙氨酸(A)距离通道计算体素式距离值。然而,针对21个氨基酸类别中的每个类别执行相同的距离计算逻辑,以生成21个氨基酸式距离通道,并且可以进一步扩展到其他原子类型,如β-碳原子和其他原子元素,如氧、氮和氢,如上文关于图1所论述的。在一些具体实施中,在距离计算之前随机旋转原子,以使得对致病性分类器的训练在原子取向上保持不变。
在图5中,体素网格522具有用索引(1,1)、(1,2)、(1,3)、(2,1)、(2,2)、(2,3)、(3,1)、(3,2)和(3,3)标识的9个体素514。体素网格522的中心定位在例如参考氨基酸序列202中位置16处的甘氨酸(G)氨基酸的α-碳原子的3D原子坐标532处,因为在替代性氨基酸序列212中,位置16经历将甘氨酸(G)氨基酸突变为丙氨酸(A)氨基酸的变体,如上文关于图2所论述的。另外,体素网格522的中心与体素(2,2)的中心重合。
居中体素网格522用于21个氨基酸式距离通道中每一者的体素式距离计算。例如,从丙氨酸(A)距离通道开始,测量9个体素514的相应中心的3D坐标与11个丙氨酸α-碳原子的3D原子坐标402之间的距离,以定位9个体素514中每一者的最接近的丙氨酸α-碳原子。然后,使用9个体素514和各自最接近的丙氨酸α-碳原子之间的9个距离的9个距离值来构建丙氨酸距离通道。得到的丙氨酸距离通道以与体素网格522中的9个体素514相同的顺序排列9个丙氨酸距离值。
对于这21个氨基酸类别中的每个类别执行上述过程。例如,居中体素网格522类似地用于计算精氨酸(R)距离通道,使得测量9个体素514的相应中心的3D坐标与35个精氨酸α-碳原子的3D原子坐标404之间的距离,以定位9个体素514中每一者的最接近的精氨酸α-碳原子。然后,使用9个体素514和各自最接近的精氨酸α-碳原子之间的9个距离的9个距离值来构建精氨酸距离通道。得到的精氨酸距离通道以与体素网格522中的9个体素514相同的顺序排列9个精氨酸距离值。对21个氨基酸式距离通道按体素方式编码,以形成距离通道张量。
具体地,在所展示的该实例中,距离512在体素网格522的体素(1,1)中心与最接近的α-碳(C
如上所述,对于具有α-碳原子的氨基酸,所述距离可以是从对应体素中心到对应氨基酸的最接近α-碳原子的最接近α-碳原子距离。此外,对于具有β-碳原子的氨基酸,所述距离可以是从对应体素中心到对应氨基酸的最接近β-碳原子的最接近β-碳原子距离。类似地,对于具有主链原子的氨基酸,所述距离可以是从对应体素中心到对应氨基酸的最接近主链原子的最接近主链原子距离。类似地,对于具有侧链原子的氨基酸,所述距离可以是从对应体素中心到对应氨基酸的最接近侧链原子的最接近侧链原子距离。在一些具体实施中,所述距离除此之外/替代性地可以包括到第二接近原子、第三接近原子、第四接近原子等的距离。
氨基酸式距离通道
图6示出了21个氨基酸式距离通道600的实例。图6中的每一列对应于21个氨基酸式距离通道602至642中相应的一个距离通道。每个氨基酸式距离通道均包括体素网格522的体素514中的每个体素的距离值。例如,丙氨酸(A)的氨基酸式距离通道602包括体素网格522的体素514中的相应体素的距离值。如上文所提及的,体素网格522是体积为3×3×3的3D网格,包括27个体素。类似地,尽管图6以二维方式展示了体素514(例如,3×3网格的九个体素),但是每个氨基酸式距离通道均可以包括3×3×3体素网格的27个体素式距离值。
方向性编码
在一些具体实施中,本发明所公开的技术使用方向性参数来指定参考氨基酸序列202中的参考氨基酸的方向性。在一些具体实施中,本发明所公开的技术使用方向性参数来指定替代性氨基酸序列212中的替代性氨基酸的方向性。在一些具体实施中,本发明所公开的技术使用方向性参数来指定蛋白质200中在氨基酸水平上经历目标变体的位置。
如上所述,在21个氨基酸式距离通道602至642中的所有距离值是从体素网格522中的相应最接近原子到体素514测量的。这些最接近原子来源于参考氨基酸序列202中的一个参考氨基酸。这些含有最接近原子的起源参考氨基酸可以分为两类:(1)在参考氨基酸序列202中经历变体的参考氨基酸204之前的那些起源参考氨基酸,以及(2)在参考氨基酸序列202中经历变体的参考氨基酸204之后的那些起源参考氨基酸。第一类中的起源参考氨基酸可以称为在先参考氨基酸。第二类中的起源参考氨基酸可以称为后续参考氨基酸。
方向性参数被应用于21个氨基酸式距离通道602至642中的那些距离值,所述距离值是从起源于在先参考氨基酸的那些最接近原子测量的。在一个具体实施中,将方向性参数与此类距离值相乘。方向性参数可以是任何数字,诸如-1。
作为应用方向性参数的结果,这21个氨基酸式距离通道600包括一些距离值,这些距离值向致病性分类器指示蛋白质200的哪一端是起始端,哪一端是终止端。这也允许致病性分类器从由距离通道以及参考通道和等位基因通道提供的3D蛋白质结构信息重建蛋白质序列。
距离通道张量
图7是距离通道张量700的示意图。距离通道张量700是来自图6的氨基酸式距离通道600的体素化表示。在距离通道张量700中,像彩色图像的RGB通道一样,21个氨基酸式距离通道602至642按体素方式连结。距离通道张量700的体素化维度是21×3×3×3(其中21表示21个氨基酸类别,3×3×3表示具有27个体素的3D体素网格);但图7是维度21×3×3的2D描绘。
独热编码
图8示出了参考氨基酸204和替代性氨基酸214的独热编码800。在图8中,左侧列是参考氨基酸甘氨酸(G)204的独热编码802,其中1指示甘氨酸氨基酸类别,0指示所有其他的氨基酸类别。在图8中,右侧列是变体/替代性氨基酸丙氨酸(A)214的独热编码804,其中1指示丙氨酸氨基酸类别,0指示所有其他的氨基酸类别。
图9是体素化独热编码的参考氨基酸902和体素化独热编码的变体/替代性氨基酸912的示意图。体素化独热编码的参考氨基酸902是来自图8的参考氨基酸甘氨酸(G)204的独热编码802的体素化表示。体素化独热编码的替代性氨基酸912是来自图8的变体/替代性氨基酸丙氨酸(A)214的独热编码804的体素化表示。体素化独热编码的参考氨基酸902的体素化维度是21×1×1×1(其中21表示21个氨基酸类别);但图9是维度21×1×1的2D描绘。类似地,体素化独热编码的替代性氨基酸912的体素化维度是21×1×1×1(其中21表示21个氨基酸类别);但图9是维度21×1×1的2D描绘。
参考等位基因张量
图10示意性地展示了按体素方式将图7的距离通道张量700与参考等位基因张量1004连结的连结过程1000。参考等位基因张量1004是来自图9的体素化独热编码的参考氨基酸902的体素式聚集(重复/克隆/复制)。即,体素化独热编码的参考氨基酸902的多个拷贝根据体素网格522中的体素514的空间布置按体素方式彼此连结,使得参考等位基因张量1004具有针对体素网格522中的体素514中的每个体素的体素化独热编码的参考氨基酸910的对应拷贝。
连结过程1000产生连结张量1010。参考等位基因张量1004的体素化维度是21×3×3×3(其中21表示21个氨基酸类别,3×3×3表示具有27个体素的3D体素网格);但图10是具有维度21×3×3的参考等位基因张量1004的2D描绘。连结张量1010的体素化维度是42×3×3x3;但图10是具有维度42×3×3的连结张量1010的2D描绘。
替代性等位基因张量
图11示意性地展示了按体素方式将图7的距离通道张量700、图10的参考等位基因张量1004与替代性等位基因张量1104连结的连结过程1100。替代性等位基因张量1104是来自图9的体素化独热编码的替代性氨基酸912的体素式聚集(重复/克隆/复制)。即,体素化独热编码的替代性氨基酸912的多个拷贝根据体素网格522中的体素514的空间布置按体素方式彼此连结,使得替代性等位基因张量1104具有针对体素网格522中的体素514中的每个体素的体素化独热编码的替代性氨基酸910的对应拷贝。
连结过程1100产生连结张量1110。替代性等位基因张量1104的体素化维度是21×3×3×3(其中21表示21个氨基酸类别,3×3×3表示具有27个体素的3D体素网格);但图11是具有维度21×3×3的替代性等位基因张量1104的2D描绘。连结张量1110的体素化维度是63×3×3×3;但图11是具有维度63×3×3的连结张量1110的2D描绘。
在一些具体实施中,运行时逻辑184通过致病性分类器处理连结张量1110,以确定变体/替代性氨基酸丙氨酸(A)214的致病性,该致病性进而被推断为产生变体/替代性氨基酸丙氨酸(A)214的基础核苷酸变体的致病性测定结果。
进化保守通道
预测变体的功能后果至少部分地依赖于以下假设:由于负选择,蛋白质家族的关键氨基酸在进化过程中是保守的(即,在这些位点处的氨基酸变化在过去是有害的),并且在这些位点处的突变增加了在人类中致病(引起疾病)的可能性。一般来讲,收集目标蛋白质的同源序列并比对,并且基于在比对中的目标位置观察到的不同氨基酸的加权频率来计算保守性度量。
因此,本发明所公开的技术将距离通道张量700、参考等位基因张量1004和替代性等位基因张量1104与进化通道连结。进化通道的一个示例是泛氨基酸保守频率。进化通道的另一个示例是每氨基酸保守频率。
在一些具体实施中,使用位置权重矩阵(PWM)来构建进化通道。在其他具体实施中,使用位置特异性频率矩阵(PSFM)来构建进化通道。在还有其他的具体实施中,使用像SIFT、PolyPhen和PANTHER-PSEC这样的计算工具来构建进化通道。在还有其他的具体实施中,这些进化通道是基于进化保存的保存通道。保存与保守性相关,因为它也反映了负选择的作用,其中负选择已经用于防止蛋白质中给定位点处的进化变化。
泛氨基酸进化谱
图12是展示根据本发明所公开技术的一个具体实施,用于确定和分配相对于体素最接近的原子的泛氨基酸保守频率(体素化)的系统的过程1200的流程图。依次讨论图12、图13、图14、图15、图16、图17和图18。
在步骤1202处,系统的相似序列查找器1204检索与参考氨基酸序列202相似(同源)的氨基酸序列。相似的氨基酸序列可以选自多个物种,如灵长类动物、哺乳动物和脊椎动物。
在步骤1212处,系统的比对器1214按位置方式将参考氨基酸序列202与相似的氨基酸序列比对,即,比对器1214进行多序列比对。图14示出了跨99个物种的参考氨基酸序列202的示例多序列比对1400。在一些具体实施中,多序列比对1400可以被划分,例如,以生成用于灵长类动物的第一位置频率矩阵1402、用于哺乳动物的第二位置频率矩阵1412和用于灵长类动物的第三位置频率矩阵1422。在其他具体实施中,跨99个物种生成单位置频率矩阵。
在步骤1222处,系统的泛氨基酸保守频率计算器1224使用多序列比对来确定参考氨基酸序列202中的参考氨基酸的泛氨基酸保守频率。
在步骤1232处,系统的最接近原子查找器1234在体素网格522中查找与体素514最接近的原子。在一些具体实施中,对体素式最接近原子的搜索可以不限于任何特定的氨基酸类别或原子类型。即,可以跨氨基酸类别和氨基酸类型选择体素式最接近原子,只要它们是与相应体素中心最接近的原子。在其他具体实施中,对体素式最接近原子的搜索可以仅限于特定的原子类别,诸如仅限于特定的原子元素(如氧、氮和氢),或者仅限于α-碳原子,或者仅限于β-碳原子,或者仅限于侧链原子,或者仅限于主链原子。
在步骤1242处,系统的氨基酸选择器1244选择参考氨基酸序列202中含有在步骤1232处识别的最接近原子的那些参考氨基酸。这种参考氨基酸可以称为最接近的参考氨基酸。图13示出了将最接近原子1302定位到体素网格522中的体素514并分别将包含最接近原子1302的最接近参考氨基酸1312映射到体素网格522中的体素514的实例。这在图13中标识为“体素到最接近氨基酸的映射1300”。
在步骤1252处,系统的体素化器1254将最接近参考氨基酸的泛氨基酸保守频率体素化。图15示出了确定体素网格522中的第一体素(1,1)的泛氨基酸保守频率序列的实例,在本文中也称为“每体素进化谱确定1500”。
转到图13,映射到第一体素(1,1)的最接近参考氨基酸是参考氨基酸序列202中位置15处的天冬氨酸(D)氨基酸。然后,在位置15处分析参考氨基酸序列202与例如99个物种的99种同源氨基酸序列的多序列比对。这种位置特异性分析和跨物种分析揭示了在跨100个比对的氨基酸序列(即,参考氨基酸序列202加上99个同源氨基酸序列)的位置15处发现了来自21个氨基酸类别中的每个类别的氨基酸实例有多少。
在图15所展示的该实例中,天冬氨酸(D)氨基酸在100个比对的氨基酸序列中的96个序列中的位置15处发现。因此,天冬氨酸氨基酸类别1504被分配了泛氨基酸保守频率0.96。类似地,在所展示的该实例中,缬氨酸(V)氨基酸在100个比对的氨基酸序列中的4个序列中的位置15处发现。因此,缬氨酸氨基酸类别1514被分配了泛氨基酸保守频率0.04。由于在位置15处没有检测到来自其他氨基酸类别的氨基酸实例,因此对剩余氨基酸类别分配为0的泛氨基酸保守频率。这样,对21个氨基酸类别中的每个类别分配相应的泛氨基酸保守频率,该泛氨基酸保守频率可以在第一体素(1,1)的泛氨基酸保守频率序列1502中编码。
图16示出了使用图15中描述的位置频率逻辑(在本文中也称为“体素到进化谱映射1600”)针对体素网格522中的体素514中的相应体素确定的相应泛氨基酸保守频率1612至1692。
然后,体素化器1254使用每体素进化谱1602来生成体素化的每体素进化谱1700,如图17所展示。通常,体素网格522中的体素514中的每个体素具有不同的泛氨基酸保守频率序列,因此具有不同的体素化每体素进化谱,这是因为体素被规则地映射到不同的最接近原子,因而被映射到不同的最接近参考氨基酸。当然,当两个或更多个体素具有相同的最接近原子并因此具有相同的最接近参考氨基酸时,相同的泛氨基酸保守频率序列和相同的体素化每体素进化谱被分配给两个或更多个体素中的每个体素。
图18描绘了进化谱张量1800的实例,其中体素化每体素进化谱1700根据体素网格522中的体素514的空间布置按体素方式彼此连结。进化谱张量1800的体素化维度是21×3×3×3(其中21表示21个氨基酸类别,3×3×3表示具有27个体素的3D体素网格);但图18是具有维度21×3×3的进化谱张量1800的2D描绘。
在步骤1262处,连结器174按体素方式将进化谱张量1800与距离通道张量700连结。在一些具体实施中,进化谱张量1800按体素方式与连结器张量1110连结,以生成维度84×3×3×3的进一步连结张量(未示出)。
在步骤1272处,运行时逻辑184通过致病性分类器处理维度84×3×3×3的进一步连结张量,以测定目标变体的致病性,该致病性进而被推断为在氨基酸水平上产生目标变体的基础核苷酸变体的致病性测定结果。
每氨基酸进化谱
图19是展示用于确定和分配相对于体素最接近的原子的每氨基酸保守频率(体素化)的系统的过程1900的流程图。在图19中,步骤1202和1212与图12中的相同。
在步骤1922处,系统的每氨基酸保守频率计算器1924使用多序列比对来确定参考氨基酸序列202中的参考氨基酸的每氨基酸保守频率。
在步骤1932处,系统的最接近原子查找器1934针对体素网格522中的体素514中的每个体素查找跨21个氨基酸类别中的每个类别的21个最接近原子。21个最接近原子中的每一者彼此不同,因为它们选自不同的氨基酸类别。这使得针对特定体素选择21个独特的最接近参考氨基酸,这进而使得针对特定体素生成21个独特的位置频率矩阵,这进而又使得针对特定体素确定21个独特的每氨基酸保守频率。
在步骤1942处,系统的氨基酸选择器1944为体素网格522中的体素514中的每个体素选择参考氨基酸序列202中的21个参考氨基酸,其包含在步骤1932中识别的21个最接近原子。这种参考氨基酸可以称为最接近的参考氨基酸。
