一种制作模拟变异数据的方法、装置、计算机可读存储介质及应用
文献发布时间:2024-04-18 19:58:53
技术领域
本发明属于生物信息学技术领域,涉及一种制作模拟变异数据的方法、装置、计算机可读存储介质及应用,尤其涉及一种制作基于杂交捕获的高通量测序模拟数据的方法、装置、计算机可读存储介质及应用。
背景技术
采用新一代测序(NGS)技术进行临床分子诊断,包括全基因组测序(WGS)、全外显子组测序和靶向基因测序等,已成为癌症诊断和治疗的重要工具。然而从NGS原始下机数据检测变异时,不同生物信息学流程的检测结果还达不到令人满意的稳定性和标准化,因此在生物信息学流程被用于临床样本的检测前,有必要先检测生物信息学流程的性能。但是,目前缺少可以用于评估生物信息学流程的标准化数据。
除去基因组的结构变异基因组的变异,基因组变异分为单碱基变异(SNV)、小片段插入缺失(InDel)、多碱基变异(MNV)、复杂变异(Complex deletion-insertion)。目前的主流模拟数据软件,都只能对基因组变异的部分形式进行模拟,并且目前还没有软件可以模拟多碱基变异和复杂变异。采用读取并编辑原始测序读段的策略有BAMSurgen、insiM和MutationMaker,这些软件都需要在现有测序数据的基础上对数据进行编辑和修改,不适合使用者没有现成可供修改的下机数据时使用;采用从头合成策略的有VarSim、SVEngine、Wessim