在步骤1952处,系统的体素化器1954将在步骤1942处针对特定体素识别的21个最接近参考氨基酸的每氨基酸保守频率体素化。这21个最接近参考氨基酸必然位于参考氨基酸序列202中的21个不同位置处,因为它们对应于不同的基础最接近原子。因此,对于特定体素,可以为这21个最接近参考氨基酸生成21个位置频率矩阵。如上文关于图12至图15所论述的,这21个位置频率矩阵可以跨其同源氨基酸序列与参考氨基酸序列202按位置方式比对的多个物种生成。
然后,使用这21个位置频率矩阵,可以针对对于特定体素识别的21个最接近参考氨基酸计算21个位置特异性保守分数。这21个位置特异性保守分数形成特定体素的每氨基酸保守频率,类似于图12中的泛氨基酸保守频率序列1502;不同的是序列1502具有许多0条目,而每氨基酸保守频率序列中的每个元素(特征)具有某个值(例如,浮点数),因为跨21个氨基酸类别的21个最接近参考氨基酸必然具有产生不同位置频率矩阵的不同位置,由此产生不同的每氨基酸保守频率。
对体素网格522中的体素514中的每个体素执行上述处理,对得到的体素式每氨基酸保守频率进行体素化、张量化、连结和处理,以便与关于图12至图18所讨论的泛氨基酸保守频率类似地进行致病性测定。
注释通道
图20示出了与距离通道张量700连结的体素化注释通道2000的各种实例。在一些具体实施中,体素化注释通道是不同蛋白质注释的独热指示符,例如,氨基酸(残基)是否为跨膜区、信号肽、活性位点或任何其他结合位点的一部分,或者残基是否经历翻译后修饰、PathRatio(参见Pei P、Zhang A:A Topological Measurement for Weighted ProteinInteraction Network.CSB 2005,268-278)等等。注释通道的附加实例可以在下文的“特定具体实施”部分和权利要求书中找到。
这些体素化注释通道按体素方式布置,使得体素可以具有相同的注释序列,如体素化参考等位基因序列和替代性等位基因序列(例如,注释通道2002、2004、2006),或者体素可以具有各自的注释序列,如体素化每体素进化谱1700(例如,注释通道2012、2014、2016(如由不同的颜色所指示))。
对这些注释通道进行体素化、张量化、连结和处理,以便与关于图12至图18所讨论的泛氨基酸保守频率类似地进行致病性测定。
结构置信度通道
本发明所公开的技术还可以将各种体素化的结构置信度通道与距离通道张量700连结。结构置信度通道的一些实例包括GMQE分数(由SwissModel提供);B-因子;同源模型的温度因素栏(表明残基满足蛋白质结构中的(物理)约束的程度);对于最接近体素中心的残基,比对模板蛋白质的归一化数目(由HHpred提供的比对,例如,体素最接近6个模板结构中的3个模板结构比对的残基,这表示该特征的值为3/6=0.5;最小、最大和平均TM分数;以及与最接近体素的残基比对的模板蛋白质结构的预测TM分数(继续以上实例,假设这3个模板结构的TM分数为0.5、0.5和1.5,则最小值为0.5,平均值为2/3,最大值为1.5)。可以由HHpred提供每个蛋白质模板的TM分数。结构置信度通道的附加实例可以在下文的“特定具体实施”部分和权利要求书中找到。
这些体素化结构置信度通道按体素方式布置,使得体素可以具有相同的结构置信度序列,如体素化参考等位基因序列和替代性等位基因序列,或者体素可以具有各自的结构置信度序列,如体素化每体素进化谱1700。
对这些结构置信度通道进行体素化、张量化、连结和处理,以便与关于图12至图18所讨论的泛氨基酸保守频率类似地进行致病性测定。
致病性分类器
图21展示了输入通道的不同组合与排列,其可以作为输入2102提供给致病性分类器2108,以便对目标变体进行致病性测定2106。输入2102中的一者可以是由距离通道生成器2272生成的距离通道2104。图22示出了计算距离通道2104的不同方法。在一个具体实施中,距离通道2104是基于体素中心与跨多个原子元素的原子之间的距离2202生成的,而与氨基酸无关。在一些具体实施中,距离2202通过最大扫描半径归一化,以生成归一化距离2202a。在另一个具体实施中,距离通道2104是基于体素中心与α-碳原子之间的距离2212在氨基酸基础上生成的。在一些具体实施中,距离2212通过最大扫描半径归一化,以生成归一化距离2212a。在又一个具体实施中,距离通道2104是基于体素中心与β-碳原子之间的距离2222在氨基酸基础上生成的。在一些具体实施中,距离2222通过最大扫描半径归一化,以生成归一化距离2222a。在又一个具体实施中,距离通道2104是基于体素中心与侧链原子之间的距离2232在氨基酸基础上生成的。在一些具体实施中,距离2232通过最大扫描半径归一化,以生成归一化距离2232a。在又一个具体实施中,距离通道2104是基于体素中心与主链原子之间的距离2242在氨基酸基础上生成的。在一些具体实施中,距离2242通过最大扫描半径归一化,以生成归一化距离2242a。在又一个具体实施中,距离通道2104是基于体素中心与相应最接近原子之间的距离2252(一个特征)而生成的,而与原子类型和氨基酸类型无关。在又一个具体实施中,距离通道2104是基于体素中心与来自非标准氨基酸的原子之间的距离2262(一个特征)而生成的。在一些具体实施中,基于体素和原子的极坐标来计算体素与原子之间的距离。极坐标由体素与原子之间的角度来参数化。在一个具体实施中,该角度信息用于生成体素的角度通道(即,独立于距离通道)。在一些具体实施中,最接近原子与邻近原子(例如,主链原子)之间的角度可以用作利用体素编码的特征。
输入2102中的另一者可以是指示在指定半径内缺失原子的特征2114。
输入2102中的另一者可以是参考氨基酸的独热编码2124。输入2102中的另一者可以是变体/替代性氨基酸的独热编码2134。
输入2102中的另一者可以是由进化谱生成器2372生成的进化通道2144,如图23所示。在一个具体实施中,进化通道2144可以基于泛氨基酸保守频率2302生成。在另一个具体实施中,进化通道2144可以基于泛氨基酸保守频率2312生成。
输入2102中的另一者可以是指示缺失残基或缺失进化谱的特征2154。
输入2102中的另一者可以是由注释生成器2472生成的注释通道2164,如图24所示。在一个具体实施中,注释通道2154可以基于分子处理注释2402生成。在另一个具体实施中,注释通道2154可以基于区域注释2412生成。在又一个具体实施中,注释通道2154可以基于位点注释2422生成。在又一个具体实施中,注释通道2154可以基于氨基酸修饰注释2432生成。在又一个具体实施中,注释通道2154可以基于二级结构注释2442生成。在又一个具体实施中,注释通道2154可以基于实验信息注释2452生成。
另一个输入2102可以是由结构置信度生成器2572生成的结构置信度通道2174,如图25所示。在一个具体实施中,结构置信度2174可以基于全局模型质量估计(GMQE)2502生成。在另一个具体实施中,结构置信度2174可以基于定性模型能量分析(QMEAN)分数2512生成。在又一个具体实施中,结构置信度2174可以基于温度因素2522生成。在又一个具体实施中,结构置信度2174可以基于模板建模分数2542生成。模板建模分数2542的实例包括最小模板建模分数2542a、平均模板建模分数2542b和最大模板建模分数2542c。
本领域的技术人员将会知道,可以将输入通道的任何排列与组合连结为输入,以便通过致病性分类器2108进行处理,从而对目标变体进行致病性测定2106。在一些具体实施中,可以只连结输入通道的子集。这些输入通道能够以任何顺序连结。在一个具体实施中,这些输入通道可以由张量生成器(输入编码器)2110连结成单个张量。然后可以将这单个张量作为输入提供给致病性分类器2108,以便对目标变体进行致病性测定2106。
在一个具体实施中,致病性分类器2108使用具有多个卷积层的卷积神经网络(CNN)。在另一个具体实施中,致病性分类器2108使用递归神经网络(RNN),诸如长短期记忆网络(LSTM)、双向LSTM(Bi-LSTM)和门控递归单元(GRU)。在又一个具体实施中,致病性分类器2108使用CNN和RNN两者。在又一个具体实施中,致病性分类器2108使用对图形结构化数据中的依赖性建模的图形卷积神经网络。在又一个具体实施中,致病性分类器2108使用变分自编码器(VAE)。在又一个具体实施中,致病性分类器2108使用生成对抗网络(GAN)。在又一个具体实施中,致病性分类器2108还可以是基于例如自注意力的语言模型,诸如由变换器和BERT实现的语言模型。
在还有其他的具体实施中,致病性分类器2108可以使用1D卷积、2D卷积、3D卷积、4D卷积、5D卷积、扩张或空洞卷积、转置卷积、深度可分离卷积、逐点卷积、1×1卷积、分组卷积、扁平卷积、空间和跨通道卷积、混洗分组卷积、空间可分离卷积,以及去卷积。该致病性分类器可以使用一种或多种损失函数,诸如逻辑回归/对数损失函数、多类交叉熵/Softmax损失函数、二元交叉熵损失函数、均方误差损失函数、L1损失函数、L2损失函数、平滑L1损失函数和Huber损失函数。该致病性分类器可以使用任何并行性、效率性和压缩方案,诸如TFRecord、压缩编码(例如,PNG)、锐化、映射图转换的并行检出、批处理、预取、模型并行性、数据并行性,以及同步/异步随机梯度下降(SGD)。该致病性分类器可以包括上采样层、下采样层、递归连接、栅极和栅极存储器单元(如LSTM或GRU)、残差块、残差连接、高速连接、跳跃连接、窥视孔连接、激活函数(例如,非线性变换函数如修正线性单元(ReLU)、泄露ReLU、指数线性单元(ELU)、S型双曲正切(tanh))、批量归一化层、正则化层、丢弃层、池化层(例如,最大或平均池化)、全局平均池化层、注意力机制,以及高斯误差线性单元。
致病性分类器2108使用基于反向传播的梯度更新技术来训练。可以用于训练致病性分类器2108的示例梯度下降技术包括随机梯度下降、批量梯度下降和微型批量梯度下降。可以用于训练致病性分类器2108的梯度下降优化算法的一些实例是Momentum、Nesterov加速梯度、Adagrad、Adadelta、RMSprop、Adam、AdaMax、Nadam和AMSGrad。在其他具体实施中,致病性分类器2108可以通过无监督学习、半监督学习、自学习、强化学习、多任务学习、多模态学习、迁移学习、知识蒸馏等来训练。
图26示出了根据本发明所公开技术的一个具体实施的致病性分类器2108的示例处理架构2600。处理架构2600包括处理模块2606、2610、2614、2618、2622、2626、2630、2634、2638和2642的级联,其中每个处理模块可以包括1D卷积(1×1×1CONV)、3D卷积(3×3×3CONV)、ReLU非线性和批量归一化(BN)。处理模块的其他实例包括全连接(FC)层、丢弃层、压平层和最终的Softmax层,该最终的Soffmax层为属于良性类别和致病性类别的目标变体产生指数归一化分数。在图26中,“64”表示由特定处理模块应用的卷积滤波器的数量。在图26中,输入体素2602的尺寸是15×15×15×8。图26还示出了由处理架构2600生成的中间输入2604、2608、2612、2616、2620、2624、2628、2632、2636和2640的相应体积维度。
图27示出了根据本发明所公开技术的一个具体实施的致病性分类器2108的示例处理架构2700。处理架构2700包括处理模块2708、2714、2720、2726、2732、2738、2744、2750、2756、2762、2768、2774和2780的级联,诸如1D卷积(CONV 1D)、3D卷积(CONV 3D)、ReLU非线性和批量归一化(BN)。处理模块的其他实例包括全连接(密集)层、丢弃层、压平层和最终的Softmax层,该最终的Softmax层为属于良性类别和致病性类别的目标变体产生指数归一化分数。在图27中,“64”和“32”表示由特定处理模块应用的卷积滤波器的数量。在图27中,由输入层2702提供的输入体素2704的尺寸是7×7×7×108。图27还示出了由处理架构2700生成的中间输入2710、2716、2722、2728、2734、2740、2746、2752、2758、2764、2770、2776和2782以及得到的中间输出2706、2712、2718、2724、2730、2736、2742、2748、2754、2760、2766、2772、2778和2784的相应体积维度。
本领域的技术人员将会知道,其他当前和未来的人工智能、机器学习和深度学习模型、数据集和训练技术可以结合到本发明所公开的变体致病性分类器中,而不偏离本发明所公开技术的实质。
性能结果作为创造性和非显而易见性的客观指标
本文所公开的变体致病性分类器基于3D蛋白质结构进行致病性预测,称之为“PrimateAI 3D”。“Primate AI”是共同拥有且先前公开的变体致病性分类器,其基于蛋白质序列进行致病性预测。关于PrimateAI的附加细节可以在共同拥有的美国专利申请号16/160,903、16/160,986、16/160,968和16/407,149,以及Sundaram,L.等人,Predicting theclinical impact of human mutation with deep neural networks.Nat.Genet.50,1161-1170(2018)中找到。
图28、图29、图30和图31使用PrimateAI作为基准模型来证明PrimateAI 3D相对于PrimateAI的分类优势。图28、图29、图30和图31中的性能结果是在跨多个验证集准确地区分良性变体与致病性变体的分类任务上生成的。在与多个验证集不同的训练集上训练PrimateAI 3D。在用作良性数据集的常见的人类变体和灵长类动物变体上训练PrimateAI3D,而将基于三核苷酸背景的模拟变体用作未标记或假致病性数据集。
新型发育迟缓障碍(新型DDD)是用于比较PrimateAI 3D与Primate AI的分类准确性的验证集的一个示例。新型DDD验证集将来自DDD个体的变体标记为致病性,而将来自DDD个体的健康亲属的相同变体标记为良性。类似的标记方案用于图31中所示的孤独症谱系障碍(ASD)验证集。
BRCA1是用于比较PrimateAI 3D与Primate AI的分类准确性的验证集的另一个示例。BRCA1验证集将合成生成的模拟BRCA1基因的蛋白质的参考氨基酸序列标记为良性变体,并将合成改变的模拟BRCA1基因的蛋白质的等位基因氨基酸序列标记为致病性变体。类似的标记方案用于TP53基因、TP53S3基因及其变体以及图31中所示的其他基因及其变体的不同验证集。
图28用蓝色水平条标识基准PrimateAI模型的表现,并且用橙色水平条标识本发明所公开的PrimateAI 3D模型的表现。绿色水平条描绘通过组合本发明所公开的PrimateAI 3D模型和基准PrimateAI模型的相应致病性预测而导出的致病性预测。在图例中,“ens10”表示10个PrimateAI 3D模型的系综,其中每个模型用不同的种子训练数据集训练并且用不同的权重和偏差来随机初始化。另外,“7×7×7×2”描绘了在训练10个PrimateAI 3D模型的系综期间用于编码输入通道的体素网格的大小。对于给定的变体,10个PrimateAI 3D模型的系综分别生成10个致病性预测,这些致病性预测随后组合(例如,通过平均)生成对于给定变体的最终致病性预测。这种逻辑类似地适用于具有不同组大小的系综。
另外,在图28中,y轴具有不同的验证集,x轴具有p值。p值越大,即水平条越长,表示区分良性变体与致病性变体的准确性越高。如图28中的p值所证实,PrimateAI 3D在大多数验证集上优于PrimateAI(仅tp53s3_A549验证集除外)。即,PrimateAI 3D的橙色水平条总是比PrimateAI的蓝色水平条长。
另外,在图28中,沿y轴的“平均值”类别计算为验证集中的每个验证集确定的p值的平均值。同样在该平均值类别中,PrimateAI 3D优于PrimateAI。
在图29中,PrimateAI由蓝色水平条表示,用大小为3×3×3的体素网格训练的20个PrimateAI 3D模型的系综由红色水平条表示,用大小为7×7×7的体素网格训练的10个PrimateAI 3D模型的系综由紫色水平条表示,用大小为7×7×7的体素网格训练的20个PrimateAI 3D模型的系综由棕色水平条表示,用大小为17×17×17的体素网格训练的20个PrimateAI 3D模型的系综由紫色水平条表示。
另外,在图29中,y轴具有不同的验证集,x轴具有p值。和之前一样,p值越大,即水平条越长,表示区分良性变体与致病性变体的准确性越高。如图20中的p值所证实,PrimateAI 3D的不同配置在大多数验证集上优于PrimateAI。即,PrimateAI 3D的红色、紫色、棕色和粉色水平条大多比PrimateAI的蓝色水平条长。
另外,在图29中,沿y轴的“平均值”类别计算为验证集中的每个验证集确定的p值的平均值。同样在该平均值类别中,PrimateAI 3D的不同配置优于PrimateAI。
在图30中,红色竖条表示PrimateAI,青色竖条表示PrimateAI 3D。在图30中,y轴具有p值,x轴具有不同的验证集。在图30中,毫无例外,在所有验证集上PrimateAI 3D始终优于PrimateAI。即,PrimateAI 3D的青色竖条总是比PrimateAI的红色竖条长。
图31用蓝色竖条标识基准PrimateAI模型的表现,并且用橙色竖条标识本发明所公开的PrimateAI 3D模型的表现。绿色竖条描绘通过组合本发明所公开的PrimateAI 3D模型和基准PrimateAI模型的相应致病性预测而导出的致病性预测。在图31中,y轴具有p值,x轴具有不同的验证集。
如图31中的p值所证实,PrimateAI 3D在大多数验证集上优于PrimateAI(仅tp53s3_A549_p53NULL_Nutlin-3验证集除外)。即,PrimateAI 3D的橙色竖条总是比PrimateAI的蓝色竖条长。
另外,在图31中,单独的“平均值”图表计算为验证集中的每个验证集确定的p值的平均值。同样在该平均值图表中,PrimateAI 3D优于PrimateAI。
平均值统计值可能由于离群值而偏移。为了解决这个问题,图31中还描绘了单独的“方法等级”图表。等级越高,表示分类准确性越差。同样在该方法等级图表中,PrimateAI3D优于PrimateAI,因为PrimateAI 3D具有计数更多的较低等级1和2,与之对比,PrimateAI的等级全都是3。
在图28至图31中,同样明显的是,将PrimateAI 3D与PrimateAI结合产生了优异的分类准确性。即,可以将蛋白质作为氨基酸序列供给PrimateAI以生成第一输出,可以将相同的蛋白质作为3D体素化蛋白质结构供给PrimateAI 3D以生成第二输出,然后可以将第一输出和第二输出组合或汇总分析,以产生蛋白质所经历变体的最终致病性预测。
高效体素化
图32是展示在逐个体素的基础上高效识别最接近原子的高效体素化过程3200的流程图。
现在重新讨论距离通道。如上文所论述的,参考氨基酸序列202可以含有不同类型的原子,诸如α-碳原子、β-碳原子、氧原子、氮原子、氢原子等。因此,如上文所论述的,距离通道可以由最接近的α-碳原子、最接近的β-碳原子、最接近的氧原子、最接近的氮原子、最接近的氢原子等布置。例如,在图6中,九个体素514中的每个体素具有用于最接近的α-碳原子的21个氨基酸式距离通道。图6可以进一步扩展为对于9个体素514中的每个体素还具有用于最接近的β-碳原子的21个氨基酸式距离通道,并且对于9个体素514中的每个体素还具有用于最接近原子的最接近的通用原子距离通道,而不考虑原子的类型和氨基酸的类型。这样,9个体素514中的每个体素可以具有43个距离通道。
讨论现在转向在逐个体素的基础上识别最接近原子以包含在距离通道中所需要的距离计算的数目。考虑图3中的实例,其描绘了分布在全部21个氨基酸类别中的总共828个α-碳原子。为了计算图6中的氨基酸式距离通道602至642,即,为了确定189个距离值,测量从9个体素514中的每个体素到828个α-碳原子中的每个α-碳原子的距离,得到9*828=7,452个距离计算值。在27个体素为3D体素的情况下,这得到27*828=22,356个距离计算值。在还包括828个β-碳原子的情况下,该数目增加至27*1656=44,712个距离计算值。
如图35A所展示,这意味着在逐个体素的基础上为单次蛋白质体素化识别最接近原子的运行时复杂度为O(#原子*#体素)。此外,当跨多种属性(例如,每体素的不同特征或通道,如注释通道和结构置信度通道)计算距离通道时,单次蛋白质体素化的运行时复杂度增加至O(#原子*#体素*#属性)。
因此,距离计算可能成为体素化过程中最消耗计算资源的部分,从而将有价值的计算资源从诸如模型训练和模型推断的关键运行时任务中抽离。例如,考虑用7,000个蛋白质的训练数据集进行模型训练的情况。为跨多个氨基酸、原子和属性的多个体素生成距离通道可以涉及每个蛋白质超过100次体素化,从而在单次训练迭代(历元)中产生约800,000次体素化。20至40次历元的训练运行(在每次历元中原子坐标的旋转)可以产生多达3200万次体素化。
3200万次体素化除计算成本高之外,数据量也太大,以至于无法容纳在主存储器中(例如,对于15×15×15的体素网格,数据量超过20TB)。考虑到用于参数优化和系综学习的重复训练运行,该体素化过程的存储器占用变得太大,以至于不能存储在盘上,使得该体素化过程成为模型训练的一部分而不是预计算步骤。
本发明所公开的技术提供了一种高效体素化过程,其在运行时复杂度为O(#原子*#体素)的情况下实现高达约100倍的加速。本发明所公开的高效体素化过程将单次蛋白质体素化的运行时复杂度降至O(#原子)。在每体素具有不同的特征或通道的情况下,本发明所公开的高效体素化过程将单次蛋白质体素化的运行时复杂度降至O(#原子*#属性)。因此,该体素化过程变得与模型训练一样快,从而将计算瓶颈从体素化转移回到在诸如GPU、ASIC、TPU、FPGA、CGRA等处理器上计算神经网络权重。
在本发明所公开的涉及大体素网格的高效体素化过程的一些具体实施中,对于每体素具有不同的特征或通道的情况,单次蛋白质体素化的运行时复杂度为O(#原子+体素)和O(#原子*#属性+体素)。当原子数量与体素数量相比微不足道时,例如,当在100×100×100体素网格中只有一个原子(即,每个原子100万个体素)时,观察到“+体素”复杂度。在这种情况下,运行时由巨量体素的开销所支配,例如,为了将存储器分配给100万个体素,将100万个体素初始化为0,等等。
讨论内容现在转向本发明所公开的高效体素化过程的细节。依次讨论图32A、图32B、图33、图34和图35B。
从图32A开始,在步骤3202处,将每个原子(例如,828个α-碳原子中的每一个和828个β-碳原子中的每一个)与包含该原子的体素(例如,9个体素514中的一个)相关联。术语“包含”是指原子的3D原子坐标位于体素中。包含原子的体素在本文中也称为“含原子体素”。
图32B和图33描述了如何选择包含特定原子的体素。图33使用2D原子坐标作为3D原子坐标的代表。需注意,体素网格522被规则地间隔开,其中体素514中的每个体素具有相同的步长大小(例如,1埃
另外,在图33中,体素网格522沿第一维度(例如,x轴)具有品红色索引[0,1,2],沿第二维度(例如,y轴)具有青色索引[0,1,2]。另外,在图33中,体素网格522中的相应体素514由绿色体素索引[体素0,体素1,体素8]和黑色体素中心索引[(1,1),(1,2),(3,3)]来标识。
另外,在图33中,沿第一维度的体素中心的中心坐标(即,第一维度体素坐标)以橙色标识。另外,在图33中,沿第二维度的体素中心的中心坐标(即,第二维度体素坐标)以红色标识。
首先,在步骤3202a(图33中的步骤1)处,将特定原子的3D原子坐标(1.7456,2.14323)量化,以生成量化的3D原子坐标(1.7,2.1)。该量化可以通过对比特进行舍入或截断来实现。
然后,在步骤3202b(图33中的步骤2)处,以维度为基础将体素514的体素坐标(或者体素中心或体素中心坐标)分配给量化的3D原子坐标。对于第一维度,将量化的原子坐标1.7分配给体素1,因为该原子坐标覆盖从1至2范围内的第一维度体素坐标,并且在第一维度中以1.5为中心。需注意,与沿第二维度具有索引0相比,体素1沿第一维度具有索引1。
对于第二维度,从体素1开始,沿第二维度遍历体素网格522。这使得量化的原子坐标2.5被分配给体素7,因为该原子坐标覆盖从2至3范围内的第二维度体素坐标,并且在第二维度中以2.5为中心。需注意,与沿第一维度具有索引1相比,体素7沿第二维度具有索引2。
然后,在步骤3202c(图33中的步骤3)处,选择对应于所分配的体素坐标的维度索引。即,对于体素1,沿第一维度选择索引1,对于体素7,沿第二维度选择索引2。本领域的技术人员将会知道,可以针对第三维度类似地执行上述步骤,以选择沿第三维度的维度索引。
然后,在步骤3202d(图33中的步骤4)处,基于用底数幂对所选择的维度索引进行位置式加权,来生成累加和。位置编号系统背后的一般思想是通过递增底数(或基数)的幂来表示数值,例如,二进制为基数2,三进制为基数3,八进制为基数8,十六进制为基数16。这通常被称为加权编号系统,因为每个位置都用底数的幂来加权。位置编号系统的有效数值集合的大小等于该系统的底数。例如,十进制系统中有10位数字,0至9,三进制系统中有3位数字,0、1、2。底数系统中的最大有效数字比底数小1(因此,在小于9的任何底数系统中,8都不是有效数字)。任何十进制整数都可以用任何其他整数基数系统来精确表示,反之亦然。
返回到图33中的实例,将所选择的维度索引1和2转换为单个整数,方法是按位置方式将这些维度索引分别乘以其各自的基数幂,然后将这些按位置方式相乘的结果相加。这里选择基数3是因为3D原子坐标有三个维度(但是为简单起见,图33仅示出了沿两个维度的2D原子坐标)。
由于索引2位于最右位(即,最低有效位),因此将其乘以3的0次幂得到2。由于索引1位于第二最右位(即,第二最低有效位),因此将其乘以3的1次幂得到3。这使得累加和为5。
然后,在步骤3202e(图33中的步骤5)处,基于累加和,选择包含特定原子的体素的体素索引。即,累加和被解释为包含特定原子的体素的体素索引。
在步骤3212处,在每个原子与含原子体素相关联之后,进一步将每个原子与在含原子体素的邻域中的一个或多个体素(在本文中也称为“邻域体素”)相关联。邻域体素可以基于在含原子体素的预定义半径(例如,5埃
需注意,不进行距离计算来确定含原子体素和邻域体素。借助体素的空间布置来选择含原子体素,这允许将量化的3D原子坐标分配给体素网格中对应的规则间隔的体素中心(不使用任何距离计算)。另外,借助与体素网格中的含原子体素在空间上邻接来选择邻域体素(同样不使用任何距离计算)。
在步骤3222处,将每个体素映射到在步骤3202和步骤3212处与之相关联的原子。在一个具体实施中,该映射被编码在基于原子到体素映射3402生成的体素到原子映射3412中(例如,通过将基于体素的排序关键字应用于原子到体素映射3402)。体素到原子的映射3412在本文中也称为“单元格到元素的映射”。在一个示例中,将第一体素映射到α-碳原子的第一子集3414,该第一子集包括在步骤3202和步骤3212处与第一体素相关联的α-碳原子。在另一个示例中,将第二体素映射到α-碳原子的第二子集3416,该第二子集包括在步骤3202和步骤3212处与第二体素相关联的α-碳原子。
在步骤3232处,对于每个体素,计算该体素与在步骤3222处映射到该体素的原子之间的距离。步骤3232的运行时复杂度为O(#原子),因为到特定原子的距离在体素到原子的映射3412中从该特定原子唯一映射到的相应体素仅测量一次。这在不考虑相邻体素时是真实的。在没有相邻体素的情况下,大O符号中所隐含的常数因子是1。在有相邻体素的情况下,大O符号等于相邻体素数目+1,因为相邻体素的数目对于每个体素是恒定的,因此运行时复杂度O(#原子)保持为真。相比之下,在图35A中,到特定原子的距离被冗余地测量与体素数目一样多的次数(例如,由于体素有27个,所以针对特定原子测量27次距离)。
在图35B中,基于体素到原子的映射3412,将每个体素映射到828个原子的相应子集(不包括计算到邻域体素的距离),如相应体素的相应椭圆所展示。相应子集基本上不重叠,但有一些例外。当多个原子被映射到同一体素时,由于一些情况而存在不明显的重叠,如图35B中由撇号和椭圆之间的黄色重叠所指示。这种最小的重叠对运行时复杂度O(#原子)有累加效应,而没有乘法效应。这种重叠是在确定包含原子的体素之后考虑相邻体素的结果。在没有相邻体素的情况下,可能没有重叠,因为一个原子仅与一个体素相关联。然而,在考虑相邻体素的情况下,每个相邻体素可以潜在地与同一原子相关联(只要同一氨基酸没有其他原子更接近相邻体素)。
在步骤3242处,对于每个体素,基于在步骤3232处计算的距离,识别对于该体素最接近的原子。在一个具体实施中,这种识别被编码在体素到最接近原子的映射3422中,本文中也称之为“单元格到最接近元素的映射”。在一个示例中,第一体素被映射到作为其最接近的α-碳原子的第2个α-碳原子3424。在另一个示例中,第二体素被映射到作为其最接近的α-碳原子的第31个α-碳原子3426。
此外,当使用上文论述的技术来计算体素式距离时,存储原子的原子类型和氨基酸类型分类以及对应的距离值,以生成分类的距离通道。
一旦使用上文论述的技术识别了到最接近原子的距离,就可以在距离通道中对这些距离进行编码,以便进行体素化并随后由致病性分类器2108进行处理。
计算机系统
图36示出了可以用于实现本发明所公开的技术的示例计算机系统3600。计算机系统3600包括经由总线子系统3655与多个外围设备通信的至少一个中央处理单元(CPU)3672。这些外围设备可以包括存储子系统3610(包括例如存储器设备和文件存储子系统3636)、用户界面输入设备3638、用户界面输出设备3676和网络接口子系统3674。输入设备和输出设备允许用户与计算机系统3600进行交互。网络接口子系统3674提供通向外部网络的接口,该接口包括通向其他计算机系统中的对应接口设备的接口。
在一个具体实施中,致病性分类器2108以能够通信的方式链接到存储子系统3610和用户界面输入设备3638。
用户界面输入设备3638可以包括:键盘;指向设备,诸如鼠标、轨迹球、触摸板或图形输入板;扫描仪;结合到显示器中的触摸屏;音频输入设备,诸如语音识别系统和麦克风;以及其他类型的输入设备。一般来讲,使用术语“输入设备”旨在包括将信息输入到计算机系统3600中的所有可能类型的设备和方式。
用户界面输出设备3676可以包括显示子系统、打印机、传真机或非视觉显示器(诸如音频输出设备)。显示子系统可包括LED显示器、阴极射线管(CRT)、平板设备诸如液晶显示器(LCD)、投影设备或用于产生可见图像的一些其他机构。显示子系统还可提供非视觉显示器,诸如音频输出设备。一般来讲,使用术语“输出设备”旨在包括将信息从计算机系统3600输出到用户或者输出到另一机器或计算机系统的所有可能类型的设备和方式。
存储子系统3610存储提供本文描述的一些或全部模块和方法的功能的编程结构和数据结构。这些软件模块通常由处理器3678来执行。
处理器3678可以是图形处理单元(GPU)、现场可编程门阵列(FPGA)、专用集成电路(ASIC)和/或粗粒度可重构架构(CGRA)。处理器3678可以由深度学习云平台(诸如GoogleCloud Platform
在存储子系统3610中使用的存储器子系统3622可以包括多个存储器,包括用于在程序执行期间存储指令和数据的主随机存取存储器(RAM)3632和其中存储固定指令的只读存储器(ROM)3634。文件存储子系统3636可以为程序文件和数据文件提供持久性存储,并且可以包括硬盘驱动器、软盘驱动器以及相关联的可移动介质、CD-ROM驱动器、光盘驱动器或可移动介质磁盘盒。实现某些具体实施的功能的模块可以由文件存储子系统3636存储在存储子系统3610中,或者存储在处理器可访问的其他机器中。
总线子系统3655提供用于使计算机系统3600的各种部件和子系统按照预期彼此通信的机构。尽管总线子系统3655被示意性地示出为单条总线,但是该总线子系统的替代性具体实施可以使用多条总线。
计算机系统3600本身可以具有不同类型,包括个人计算机、便携式计算机、工作站、计算机终端、网络计算机、电视机、主机、服务器群、一组广泛分布的松散联网计算机,或者任何其他数据处理系统或用户设备。由于计算机和网络的性质不断变化,对图36中描绘的计算机系统3600的描述仅旨在作为用于展示本发明的优选具体实施的具体实例。计算机系统3600也可能具有许多其他配置,其中的部件相比图36中所描绘的计算机系统更多或更少。
特定具体实施1
以下具体实施可以作为系统、方法或制品来实践。具体实施的一个或多个特征可与基本具体实施组合。不互相排斥的具体实施被教导为可组合的。具体实施的一个或多个特征可与其他具体实施组合。本公开周期性地提醒用户这些选项。从一些具体实施中省略重复这些选项的表述不应被视为限制前述部分中教导的组合,这些表述将据此以引用方式并入以下具体实施中的每个具体实施中。
尽管本发明所公开的技术使用3D数据作为输入,但是在其他具体实施中,可以类似地使用1D数据、2D数据(例如,像素和2D原子坐标)、4D数据、5D数据等。
在一些具体实施中,系统包括存储蛋白质中多个氨基酸的氨基酸式距离通道的存储器。氨基酸式距离通道中的每个氨基酸式距离通道具有多个体素中的体素的体素式距离值。体素式距离值指定从多个体素中的对应体素到多个氨基酸中的对应氨基酸的原子的距离。该系统还包括致病性测定引擎,其被配置为处理包括氨基酸式距离通道和由变体表达的蛋白质的替代性等位基因的张量。致病性测定引擎还可以被配置为至少部分地基于张量来确定变体的致病性。
在一些具体实施中,该系统还包括距离通道生成器,其将体素的体素网格中心定位在氨基酸的相应残基的α碳原子上。距离通道生成器可以将体素网格中心定位在位于蛋白质中变体氨基酸处的特定氨基酸残基的α-碳原子上。
该系统可以被配置为通过将特定氨基酸之前的那些氨基酸的体素式距离值乘以方向性参数,来在该张量中编码氨基酸的方向性和特定氨基酸的位置。所述距离可以是从体素网格中的对应体素中心到对应氨基酸的最接近原子的最接近原子距离。在一些具体实施中,最接近原子距离可以是欧几里得距离。最接近原子距离可以通过将欧几里得距离除以最大最接近原子距离来归一化。氨基酸可以具有α-碳原子,并且在一些具体实施中,所述距离可以是从对应体素中心到对应氨基酸的最接近α-碳原子的最接近α-碳原子距离。氨基酸可以具有β-碳原子,并且在一些具体实施中,所述距离可以是从对应体素中心到对应氨基酸的最接近β-碳原子的最接近β-碳原子距离。氨基酸可以具有主链原子,并且在一些具体实施中,所述距离可以是从对应体素中心到对应氨基酸的最接近主链原子的最接近主链原子距离。氨基酸具有侧链原子,并且在一些具体实施中,所述距离可以是从对应体素中心到对应氨基酸的最接近侧链原子的最接近侧链原子距离。
该系统可以进一步被配置为在张量中编码最接近原子通道,该最接近原子通道指定从每个体素到最接近原子的距离。可以选择最接近原子而不考虑氨基酸和氨基酸的原子元素。在一些具体实施中,该距离是欧几里得距离。该距离可以通过将欧几里得距离除以最大距离来归一化。氨基酸可以包括非标准氨基酸。张量可以包括指定未在体素中心的预定义半径内找到的原子的缺席原子通道,并且该缺席原子通道可以是独热编码的。在一些具体实施中,张量可以还包括按体素方式编码为氨基酸式距离通道中的每个氨基酸式距离通道的替代性等位基因的独热编码。该张量可以还包括蛋白质的参考等位基因。在一些具体实施中,张量可以还包括按体素方式编码为氨基酸式距离通道中的每个氨基酸式距离通道的参考等位基因的独热编码。张量可以还包括指定跨多个物种的氨基酸的保守水平的进化谱。
该系统可以还包括进化谱生成器,该进化谱生成器为体素中的每个体素选择跨氨基酸类别和原子类别的最接近原子、为包括该最接近原子的氨基酸残基选择泛氨基酸保守频率序列,并且使该泛氨基酸保守频率序列可用作进化谱之一。可以为在多个物种中观察到的残基的特定位置配置泛氨基酸保守频率序列。泛氨基酸保守频率序列可以指定对于特定氨基酸是否存在缺失的保守频率。在一些具体实施中,对于体素中的每个体素,该进化谱生成器可以选择氨基酸中的相应氨基酸中的相应最接近原子、可以选择氨基酸中包括这些最接近原子的相应残基的相应每氨基酸保守频率,并且可以使得这些每氨基酸保守频率可用作进化谱之一。可以为在多个物种中观察到的残基的特定位置配置每氨基酸保守频率。每氨基酸保守频率可以指定对于特定氨基酸是否存在缺失的保守频率。
在该系统的一些具体实施中,张量可以还包括用于氨基酸的注释通道。这些注释通道可以在张量中进行独热编码。注释通道可以是分子处理注释,这些分子处理注释包括起始甲硫氨酸、信号、转运肽、前肽、链和肽。注释通道可以是区域注释,这些区域注释包括拓扑结构域、跨膜、膜内、结构域、重复序列、钙结合、锌指、脱氧核糖核酸(DNA)结合、核苷酸结合、区域、卷曲螺旋、基序和组成偏倚。注释通道可以是位点注释,这些位点注释包括活性位点、金属结合、结合位点和位点。注释通道可以是氨基酸修饰注释,这些氨基酸修饰注释包括非标准残基、经修饰残基、脂化、糖基化、二硫键和交联。注释通道可以是二级结构注释,这些二级结构注释包括螺旋、转角和β链。注释通道可以是实验信息注释,这些实验信息注释包括诱变、序列不确定性、序列冲突、非相邻残基和非末端残基。
在该系统的一些具体实施中,张量还包括用于氨基酸的结构置信度通道,这些结构置信度通道指定氨基酸相应结构的质量。结构置信度通道可以是全局模型质量估计(GMQE)。结构置信度通道可以包括定性模型能量分析(QMEAN)分数。结构置信度通道可以是指定残基满足相应蛋白质结构的物理约束的程度的温度因素。结构置信度通道可以是指定最接近体素的原子的残基具有对准的模板结构的程度的模板结构比对。结构置信度通道可以是对准的模板结构的模板建模分数。结构置信度通道可以是模板建模分数中最小的一个、模板建模分数的平均值,以及模板建模分数中最大的一个。
在一些具体实施中,该系统可以还包括张量生成器,该张量生成器按体素方式将用于α-碳原子的氨基酸式距离通道与替代性等位基因的独热编码连结以生成张量。张量生成器可以按体素方式将用于β-碳原子的氨基酸式距离通道与替代性等位基因的独热编码连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道与替代性等位基因的独热编码连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码与泛氨基酸保守频率连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、泛氨基酸保守频率与注释通道连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、泛氨基酸保守频率、注释通道与结构置信度通道连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码与氨基酸中的每种氨基酸的每氨基酸保守频率连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、氨基酸中的每种氨基酸的每氨基酸保守频率与注释通道连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、氨基酸中的每种氨基酸的每氨基酸保守频率、注释通道与结构置信度通道连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码与参考等位基因的独热编码连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、参考等位基因的独热编码与泛氨基酸保守频率连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、参考等位基因的独热编码、泛氨基酸保守频率与注释通道连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、参考等位基因的独热编码、泛氨基酸保守频率、注释通道与结构置信度通道连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、参考等位基因的独热编码与氨基酸中的每种氨基酸的每氨基酸保守频率连结以生成张量。张量生成器可以按体素方式将用于α-碳原子的氨基酸式距离通道、用于β-碳原子的氨基酸式距离通道、替代性等位基因的独热编码、参考等位基因的独热编码、氨基酸中的每种氨基酸的每氨基酸保守频率与注释通道连结以生成张量。张量生成器可以按体素方式将用于α碳原子的氨基酸式距离通道、用于β碳原子的所述氨基酸式距离通道、替代性等位基因的独热编码、参考等位基因的独热编码、氨基酸中的每种氨基酸的每氨基酸保守频率、注释通道与结构置信度通道连结以生成张量。
在一些具体实施中,该系统可以还包括原子旋转引擎,该原子旋转引擎在氨基酸式距离通道生成之前旋转氨基酸的原子。致病性测定引擎可以是神经网络。在特定具体实施中,致病性测定引擎可以是卷积神经网络。卷积神经网络可以使用1×1×1卷积、3×3×3卷积、矫正线性单元激活层、批量归一化层、全连接层、Dropout正则化层和Softmax分类层。1×1×1卷积和3×3×3卷积可以是三维卷积。
在一些具体实施中,1×1×1卷积的层可以处理该张量并且产生作为该张量的卷积表示的中间输出。3×3×3卷积的层序列可以处理中间输出并且产生扁平化输出。全连接层可以处理扁平化输出并且产生非归一化输出。Softmax分类层可以处理非归一化输出并且产生指数归一化输出,这些指数归一化输出识别变体是致病性变体和良性变体的可能性。S型层可以处理非归一化输出并且产生归一化输出,该归一化输出识别变体是致病性变体的可能性。体素、原子和距离可以具有三维坐标。张量可以具有至少三个维度,中间输出可以具有至少三个维度,并且扁平化输出可以具有一个维度。
在一些具体实施中,致病性测定引擎是递归神经网络。在其他具体实施中,致病性测定引擎是基于注意力的神经网络。在还有其他具体实施中,致病性测定引擎是梯度提升树。在还有其他具体实施中,致病性测定引擎是状态向量机。
在其他具体实施中,系统可以包括存储蛋白质中氨基酸的原子类别式距离通道的存储器。这些氨基酸可以具有多个原子类别的原子,而所述多个原子类别中的原子类别可以指定这些氨基酸的原子元素。原子类别式距离通道可以具有多个体素中的体素的体素式距离值。体素式距离值可以指定从多个体素中的对应体素到多个原子类别中的对应原子类别中的原子的距离。该系统可以还包括致病性测定引擎,其被配置为处理包括原子类别式距离通道和由变体表达的蛋白质的替代性等位基因的张量,并且至少部分地基于该张量来测定变体的致病性。
该系统可以还包括距离通道生成器,其将体素的体素网格中心定位在多个原子类别中的相应原子类别的相应原子上。距离通道生成器可以将体素网格中心定位在蛋白质中至少一个变体氨基酸的残基的α-碳原子上。所述距离可以是从体素网格中的对应体素中心到对应原子类别中的最接近原子的最接近原子距离。最接近原子距离可以是欧几里得距离。最接近原子距离可以通过将欧几里得距离除以最大最接近原子距离来归一化。该距离可以是从体素网格中的对应体素中心到最接近原子的最接近原子距离,而与氨基酸和氨基酸的原子种类无关。最接近原子距离可以是欧几里得距离。最接近原子距离可以通过将欧几里得距离除以最大最接近原子距离来归一化。
在本部分中描述的方法的其他具体实施可包括存储指令的非暂态计算机可读存储介质,这些指令可由处理器执行以执行上述方法中的任一种方法。在本部分中描述的方法的又一具体实施可包括一种系统,该系统包括存储器和一个或多个处理器,该一个或多个处理器可操作以执行存储在存储器中的指令,以执行上述方法中的任一种方法。
条款组1
1.一种计算机实现的方法,包括:
存储蛋白质中多个氨基酸的氨基酸式距离通道,
其中所述氨基酸式距离通道中的每个氨基酸式距离通道具有多个体素中的体素的体素式距离值,并且
其中所述体素式距离值指定从所述多个体素中的对应体素到所述多个氨基酸中的对应氨基酸的原子的距离;
处理包括所述氨基酸式距离通道和由变体表达的所述蛋白质的替代性等位基因的张量;以及
至少部分地基于所述张量来确定所述变体的致病性。
2.根据条款1所述的计算机实现的方法,还包括将所述体素的体素网格中心定位在所述氨基酸的相应残基的α碳原子上。
3.根据条款2所述的计算机实现的方法,还包括将所述体素网格中心定位在特定氨基酸中对应于所述蛋白质中的至少一个变体氨基酸的残基的α碳原子上。
4.根据条款3所述的计算机实现的方法,还包括通过将所述特定氨基酸之前的那些氨基酸的体素式距离值乘以方向性参数,来在所述张量中编码所述氨基酸的方向性和所述特定氨基酸的位置。
5.根据条款3所述的计算机实现的方法,其中所述距离是从所述体素网格中的对应体素中心到所述对应氨基酸的最接近原子的最接近原子距离。
6.根据条款5所述的计算机实现的方法,其中所述最接近原子距离是欧几里得距离。
7.根据条款6所述的计算机实现的方法,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
8.根据条款5所述的计算机实现的方法,其中所述氨基酸具有α碳原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近α碳原子的最接近α碳原子距离。
9.根据条款5所述的计算机实现的方法,其中所述氨基酸具有β碳原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近β碳原子的最接近β碳原子距离。
10.根据条款5所述的计算机实现的方法,其中所述氨基酸具有主链原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近主链原子的最接近主链原子距离。
11.根据条款5所述的计算机实现的方法,其中所述氨基酸具有侧链原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近侧链原子的最接近侧链原子距离。
12.根据条款3所述的计算机实现的方法,还包括在所述张量中编码最接近原子通道,所述最接近原子通道指定从每个体素到最接近原子的距离,其中选择所述最接近原子而不考虑所述氨基酸和所述氨基酸的原子元素。
13.根据条款12所述的计算机实现的方法,其中所述距离是欧几里得距离。
14.根据条款13所述的计算机实现的方法,其中所述距离通过将所述欧几里得距离除以最大距离来归一化。
15.根据条款12所述的计算机实现的方法,其中所述氨基酸包括非标准氨基酸。
16.根据条款1所述的计算机实现的方法,其中所述张量还包括指定未在体素中心的预定义半径内找到的原子的缺席原子通道,并且其中所述缺席原子通道是独热编码的。
17.根据条款1所述的计算机实现的方法,其中所述张量还包括按体素方式编码为所述氨基酸式距离通道中的每个氨基酸式距离通道的所述替代性等位基因的独热编码。
18.根据条款1所述的计算机实现的方法,其中所述张量还包括所述蛋白质的参考等位基因。
19.根据条款18所述的计算机实现的方法,其中所述张量还包括按体素方式编码为所述氨基酸式距离通道中的每个氨基酸式距离通道的所述参考等位基因的独热编码。
20.根据条款1所述的计算机实现的方法,其中所述张量还包括指定跨多个物种的所述氨基酸的保守水平的进化谱。
21.根据条款20所述的计算机实现的方法,还包括:对于所述体素中的每个体素,
跨所述氨基酸和所述原子类别选择最接近原子,
为包括所述最接近原子的氨基酸残基选择泛氨基酸保守频率序列,以及
使所述泛氨基酸保守频率序列可用作所述进化谱之一。
22.根据条款21所述的计算机实现的方法,其中为在所述多个物种中观察到的所述残基的特定位置配置所述泛氨基酸保守频率序列。
23.根据条款21所述的计算机实现的方法,其中所述泛氨基酸保守频率序列指定对于特定氨基酸是否存在缺失的保守频率。
24.根据条款21所述的计算机实现的方法,还包括:对于所述体素中的每个体素,
在所述氨基酸中的相应氨基酸中选择相应的最接近原子,
为包括所述最接近原子的所述氨基酸的相应残基选择相应的每氨基酸保守频率,以及
使所述每氨基酸保守频率可用作所述进化谱之一。
25.根据条款24所述的计算机实现的方法,其中为在所述多个物种中观察到的所述残基的特定位置配置所述每氨基酸保守频率。
26.根据条款24所述的计算机实现的方法,其中所述每氨基酸保守频率指定对于特定氨基酸是否存在缺失的保守频率。
27.根据条款1所述的计算机实现的方法,其中所述张量还包括用于所述氨基酸的注释通道,其中所述注释通道在所述张量中进行独热编码。
28.根据条款27所述的计算机实现的方法,其中所述注释通道是分子处理注释,所述分子处理注释包括起始甲硫氨酸、信号、转运肽、前肽、链和肽。
29.根据条款27所述的计算机实现的方法,其中所述注释通道是区域注释,所述区域注释包括拓扑结构域、跨膜、膜内、结构域、重复序列、钙结合、锌指、脱氧核糖核酸(DNA)结合、核苷酸结合、区域、卷曲螺旋、基序和组成偏倚。
30.根据条款27所述的计算机实现的方法,其中所述注释通道是位点注释,所述位点注释包括活性位点、金属结合、结合位点和位点。
31.根据条款27所述的计算机实现的方法,其中所述注释通道是氨基酸修饰注释,所述氨基酸修饰注释包括非标准残基、经修饰残基、脂化、糖基化、二硫键和交联。
32.根据条款27所述的计算机实现的方法,其中所述注释通道是二级结构注释,所述二级结构注释包括螺旋、转角和β链。
33.根据条款27所述的计算机实现的方法,其中所述注释通道是实验信息注释,所述实验信息注释包括诱变、序列不确定性、序列冲突、非相邻残基和非末端残基。
34.根据条款1所述的计算机实现的方法,其中所述张量还包括用于所述氨基酸的结构置信度通道,所述结构置信度通道指定所述氨基酸的相应结构的质量。
35.根据条款34所述的计算机实现的方法,其中所述结构置信度通道是全局模型质量估计(GMQE)。
36.根据条款34所述的计算机实现的方法,其中所述结构置信度通道包括定性模型能量分析(QMEAN)分数。
37.根据条款34所述的计算机实现的方法,其中所述结构置信度通道是指定所述残基满足相应蛋白质结构的物理约束的程度的温度因素。
38.根据条款34所述的计算机实现的方法,其中所述结构置信度通道是指定最接近所述体素的原子的残基具有对准的模板结构的程度的模板结构比对。
39.根据条款38所述的计算机实现的方法,其中所述结构置信度通道是所述对准的模板结构的模板建模分数。
40.根据条款39所述的计算机实现的方法,其中所述结构置信度通道是所述模板建模分数中最小的一个、所述模板建模分数的平均值,以及所述模板建模分数中最大的一个。
41.根据条款1所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的氨基酸式距离通道与所述替代性等位基因的所述独热编码连结以生成所述张量。
42.根据条款41所述的计算机实现的方法,还包括按体素方式将用于所述β碳原子的氨基酸式距离通道与所述替代性等位基因的所述独热编码连结以生成所述张量。
43.根据条款42所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道与所述替代性等位基因的所述独热编码连结以生成所述张量。
44.根据条款43所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码与泛氨基酸保守频率序列连结以生成所述张量。
45.根据条款44所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述泛氨基酸保守频率序列与所述注释通道连结以生成所述张量。
46.根据条款45所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述泛氨基酸保守频率序列、所述注释通道与所述结构置信度通道连结以生成所述张量。
47.根据条款46所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码与所述氨基酸中的每种氨基酸的每氨基酸保守频率连结以生成所述张量。
48.根据条款47所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述氨基酸中的每种氨基酸的每氨基酸保守频率与所述注释通道连结以生成所述张量。
49.根据条款48所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述氨基酸中的每种氨基酸的每氨基酸保守频率、所述注释通道与所述结构置信度通道连结以生成所述张量。
50.根据条款49所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码与所述参考等位基因的所述独热编码连结以生成所述张量。
51.根据条款50所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码与所述泛氨基酸保守频率序列连结以生成所述张量。
52.根据条款51所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述泛氨基酸保守频率序列与所述注释通道连结以生成所述张量。
53.根据条款52所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述泛氨基酸保守频率序列、所述注释通道与所述结构置信度通道连结以生成所述张量。
54.根据条款53所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码与所述氨基酸中的每种氨基酸的所述每氨基酸保守频率连结以生成所述张量。
55.根据条款54所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述氨基酸中的每种氨基酸的所述每氨基酸保守频率与所述注释通道连结以生成所述张量。
56.根据条款55所述的计算机实现的方法,还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述氨基酸中的每种氨基酸的所述每氨基酸保守频率、所述注释通道与所述结构置信度通道连结以生成所述张量。
57.根据条款1所述的计算机实现的方法,还包括在所述氨基酸式距离通道生成之前旋转所述氨基酸的原子。
58.根据条款1所述的计算机实现的方法,还包括在卷积神经网络中使用1×1×1卷积、3×3×3卷积、矫正线性单元激活层、批量归一化层、全连接层、Dropout正则化层和Softmax分类层。
59.根据条款58所述的计算机实现的方法,其中所述1×1×1卷积和所述3×3×3卷积是三维卷积。
60.根据条款58所述的计算机实现的方法,其中所述1×1×1卷积的层处理所述张量并且产生作为所述张量的卷积表示的中间输出,其中所述3×3×3卷积的层序列处理所述中间输出并且产生扁平化输出,其中所述全连接层处理所述扁平化输出并且产生非归一化输出,并且其中所述Softmax分类层处理所述非归一化输出并且产生指数归一化输出,所述指数归一化输出识别所述变体是致病性变体和良性变体的可能性。
61.根据条款60所述的计算机实现的方法,其中S型层处理所述非归一化输出并且产生归一化输出,所述归一化输出识别所述变体是致病性变体的可能性。
62.根据条款60所述的计算机实现的方法,其中所述体素、所述原子和所述距离具有三维坐标,其中所述张量具有至少三个维度,其中所述中间输出具有至少三个维度,并且其中所述扁平化输出具有一个维度。
63.一种计算机实现的方法,包括:
存储蛋白质中氨基酸的原子类别式距离通道,
其中所述氨基酸具有多个原子类别的原子,
其中所述多个原子类别中的原子类别指定所述氨基酸的原子元素,
其中所述原子类别式距离通道中的每个原子类别式距离通道具有多个体素中的体素的体素式距离值,并且
其中所述体素式距离值指定从所述多个体素中的对应体素到所述多个原子类别中的对应原子类别中的原子的距离;
处理包括所述原子类别式距离通道和由变体表达的所述蛋白质的替代性等位基因的张量;以及
至少部分地基于所述张量来确定所述变体的致病性。
64.根据条款63所述的计算机实现的方法,还包括将所述体素的体素网格中心定位在所述多个原子类别中的相应原子类别的相应原子上。
65.根据条款64所述的计算机实现的方法,还包括将所述体素网格中心定位在所述蛋白质中的至少一个变体氨基酸的残基的α碳原子上。
66.根据条款65所述的计算机实现的方法,其中所述距离是从所述体素网格中的对应体素中心到所述对应原子类别中的最接近原子的最接近原子距离。
67.根据条款66所述的计算机实现的方法,其中所述最接近原子距离是欧几里得距离。
68.根据条款67所述的计算机实现的方法,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
69.根据条款68所述的计算机实现的方法,其中所述距离是从所述体素网格中的所述对应体素中心到最接近原子的最接近原子距离,而与所述氨基酸和所述氨基酸的所述原子类别无关。
70.根据条款69所述的计算机实现的方法,其中所述最接近原子距离是欧几里得距离。
71.根据条款70所述的计算机实现的方法,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
条款组2
1.一种或多种存储计算机可执行指令的计算机可读介质,所述计算机可执行指令当在一个或多个处理器上执行时,将计算机配置为执行包括以下项的操作:
存储蛋白质中多个氨基酸的氨基酸式距离通道,
其中所述氨基酸式距离通道中的每个氨基酸式距离通道具有多个体素中的体素的体素式距离值,并且
其中所述体素式距离值指定从所述多个体素中的对应体素到所述多个氨基酸中的对应氨基酸的原子的距离;
处理包括所述氨基酸式距离通道和由变体表达的所述蛋白质的替代性等位基因的张量;以及
至少部分地基于所述张量来确定所述变体的致病性。
2.根据条款1所述的计算机可读介质,所述操作还包括将所述体素的体素网格中心定位在所述氨基酸的相应残基的α碳原子上。
3.根据条款2所述的计算机可读介质,所述操作还包括将所述体素网格中心定位在特定氨基酸中对应于所述蛋白质中的至少一个变体氨基酸的残基的α碳原子上。
4.根据条款3所述的计算机可读介质,所述操作还包括通过将所述特定氨基酸之前的那些氨基酸的体素式距离值乘以方向性参数,来在所述张量中编码所述氨基酸的方向性和所述特定氨基酸的位置。
5.根据条款3所述的计算机可读介质,其中所述距离是从所述体素网格中的对应体素中心到所述对应氨基酸的最接近原子的最接近原子距离。
6.根据条款5所述的计算机可读介质,其中所述最接近原子距离是欧几里得距离。
7.根据条款6所述的计算机可读介质,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
8.根据条款5所述的计算机可读介质,其中所述氨基酸具有α碳原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近α碳原子的最接近α碳原子距离。
9.根据条款5所述的计算机可读介质,其中所述氨基酸具有β碳原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近β碳原子的最接近β碳原子距离。
10.根据条款5所述的计算机可读介质,其中所述氨基酸具有主链原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近主链原子的最接近主链原子距离。
11.根据条款5所述的计算机可读介质,其中所述氨基酸具有侧链原子,并且其中所述距离是从所述对应体素中心到所述对应氨基酸的最接近侧链原子的最接近侧链原子距离。
12.根据条款3所述的计算机可读介质,所述操作还包括在所述张量中编码最接近原子通道,所述最接近原子通道指定从每个体素到最接近原子的距离,其中选择所述最接近原子而不考虑所述氨基酸和所述氨基酸的原子元素。
13.根据条款12所述的计算机可读介质,其中所述距离是欧几里得距离。
14.根据条款13所述的计算机可读介质,其中所述距离通过将所述欧几里得距离除以最大距离来归一化。
15.根据条款12所述的计算机可读介质,其中所述氨基酸包括非标准氨基酸。
16.根据条款1所述的计算机可读介质,其中所述张量还包括指定未在体素中心的预定义半径内找到的原子的缺席原子通道,并且其中所述缺席原子通道是独热编码的。
17.根据条款1所述的计算机可读介质,其中所述张量还包括按体素方式编码为所述氨基酸式距离通道中的每个氨基酸式距离通道的所述替代性等位基因的独热编码。
18.根据条款1所述的计算机可读介质,其中所述张量还包括所述蛋白质的参考等位基因。
19.根据条款18所述的计算机可读介质,其中所述张量还包括按体素方式编码为所述氨基酸式距离通道中的每个氨基酸式距离通道的所述参考等位基因的独热编码。
20.根据条款1所述的计算机可读介质,其中所述张量还包括指定跨多个物种的所述氨基酸的保守水平的进化谱。
21.根据条款20所述的计算机可读介质,所述操作还包括:对于所述体素中的每个体素,
跨所述氨基酸和所述原子类别选择最接近原子,
为包括所述最接近原子的氨基酸残基选择泛氨基酸保守频率序列,以及
使所述泛氨基酸保守频率序列可用作所述进化谱之一。
22.根据条款21所述的计算机可读介质,其中为在所述多个物种中观察到的所述残基的特定位置配置所述泛氨基酸保守频率序列。
23.根据条款21所述的计算机可读介质,其中所述泛氨基酸保守频率序列指定对于特定氨基酸是否存在缺失的保守频率。
24.根据条款21所述的计算机可读介质,所述操作还包括:对于所述体素中的每个体素,
在所述氨基酸中的相应氨基酸中选择相应的最接近原子,
为包括所述最接近原子的所述氨基酸的相应残基选择相应的每氨基酸保守频率,以及
使所述每氨基酸保守频率可用作所述进化谱之一。
25.根据条款24所述的计算机可读介质,其中为在所述多个物种中观察到的所述残基的特定位置配置所述每氨基酸保守频率。
26.根据条款24所述的计算机可读介质,其中所述每氨基酸保守频率指定对于特定氨基酸是否存在缺失的保守频率。
27.根据条款1所述的计算机可读介质,其中所述张量还包括用于所述氨基酸的注释通道,其中所述注释通道在所述张量中进行独热编码。
28.根据条款27所述的计算机可读介质,其中所述注释通道是分子处理注释,所述分子处理注释包括起始甲硫氨酸、信号、转运肽、前肽、链和肽。
29.根据条款27所述的计算机可读介质,其中所述注释通道是区域注释,所述区域注释包括拓扑结构域、跨膜、膜内、结构域、重复序列、钙结合、锌指、脱氧核糖核酸(DNA)结合、核苷酸结合、区域、卷曲螺旋、基序和组成偏倚。
30.根据条款27所述的计算机可读介质,其中所述注释通道是位点注释,所述位点注释包括活性位点、金属结合、结合位点和位点。
31.根据条款27所述的计算机可读介质,其中所述注释通道是氨基酸修饰注释,所述氨基酸修饰注释包括非标准残基、经修饰残基、脂化、糖基化、二硫键和交联。
32.根据条款27所述的计算机可读介质,其中所述注释通道是二级结构注释,所述二级结构注释包括螺旋、转角和β链。
33.根据条款27所述的计算机可读介质,其中所述注释通道是实验信息注释,所述实验信息注释包括诱变、序列不确定性、序列冲突、非相邻残基和非末端残基。
34.根据条款1所述的计算机可读介质,其中所述张量还包括用于所述氨基酸的结构置信度通道,所述结构置信度通道指定所述氨基酸的相应结构的质量。
35.根据条款34所述的计算机可读介质,其中所述结构置信度通道是全局模型质量估计(GMQE)。
36.根据条款34所述的计算机可读介质,其中所述结构置信度通道包括定性模型能量分析(QMEAN)分数。
37.根据条款34所述的计算机可读介质,其中所述结构置信度通道是指定所述残基满足相应蛋白质结构的物理约束的程度的温度因素。
38.根据条款34所述的计算机可读介质,其中所述结构置信度通道是指定最接近所述体素的原子的残基具有对准的模板结构的程度的模板结构比对。
39.根据条款38所述的计算机可读介质,其中所述结构置信度通道是所述对准的模板结构的模板建模分数。
40.根据条款39所述的计算机可读介质,其中所述结构置信度通道是所述模板建模分数中最小的一个、所述模板建模分数的平均值,以及所述模板建模分数中最大的一个。
41.根据条款1所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的氨基酸式距离通道与所述替代性等位基因的所述独热编码连结以生成所述张量。
42.根据条款41所述的计算机可读介质,所述操作还包括按体素方式将用于所述β碳原子的氨基酸式距离通道与所述替代性等位基因的所述独热编码连结以生成所述张量。
43.根据条款42所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道与所述替代性等位基因的所述独热编码连结以生成所述张量。
44.根据条款43所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码与泛氨基酸保守频率序列连结以生成所述张量。
45.根据条款44所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述泛氨基酸保守频率序列与所述注释通道连结以生成所述张量。
46.根据条款45所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述泛氨基酸保守频率序列、所述注释通道与所述结构置信度通道连结以生成所述张量。
47.根据条款46所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码与所述氨基酸中的每种氨基酸的每氨基酸保守频率连结以生成所述张量。
48.根据条款47所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述氨基酸中的每种氨基酸的每氨基酸保守频率与所述注释通道连结以生成所述张量。
49.根据条款48所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述氨基酸中的每种氨基酸的每氨基酸保守频率、所述注释通道与所述结构置信度通道连结以生成所述张量。
50.根据条款49所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码与所述参考等位基因的所述独热编码连结以生成所述张量。
51.根据条款50所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码与所述泛氨基酸保守频率序列连结以生成所述张量。
52.根据条款51所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述泛氨基酸保守频率序列与所述注释通道连结以生成所述张量。
53.根据条款52所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述泛氨基酸保守频率序列、所述注释通道与所述结构置信度通道连结以生成所述张量。
54.根据条款53所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码与所述氨基酸中的每种氨基酸的所述每氨基酸保守频率连结以生成所述张量。
55.根据条款54所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述氨基酸中的每种氨基酸的所述每氨基酸保守频率与所述注释通道连结以生成所述张量。
56.根据条款55所述的计算机可读介质,所述操作还包括按体素方式将用于所述α碳原子的所述氨基酸式距离通道、用于所述β碳原子的所述氨基酸式距离通道、所述替代性等位基因的所述独热编码、所述参考等位基因的所述独热编码、所述氨基酸中的每种氨基酸的所述每氨基酸保守频率、所述注释通道与所述结构置信度通道连结以生成所述张量。
57.根据条款1所述的计算机可读介质,所述操作还包括在所述氨基酸式距离通道生成之前旋转所述氨基酸的原子。
58.根据条款1所述的计算机可读介质,所述操作还包括在卷积神经网络中使用1×1×1卷积、3×3×3卷积、矫正线性单元激活层、批量归一化层、全连接层、Dropout正则化层和Softmax分类层。
59.根据条款58所述的计算机可读介质,其中所述1×1×1卷积和所述3×3×3卷积是三维卷积。
60.根据条款58所述的计算机可读介质,其中所述1×1×1卷积的层处理所述张量并且产生作为所述张量的卷积表示的中间输出,其中所述3×3×3卷积的层序列处理所述中间输出并且产生扁平化输出,其中所述全连接层处理所述扁平化输出并且产生非归一化输出,并且其中所述Softmax分类层处理所述非归一化输出并且产生指数归一化输出,所述指数归一化输出识别所述变体是致病性变体和良性变体的可能性。
61.根据条款60所述的计算机可读介质,其中S型层处理所述非归一化输出并且产生归一化输出,所述归一化输出识别所述变体是致病性变体的可能性。
62.根据条款60所述的计算机可读介质,其中所述体素、所述原子和所述距离具有三维坐标,其中所述张量具有至少三个维度,其中所述中间输出具有至少三个维度,并且其中所述扁平化输出具有一个维度。
63.一种或多种存储计算机可执行指令的计算机可读介质,所述计算机可执行指令当在一个或多个处理器上执行时,将计算机配置为执行包括以下项的操作:
存储蛋白质中氨基酸的原子类别式距离通道,
其中所述氨基酸具有多个原子类别的原子,
其中所述多个原子类别中的原子类别指定所述氨基酸的原子元素,
其中所述原子类别式距离通道中的每个原子类别式距离通道具有多个体素中的体素的体素式距离值,并且
其中所述体素式距离值指定从所述多个体素中的对应体素到所述多个原子类别中的对应原子类别中的原子的距离;
处理包括所述原子类别式距离通道和由变体表达的所述蛋白质的替代性等位基因的张量;以及
至少部分地基于所述张量来确定所述变体的致病性。
64.根据条款63所述的计算机可读介质,所述操作还包括将所述体素的体素网格中心定位在所述多个原子类别中的相应原子类别的相应原子上。
65.根据条款64所述的计算机可读介质,所述操作还包括将所述体素网格中心定位在所述蛋白质中的至少一个变体氨基酸的残基的α碳原子上。
66.根据条款65所述的计算机可读介质,其中所述距离是从所述体素网格中的对应体素中心到所述对应原子类别中的最接近原子的最接近原子距离。
67.根据条款66所述的计算机可读介质,其中所述最接近原子距离是欧几里得距离。
68.根据条款67所述的计算机可读介质,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
69.根据条款68所述的计算机可读介质,其中所述距离是从所述体素网格中的所述对应体素中心到最接近原子的最接近原子距离,而与所述氨基酸和所述氨基酸的所述原子类别无关。
70.根据条款69所述的计算机可读介质,其中所述最接近原子距离是欧几里得距离。
71.根据条款70所述的计算机可读介质,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
特定具体实施2
在一些具体实施中,系统包括体素化器,该体素化器访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于该三维结构中的原子的三维体素网格拟合,以生成氨基酸式距离通道。氨基酸式距离通道中的每个氨基酸式距离通道具有三维体素网格中每个体素的三维距离值。该三维距离值指定从三维体素网格中的对应体素到参考氨基酸序列中的对应参考氨基酸的原子的距离。该系统还包括替代性等位基因编码器,其将替代性等位基因氨基酸编码为三维体素网格中的每个体素。替代性等位基因氨基酸是由变体核苷酸表达的变体氨基酸的独热编码的三维表示。该系统还包括进化保守编码器,其将进化保守序列编码为三维体素网格中的每个体素。进化保守序列可以是跨多个物种的氨基酸特异性保守频率的三维表示。氨基酸特异性保守频率可以依据氨基酸与对应体素的接近度来选择。该系统还包括卷积神经网络,其被配置为将三维卷积应用于包括用替代性等位基因氨基酸和相应进化保守序列编码的氨基酸式距离通道的张量。卷积神经网络还可以被配置为至少部分地基于该张量来确定变体核苷酸的致病性。
体素化器可以将三维体素网格中心定位在参考氨基酸序列中的相应参考氨基酸残基的α-碳原子上。体素化器可以将三维体素网格中心定位在特定参考氨基酸残基中定位于变体氨基酸处的α-碳原子上。
在一些具体实施中,该系统可以进一步被配置为通过将特定参考氨基酸之前的那些参考氨基酸的三维距离值乘以方向性参数,来在张量中编码参考氨基酸序列中的参考氨基酸的方向性和特定参考氨基酸的位置。所述距离可以是从三维体素网格中的对应体素中心到对应参考氨基酸的最接近原子的最接近原子距离。最接近原子距离可以是欧几里得距离,并且可以通过将欧几里得距离除以最大最接近原子距离来归一化。
在一些具体实施中,参考氨基酸可以具有α-碳原子,并且所述距离可以是从对应体素中心到对应参考氨基酸的最接近α-碳原子的最接近α-碳原子距离。在一些具体实施中,参考氨基酸可以具有β-碳原子,并且所述距离可以是从对应体素中心到对应参考氨基酸的最接近β-碳原子的最接近β-碳原子距离。在一些具体实施中,参考氨基酸可以具有主链原子,并且所述距离可以是从对应体素中心到对应参考氨基酸的最接近主链原子的最接近主链原子距离。在一些具体实施中,参考氨基酸可以具有侧链原子,并且所述距离可以是从对应体素中心到对应参考氨基酸的最接近侧链原子的最接近侧链原子距离。
在一些具体实施中,该系统可以进一步被配置为在张量中编码最接近原子通道,该最接近原子通道指定从每个体素到最接近原子的距离。可以选择最接近原子而不考虑氨基酸和氨基酸的原子元素。该距离可以是欧几里得距离,并且可以通过将欧几里得距离除以最大距离来归一化。氨基酸可以包括非标准氨基酸。张量可以还包括指定未在体素中心的预定义半径内找到的原子的缺席原子通道。缺席原子通道可以是独热编码的。
在一些具体实施中,该系统可以还包括参考等位基因编码器,该参考等位基因编码器在氨基酸位置的基础上按体素方式将参考等位基因氨基酸编码为每个三维距离值。参考等位基因氨基酸可以是参考氨基酸序列的独热编码的三维表示。氨基酸特异性保守频率可以指定跨多个物种的相应氨基酸的保守水平。
在一些具体实施中,进化保守编码器可以跨参考氨基酸和原子类别选择对应体素的最接近原子,可以为包括该最接近原子的参考氨基酸的残基选择泛氨基酸保守频率,并且可以使用泛氨基酸保守频率的三维表示作为进化保守序列。可以为在多个物种中观察到的残基的特定位置配置泛氨基酸保守频率。泛氨基酸保守频率可以指定对于特定参考氨基酸是否存在缺失的保守频率。
在一些具体实施中,进化保守编码器可以在参考氨基酸中的相应参考氨基酸中选择对于对应体素的相应最接近原子,可以为包括最接近原子的参考氨基酸的相应残基选择相应的每氨基酸保守频率,并且可以使用每氨基酸保守频率的三维表示作为进化保守序列。可以为在多个物种中观察到的残基的特定位置配置每氨基酸保守频率。每氨基酸保守频率可以指定对于特定参考氨基酸是否存在缺失的保守频率。
在一些具体实施中,该系统可以还包括注释编码器,该注释编码器按体素方式将一个或多个注释通道编码为每个三维距离值。注释通道可以是残基注释独热编码的三维表示,并且可以是包括起始甲硫氨酸、信号、转运肽、前肽、链和肽的分子处理注释。在一些具体实施中,注释通道可以是区域注释,这些区域注释包括拓扑结构域、跨膜、膜内、结构域、重复序列、钙结合、锌指、脱氧核糖核酸(DNA)结合、核苷酸结合、区域、卷曲螺旋、基序和组成偏倚,或者可以是位点注释,这些位点注释包括活性位点、金属结合、结合位点和位点。在一些具体实施中,注释通道可以是氨基酸修饰注释,这些氨基酸修饰注释包括非标准残基、经修饰残基、脂化、糖基化、二硫键和交联,或者可以是二级结构注释,这些二级结构注释包括螺旋、转角和β链。注释通道可以是实验信息注释,这些实验信息注释包括诱变、序列不确定性、序列冲突、非相邻残基和非末端残基。
在一些具体实施中,该系统可以还包括结构置信度编码器,结构置信度编码器按体素方式将一个或多个结构置信度通道编码为每个三维距离值。结构置信度通道可以是置信度分数的三维表示,其指定相应残基结构的质量。结构置信度通道可以是全局模型质量估计(GMQE),可以是定性模型能量分析(QMEAN)分数,可以是指定残基满足相应蛋白质结构的物理约束的程度的温度因素,可以是指定最接近体素的原子的残基具有对准的模板结构的程度的模板结构比对,可以是对准的模板结构的模板建模分数,或者可以是模板建模分数中最小的一个、模板建模分数的平均值,以及模板建模分数中最大的一个。
在一些具体实施中,该系统可以还包括原子旋转引擎,该原子旋转引擎在氨基酸式距离通道生成之前对原子进行旋转。
卷积神经网络可以使用1×1×1卷积、3×3×3卷积、矫正线性单元激活层、批量归一化层、全连接层、Dropout正则化层和Softmax分类层。1×1×1卷积和3×3×3卷积可以是三维卷积。在一些具体实施中,1×1×1卷积的层可以处理该张量并且产生作为该张量的卷积表示的中间输出。3×3×3卷积的层序列可以处理中间输出并且产生扁平化输出。全连接层可以处理扁平化输出并且产生非归一化输出。Softmax分类层可以处理非归一化输出并且产生指数归一化输出,这些指数归一化输出识别变体核苷酸是致病性变体核苷酸和良性变体核苷酸的可能性。
在一些具体实施中,S型层可以处理非归一化输出并且产生归一化输出,该归一化输出识别变体核苷酸是致病性变体核苷酸的可能性。卷积神经网络可以是基于注意力的神经网络。张量可以包括用参考等位基因氨基酸进一步编码的氨基酸式距离通道,可以包括用注释通道进一步编码的氨基酸式距离通道,或者可以包括用结构置信度通道进一步编码的氨基酸式距离通道。
在一些具体实施中,系统可以包括体素化器,该体素化器访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于该三维结构中的原子的三维体素网格拟合,以生成原子类别式距离通道。这些原子跨越多个原子类别,这些原子类别指定氨基酸的原子元素。原子类别式距离通道中的每个原子类别式距离通道具有三维体素网格中每个体素的三维距离值。该三维距离值指定从三维体素网格中的对应体素到多个原子类别中的对应原子类别的原子的距离。该系统还包括替代性等位基因编码器,其将替代性等位基因氨基酸编码为三维体素网格中的每个体素。替代性等位基因氨基酸是由变体核苷酸表达的变体氨基酸的独热编码的三维表示。该系统还包括进化保守编码器,其将进化保守序列编码为三维体素网格中的每个体素。进化保守序列可以是跨多个物种的氨基酸特异性保守频率的三维表示。氨基酸特异性保守频率可以依据氨基酸与对应体素的接近度来选择。该系统还包括卷积神经网络,其被配置为将三维卷积应用于包括用替代性等位基因氨基酸和相应进化保守序列编码的原子类别式距离通道的张量,然后至少部分地基于该张量来确定变体核苷酸的致病性。
在一些具体实施中,系统包括体素化器,该体素化器访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于该三维结构中的原子的三维体素网格拟合,以生成氨基酸式距离通道。氨基酸式距离通道中的每个氨基酸式距离通道可以具有三维体素网格中每个体素的三维距离值。该三维距离值可以指定从三维体素网格中的对应体素到参考氨基酸序列中的对应参考氨基酸的原子的距离。该系统还包括替代性等位基因编码器,其将替代性等位基因氨基酸编码为三维体素网格中的每个体素。替代性等位基因氨基酸是由变体核苷酸表达的变体氨基酸的独热编码的三维表示。该系统还包括进化保守编码器,其将进化保守序列编码为三维体素网格中的每个体素。进化保守序列可以是跨多个物种的氨基酸特异性保守频率的三维表示。氨基酸特异性保守频率可以依据氨基酸与对应体素的接近度来选择。该系统还包括张量生成器,其被配置为生成包括用替代性等位基因氨基酸和相应进化保守序列编码的氨基酸式距离通道的张量。
在一些具体实施中,系统包括体素化器,该体素化器访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于该三维结构中的原子的三维体素网格拟合,以生成原子类别式距离通道。这些原子可以跨越多个原子类别,这些原子类别指定氨基酸的原子元素。原子类别式距离通道中的每个原子类别式距离通道可以具有三维体素网格中每个体素的三维距离值。该三维距离值可以指定从三维体素网格中的对应体素到多个原子类别中的对应原子类别的原子的距离。该系统还包括替代性等位基因编码器,其将替代性等位基因氨基酸编码为三维体素网格中的每个体素。替代性等位基因氨基酸是由变体核苷酸表达的变体氨基酸的独热编码的三维表示。该系统还包括进化保守编码器,其将进化保守序列编码为三维体素网格中的每个体素。进化保守序列可以是跨多个物种的氨基酸特异性保守频率的三维表示。氨基酸特异性保守频率可以依据氨基酸与对应体素的接近度来选择。该系统还包括张量生成器,其被配置为生成包括用替代性等位基因氨基酸和相应进化保守序列编码的原子类别式距离通道的张量。
条款组1
1.一种计算机实现的方法,包括:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成氨基酸式距离通道,
其中所述氨基酸式距离通道中的每个氨基酸式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述参考氨基酸序列中的对应参考氨基酸的原子的距离;
将替代性等位基因通道编码为所述三维体素网格中的每个体素,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述氨基酸式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;
将三维卷积应用于包括用所述替代性等位基因通道和相应进化保守通道编码的所述氨基酸式距离通道的张量;以及
至少部分地基于所述张量来确定所述变体核苷酸的致病性。
2.根据条款1所述的计算机实现的方法,还包括将所述三维体素网格中心定位在所述参考氨基酸序列中的相应参考氨基酸残基的α碳原子上。
3.根据条款2所述的计算机实现的方法,还包括将所述三维体素网格中心定位在特定参考氨基酸中对应于所述变体氨基酸的残基的α碳原子上。
4.根据条款3所述的计算机实现的方法,还包括通过将所述特定参考氨基酸之前的那些参考氨基酸的三维距离值乘以方向性参数,来在所述张量中编码所述参考氨基酸序列中的所述参考氨基酸的方向性和所述特定参考氨基酸的位置。
5.根据条款4所述的计算机实现的方法,其中所述距离是从所述三维体素网格中的对应体素中心到所述对应参考氨基酸的最接近原子的最接近原子距离。
6.根据条款5所述的计算机实现的方法,其中所述最接近原子距离是欧几里得距离。
7.根据条款6所述的计算机实现的方法,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
8.根据条款5所述的计算机实现的方法,其中所述参考氨基酸具有α碳原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近α碳原子的最接近α碳原子距离。
9.根据条款5所述的计算机实现的方法,其中所述参考氨基酸具有β碳原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近β碳原子的最接近β碳原子距离。
10.根据条款5所述的计算机实现的方法,其中所述参考氨基酸具有主链原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近主链原子的最接近主链原子距离。
11.根据条款5所述的计算机实现的方法,其中所述氨基酸具有侧链原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近侧链原子的最接近侧链原子距离。
12.根据条款3所述的计算机实现的方法,还包括在所述张量中编码最接近原子通道,所述最接近原子通道指定从每个体素到最接近原子的距离,其中选择所述最接近原子而不考虑所述氨基酸和所述氨基酸的原子元素。
13.根据条款12所述的计算机实现的方法,其中所述距离是欧几里得距离。
14.根据条款13所述的计算机实现的方法,其中所述距离通过将所述欧几里得距离除以最大距离来归一化。
15.根据条款12所述的计算机实现的方法,其中所述氨基酸包括非标准氨基酸。
16.根据条款1所述的计算机实现的方法,其中所述张量还包括指定未在体素中心的预定义半径内找到的原子的缺席原子通道。
17.根据条款16所述的计算机实现的方法,其中所述缺席原子通道是独热编码的。
18.根据条款1所述的计算机实现的方法,还包括按体素方式将参考等位基因通道编码为所述三维体素网格中的每个体素。
19.根据条款18所述的计算机实现的方法,所述参考等位基因氨基酸是经历所述变体氨基酸的参考氨基酸独热编码的三维表示。
20.根据条款1所述的计算机实现的方法,其中所述氨基酸特异性保守频率指定跨所述多个物种的相应氨基酸的保守水平。
21.根据条款20所述的计算机实现的方法,还包括:
跨所述参考氨基酸和所述原子类别选择对于所述对应体素的最接近原子,
为包括所述最接近原子的参考氨基酸残基选择泛氨基酸保守频率,以及
使用所述泛氨基酸保守频率的三维表示作为所述进化保守通道。
22.根据条款21所述的计算机实现的方法,其中为在所述多个物种中观察到的所述残基的特定位置配置所述泛氨基酸保守频率。
23.根据条款21所述的计算机实现的方法,其中所述泛氨基酸保守频率指定对于特定参考氨基酸是否存在缺失的保守频率。
24.根据条款21所述的计算机实现的方法,还包括:
在所述参考氨基酸中的相应参考氨基酸中选择对于所述对应体素的相应最接近原子,
为包括所述最接近原子的所述参考氨基酸的相应残基选择相应的每氨基酸保守频率,以及
使用所述每氨基酸保守频率的三维表示作为所述进化保守通道。
25.根据条款24所述的计算机实现的方法,其中为在所述多个物种中观察到的所述残基的特定位置配置所述每氨基酸保守频率。
26.根据条款24所述的计算机实现的方法,其中所述每氨基酸保守频率指定对于特定参考氨基酸是否存在缺失的保守频率。
27.根据条款1所述的计算机实现的方法,还包括按体素方式将一个或多个注释通道编码为所述三维体素网格中的每个体素,其中所述注释通道是残基注释的独热编码的三维表示。
28.根据条款27所述的计算机实现的方法,其中所述注释通道是分子处理注释,所述分子处理注释包括起始甲硫氨酸、信号、转运肽、前肽、链和肽。
29.根据条款27所述的计算机实现的方法,其中所述注释通道是区域注释,所述区域注释包括拓扑结构域、跨膜、膜内、结构域、重复序列、钙结合、锌指、脱氧核糖核酸(DNA)结合、核苷酸结合、区域、卷曲螺旋、基序和组成偏倚。
30.根据条款27所述的计算机实现的方法,其中所述注释通道是位点注释,所述位点注释包括活性位点、金属结合、结合位点和位点。
31.根据条款27所述的计算机实现的方法,其中所述注释通道是氨基酸修饰注释,所述氨基酸修饰注释包括非标准残基、经修饰残基、脂化、糖基化、二硫键和交联。
32.根据条款27所述的计算机实现的方法,其中所述注释通道是二级结构注释,所述二级结构注释包括螺旋、转角和β链。
33.根据条款27所述的计算机实现的方法,其中所述注释通道是实验信息注释,所述实验信息注释包括诱变、序列不确定性、序列冲突、非相邻残基和非末端残基。
34.根据条款1所述的计算机实现的方法,还包括按体素方式将一个或多个结构置信度通道编码为所述三维体素网格中的每个体素,其中所述结构置信度通道是指定相应残基结构的质量的置信度分数的三维表示。
35.根据条款34所述的计算机实现的方法,其中所述结构置信度通道是全局模型质量估计(GMQE)。
36.根据条款34所述的计算机实现的方法,其中所述结构置信度通道是定性模型能量分析(QMEAN)分数。
37.根据条款34所述的计算机实现的方法,其中所述结构置信度通道是指定所述残基满足相应蛋白质结构的物理约束的程度的温度因素。
38.根据条款34所述的计算机实现的方法,其中所述结构置信度通道是指定最接近所述体素的原子的残基具有对准的模板结构的程度的模板结构比对。
39.根据条款38所述的计算机实现的方法,其中所述结构置信度通道是所述对准的模板结构的模板建模分数。
40.根据条款39所述的计算机实现的方法,其中所述结构置信度通道是所述模板建模分数中最小的一个、所述模板建模分数的平均值,以及所述模板建模分数中最大的一个。
41.根据条款1所述的计算机实现的方法,还包括在所述氨基酸式距离通道生成之前旋转所述原子。
42.根据条款1所述的计算机实现的方法,还包括在卷积神经网络中使用1×1×1卷积、3×3×3卷积、矫正线性单元激活层、批量归一化层、全连接层、Dropout正则化层和Softmax分类层。
43.根据条款42所述的计算机实现的方法,其中所述1×1×1卷积和所述3×3×3卷积是所述三维卷积。
44.根据条款42所述的计算机实现的方法,其中所述1×1×1卷积的层处理所述张量并且产生作为所述张量的卷积表示的中间输出,其中所述3×3×3卷积的层序列处理所述中间输出并且产生扁平化输出,其中所述全连接层处理所述扁平化输出并且产生非归一化输出,并且其中所述Softmax分类层处理所述非归一化输出并且产生指数归一化输出,所述指数归一化输出识别所述变体核苷酸是致病性变体核苷酸和良性变体核苷酸的可能性。
45.根据条款44所述的计算机实现的方法,其中S型层处理所述非归一化输出并且产生归一化输出,所述归一化输出识别所述变体核苷酸是致病性变体核苷酸的可能性。
46.根据条款1所述的计算机实现的方法,其中所述卷积神经网络是基于注意力的神经网络。
47.根据条款1所述的计算机实现的方法,其中所述张量包括用所述参考等位基因通道进一步编码的所述氨基酸式距离通道。
48.根据条款1所述的计算机实现的方法,其中所述张量包括用所述注释通道进一步编码的所述氨基酸式距离通道。
49.根据条款1所述的计算机实现的方法,其中所述张量包括用所述结构置信度通道进一步编码的所述氨基酸式距离通道。
50.一种计算机实现的方法,包括:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成原子类别式距离通道,
其中所述原子跨越多个原子类别,
其中所述多个原子类别中的原子类别指定所述氨基酸的原子元素,
其中所述原子类别式距离通道中的每个原子类别式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述多个原子类别中的对应原子类别的原子的距离;
将替代性等位基因通道编码为所述三维体素网格中的每个体素,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述原子类别式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;
将三维卷积应用于包括用所述替代性等位基因通道和相应进化保守通道编码的所述原子类别式距离通道的张量;以及
至少部分地基于所述张量来确定所述变体核苷酸的致病性。
51.一种计算机实现的方法,包括:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成氨基酸式距离通道,
其中所述氨基酸式距离通道中的每个氨基酸式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述参考氨基酸序列中的对应参考氨基酸的原子的距离;
将替代性等位基因通道编码为所述三维体素网格中的每个体素,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述氨基酸式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;以及
生成包括用所述替代性等位基因通道和相应进化保守通道编码的所述氨基酸式距离通道的张量。
52.一种计算机实现的方法,包括:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成原子类别式距离通道,
其中所述原子跨越多个原子类别,
其中所述多个原子类别中的原子类别指定所述氨基酸的原子元素,
其中所述原子类别式距离通道中的每个原子类别式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述多个原子类别中的对应原子类别的原子的距离;
将替代性等位基因通道编码为所述三维体素网格中的每个体素,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述原子类别式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;以及
生成包括用所述替代性等位基因通道和相应进化保守通道编码的所述原子类别式距离通道的张量。
条款组2
1.一种或多种存储计算机可执行指令的计算机可读介质,所述计算机可执行指令当在一个或多个处理器上执行时,将计算机配置为执行包括以下项的操作:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成氨基酸式距离通道,
其中所述氨基酸式距离通道中的每个氨基酸式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述参考氨基酸序列中的对应参考氨基酸的原子的距离;
将替代性等位基因通道编码为所述三维体素网格中的每个体素,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述氨基酸式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;
将三维卷积应用于包括用所述替代性等位基因通道和相应进化保守通道编码的所述氨基酸式距离通道的张量;以及
至少部分地基于所述张量来确定所述变体核苷酸的致病性。
2.根据条款1所述的计算机可读介质,所述操作还包括将所述三维体素网格中心定位在所述参考氨基酸序列中的相应参考氨基酸残基的α碳原子上。
3.根据条款2所述的计算机可读介质,所述操作还包括将所述三维体素网格中心定位在特定参考氨基酸残基中对应于所述变体氨基酸的α碳原子上。
4.根据条款3所述的计算机可读介质,所述操作还包括通过将所述特定参考氨基酸之前的那些参考氨基酸的三维距离值乘以方向性参数,来在所述张量中编码所述参考氨基酸序列中的所述参考氨基酸的方向性和所述特定参考氨基酸的位置。
5.根据条款4所述的计算机可读介质,其中所述距离是从所述三维体素网格中的对应体素中心到所述对应参考氨基酸的最接近原子的最接近原子距离。
6.根据条款5所述的计算机可读介质,其中所述最接近原子距离是欧几里得距离。
7.根据条款6所述的计算机可读介质,其中所述最接近原子距离通过将所述欧几里得距离除以最大最接近原子距离来归一化。
8.根据条款5所述的计算机可读介质,其中所述参考氨基酸具有α碳原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近α碳原子的最接近α碳原子距离。
9.根据条款5所述的计算机可读介质,其中所述参考氨基酸具有β碳原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近β碳原子的最接近β碳原子距离。
10.根据条款5所述的计算机可读介质,其中所述参考氨基酸具有主链原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近主链原子的最接近主链原子距离。
11.根据条款5所述的计算机可读介质,其中所述氨基酸具有侧链原子,并且其中所述距离是从所述对应体素中心到所述对应参考氨基酸的最接近侧链原子的最接近侧链原子距离。
12.根据条款3所述的计算机可读介质,所述操作还包括在所述张量中编码最接近原子通道,所述最接近原子通道指定从每个体素到最接近原子的距离,其中选择所述最接近原子而不考虑所述氨基酸和所述氨基酸的原子元素。
13.根据条款12所述的计算机可读介质,其中所述距离是欧几里得距离。
14.根据条款13所述的计算机可读介质,其中所述距离通过将所述欧几里得距离除以最大距离来归一化。
15.根据条款12所述的计算机可读介质,其中所述氨基酸包括非标准氨基酸。
16.根据条款1所述的计算机可读介质,其中所述张量还包括指定未在体素中心的预定义半径内找到的原子的缺席原子通道。
17.根据条款16所述的计算机可读介质,其中所述缺席原子通道是独热编码的。
18.根据条款1所述的计算机可读介质,所述操作还包括按体素方式将参考等位基因通道编码为所述三维体素网格中的每个体素。
19.根据条款18所述的计算机可读介质,所述参考等位基因氨基酸是经历所述变体氨基酸的参考氨基酸独热编码的三维表示。
20.根据条款1所述的计算机可读介质,其中所述氨基酸特异性保守频率指定跨所述多个物种的相应氨基酸的保守水平。
21.根据条款20所述的计算机可读介质,所述操作还包括:跨所述参考氨基酸和所述原子类别选择对于所述对应体素的最接近原子,
为包括所述最接近原子的参考氨基酸残基选择泛氨基酸保守频率,以及
使用所述泛氨基酸保守频率的三维表示作为所述进化保守通道。
22.根据条款21所述的计算机可读介质,其中为在所述多个物种中观察到的所述残基的特定位置配置所述泛氨基酸保守频率。
23.根据条款21所述的计算机可读介质,其中所述泛氨基酸保守频率指定对于特定参考氨基酸是否存在缺失的保守频率。
24.根据条款21所述的计算机可读介质,所述操作还包括:
在所述参考氨基酸中的相应参考氨基酸中选择对于所述对应体素的相应最接近原子,
为包括所述最接近原子的所述参考氨基酸的相应残基选择相应的每氨基酸保守频率,以及
使用所述每氨基酸保守频率的三维表示作为所述进化保守通道。
25.根据条款24所述的计算机可读介质,其中为在所述多个物种中观察到的所述残基的特定位置配置所述每氨基酸保守频率。
26.根据条款24所述的计算机可读介质,其中所述每氨基酸保守频率指定对于特定参考氨基酸是否存在缺失的保守频率。
27.根据条款1所述的计算机可读介质,所述操作还包括按体素方式将一个或多个注释通道编码为所述三维体素网格中的每个体素,其中所述注释通道是残基注释的独热编码的三维表示。
28.根据条款27所述的计算机可读介质,其中所述注释通道是分子处理注释,所述分子处理注释包括起始甲硫氨酸、信号、转运肽、前肽、链和肽。
29.根据条款27所述的计算机可读介质,其中所述注释通道是区域注释,所述区域注释包括拓扑结构域、跨膜、膜内、结构域、重复序列、钙结合、锌指、脱氧核糖核酸(DNA)结合、核苷酸结合、区域、卷曲螺旋、基序和组成偏倚。
30.根据条款27所述的计算机可读介质,其中所述注释通道是位点注释,所述位点注释包括活性位点、金属结合、结合位点和位点。
31.根据条款27所述的计算机可读介质,其中所述注释通道是氨基酸修饰注释,所述氨基酸修饰注释包括非标准残基、经修饰残基、脂化、糖基化、二硫键和交联。
32.根据条款27所述的计算机可读介质,其中所述注释通道是二级结构注释,所述二级结构注释包括螺旋、转角和β链。
33.根据条款27所述的计算机可读介质,其中所述注释通道是实验信息注释,所述实验信息注释包括诱变、序列不确定性、序列冲突、非相邻残基和非末端残基。
34.根据条款1所述的计算机可读介质,所述操作还包括按体素方式将一个或多个结构置信度通道编码为所述三维体素网格中的每个体素,其中所述结构置信度通道是指定相应残基结构的质量的置信度分数的三维表示。
35.根据条款34所述的计算机可读介质,其中所述结构置信度通道是全局模型质量估计(GMQE)。
36.根据条款34所述的计算机可读介质,其中所述结构置信度通道是定性模型能量分析(QMEAN)分数。
37.根据条款34所述的计算机可读介质,其中所述结构置信度通道是指定所述残基满足相应蛋白质结构的物理约束的程度的温度因素。
38.根据条款34所述的计算机可读介质,其中所述结构置信度通道是指定最接近所述体素的原子的残基具有对准的模板结构的程度的模板结构比对。
39.根据条款38所述的计算机可读介质,其中所述结构置信度通道是所述对准的模板结构的模板建模分数。
40.根据条款39所述的计算机可读介质,其中所述结构置信度通道是所述模板建模分数中最小的一个、所述模板建模分数的平均值,以及所述模板建模分数中最大的一个。
41.根据条款1所述的计算机可读介质,所述操作还包括在所述氨基酸式距离通道生成之前旋转所述原子。
42.根据条款1所述的计算机可读介质,所述操作还包括在卷积神经网络中使用1×1×1卷积、3×3×3卷积、矫正线性单元激活层、批量归一化层、全连接层、Dropout正则化层和Softmax分类层。
43.根据条款42所述的计算机可读介质,其中所述1×1×1卷积和所述3×3×3卷积是所述三维卷积。
44.根据条款42所述的计算机可读介质,其中所述1×1×1卷积的层处理所述张量并且产生作为所述张量的卷积表示的中间输出,其中所述3×3×3卷积的层序列处理所述中间输出并且产生扁平化输出,其中所述全连接层处理所述扁平化输出并且产生非归一化输出,并且其中所述Softmax分类层处理所述非归一化输出并且产生指数归一化输出,所述指数归一化输出识别所述变体核苷酸是致病性变体核苷酸和良性变体核苷酸的可能性。
45.根据条款44所述的计算机可读介质,其中S型层处理所述非归一化输出并且产生归一化输出,所述归一化输出识别所述变体核苷酸是致病性变体核苷酸的可能性。
46.根据条款1所述的计算机可读介质,其中所述卷积神经网络是基于注意力的神经网络。
47.根据条款1所述的计算机可读介质,其中所述张量包括用所述参考等位基因通道进一步编码的所述氨基酸式距离通道。
48.根据条款1所述的计算机可读介质,其中所述张量包括用所述注释通道进一步编码的所述氨基酸式距离通道。
49.根据条款1所述的计算机可读介质,其中所述张量包括用所述结构置信度通道进一步编码的所述氨基酸式距离通道。
50.一种或多种存储计算机可执行指令的计算机可读介质,所述计算机可执行指令当在一个或多个处理器上执行时,将计算机配置为执行包括以下项的操作:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成原子类别式距离通道,
其中所述原子跨越多个原子类别,
其中所述多个原子类别中的原子类别指定所述氨基酸的原子元素,
其中所述原子类别式距离通道中的每个原子类别式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述多个原子类别中的对应原子类别的原子的距离;
将替代性等位基因通道编码为所述三维体素网格中的每个体素,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述原子类别式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;
将三维卷积应用于包括用所述替代性等位基因通道和相应进化保守通道编码的所述原子类别式距离通道的张量;以及
至少部分地基于所述张量来确定所述变体核苷酸的致病性。
51.一种或多种存储计算机可执行指令的计算机可读介质,所述计算机可执行指令当在一个或多个处理器上执行时,将计算机配置为执行包括以下项的操作:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成氨基酸式距离通道,
其中所述氨基酸式距离通道中的每个氨基酸式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述参考氨基酸序列中的对应参考氨基酸的原子的距离;
在氨基酸位置基础上将替代性等位基因通道编码为所述氨基酸式距离通道中的每个氨基酸式距离通道中的每个三维距离值,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述氨基酸式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;以及
生成包括用所述替代性等位基因通道和相应进化保守通道编码的所述氨基酸式距离通道的张量。
52.一种或多种存储计算机可执行指令的计算机可读介质,所述计算机可执行指令当在一个或多个处理器上执行时,将计算机配置为执行包括以下项的操作:
访问蛋白质的参考氨基酸序列的三维结构,并且在氨基酸基础上将关于所述三维结构中的原子的三维体素网格拟合,以生成原子类别式距离通道,
其中所述原子跨越多个原子类别,
其中所述多个原子类别中的原子类别指定所述氨基酸的原子元素,
其中所述原子类别式距离通道中的每个原子类别式距离通道具有所述三维体素网格中的每个体素的三维距离值,并且
其中所述三维距离值指定从所述三维体素网格中的对应体素到所述多个原子类别中的对应原子类别的原子的距离;
将替代性等位基因通道编码为所述三维体素网格中的每个体素,
其中所述替代性等位基因通道是由变体核苷酸表达的变体氨基酸的独热编码的三维表示;
在体素位置基础上将进化保守通道编码为跨所述原子类别式距离通道的三维距离值的每个序列,
其中所述进化保守通道是跨多个物种的氨基酸特异性保守频率的三维表示,并且
其中所述氨基酸特异性保守频率依据氨基酸与所述对应体素的接近度来选择;以及
生成包括用所述替代性等位基因通道和相应进化保守通道编码的所述原子类别式距离通道的张量。
在本部分中描述的方法的其他具体实施可包括存储指令的非暂态计算机可读存储介质,这些指令可由处理器执行以执行上述方法中的任一种方法。在本部分中描述的方法的又一具体实施可包括一种系统,该系统包括存储器和一个或多个处理器,该一个或多个处理器可操作以执行存储在存储器中的指令,以执行上述方法中的任一种方法。
特定具体实施3
条
1.一种高效确定序列中哪些元素最接近网格中均匀间隔的单元格的计算机实现的方法,其中所述元素具有元素坐标,并且所述单元格具有维度式单元格索引和单元格坐标,所述方法包括:
生成将所述单元格的子集映射到所述元素中的每个元素的元素到单元格映射,
其中映射到所述序列中的特定元素的所述单元格的所述子集包括所述网格中的最接近单元格和所述网格中的一个或多个邻域单元格,
其中基于将所述特定元素的元素坐标匹配到所述单元格坐标来选择所述最接近单元格,并且
其中所述邻域单元格与所述最接近单元格连续相邻,并且基于在相对所述特定元素处于距离接近范围内来选择;
生成单元格到元素映射,其将所述元素的子集映射到所述单元格中的每个单元格,
其中映射到所述网格中的特定单元格的所述元素的所述子集包括所述序列中通过所述元素到单元格映射而映射到所述特定单元格的那些元素;以及
使用所述单元格到元素映射来为所述单元格中的每个单元格确定所述序列中的最接近元素,
其中基于所述特定单元格与所述元素子集中的所述元素之间的距离来确定所述特定单元格的所述最接近元素。
2.根据条款1所述的计算机实现的方法,其中将所述特定元素的所述元素坐标匹配到所述单元格坐标还包括截断所述元素坐标的小数部分以生成截断的元素坐标。
3.根据条款2所述的计算机实现的方法,其中将所述特定元素的所述元素坐标匹配到所述单元格坐标还包括:
对于第一维度,将所述截断的元素坐标中的第一截断的元素坐标与所述网格中第一单元格的第一单元格坐标进行匹配,并且选择所述第一单元格的第一维度索引;
对于第二维度,将所述截断的元素坐标中的第二截断的元素坐标与所述网格中第二单元格的第二单元格坐标进行匹配,并且选择所述第二单元格的第二维度索引;
对于第三维度,将所述截断的元素坐标中的第三截断的元素坐标与所述网格中第三单元格的第三单元格坐标进行匹配,并且选择所述第三单元格的第三维度索引;
基于用底数幂对所选择的第一维度索引、第二维度索引和第三维度索引进行位置式加权,使用所选择的第一维度索引、第二维度索引和第三维度索引来生成累加和;以及
使用所述累加和作为用于选择所述最接近单元格的单元格索引。
4.根据条款1所述的计算机实现的方法,其中计算所述特定单元格的单元格坐标与所述元素子集中的所述元素的元素坐标之间的所述距离。
5.根据条款1所述的计算机实现的方法,其中所述序列是氨基酸的蛋白质序列。
6.根据条款5所述的计算机实现的方法,其中所述元素是所述氨基酸的原子。
7.根据条款6所述的计算机实现的方法,其中生成所述元素到单元格映射、生成所述单元格到元素映射,以及使用所述单元格到元素映射来为所述单元格中的每个单元格确定所述最接近元素的所述步骤的运行时复杂度为O(a*f+v),其中
a是所述原子的数目,
f是所述氨基酸的数目,
v是所述单元格的数目,并且
*是乘法运算。
8.根据条款7所述的计算机实现的方法,其中所述原子包括α碳原子。
9.根据条款7所述的计算机实现的方法,其中所述原子包括β碳原子。
10.根据条款7所述的计算机实现的方法,其中所述原子包括非碳原子。
11.根据条款1所述的计算机实现的方法,其中所述单元格是三维体素。
12.根据条款11所述的计算机实现的方法,其中所述单元格坐标是三维坐标。
13.根据条款12所述的计算机实现的方法,其中所述元素坐标是三维坐标。
14.根据条款1所述的计算机实现的方法,其中所述邻域单元格基于在相对于所述最接近单元格的索引邻接范围内来选择。
15.根据条款1所述的计算机实现的方法,其中所述邻域单元格基于在所述网格中包括所述最接近单元格的单元格邻域内来选择。
16.根据条款1所述的计算机实现的方法,其中所述序列包括M个元素,其中所述元素的所述子集包括N个元素,并且其中M>>N。
17.一种有效确定蛋白质中哪些原子最接近网格中的体素的计算机实现的方法,其中所述原子具有三维(3D)原子坐标,并且所述体素具有3D体素坐标,所述方法包括:
生成原子到体素映射,其将基于所述蛋白质的特定原子的3D原子坐标与所述网格中的所述3D体素坐标匹配而选择的包含体素映射到所述原子中的每个原子;
生成体素到原子映射,其将所述原子的子集映射到所述体素中的每个体素,其中映射到所述网格中的特定体素的所述原子的子集包括所述蛋白质中通过所述原子到体素映射而映射到所述特定体素的那些原子;以及使用所述体素到原子映射来为所述体素中的每个体素确定所述蛋白质中的最接近原子。
18.根据条款17所述的计算机实现的方法,其中条款17所述步骤的运行时复杂度为O(原子数目)。
在本部分中描述的方法的其他具体实施可包括存储指令的非暂态计算机可读存储介质,这些指令可由处理器执行以执行上述方法中的任一种方法。在本部分中描述的方法的又一具体实施可包括一种系统,该系统包括存储器和一个或多个处理器,该一个或多个处理器可操作以执行存储在存储器中的指令,以执行上述方法中的任一种方法。
虽然通过参考上文详细描述的优选实施方式和示例公开了本发明,但是应当理解,这些示例旨在进行说明而非进行限制。可以预期,本领域的技术人员将容易想到修改和组合,这些修改和组合将在本发明的实质和以下权利要求书的范围之内。
- 一种可重参数化的轻量级体素深度学习方法
- 通过一体化酶生产进行的高效木质纤维素水解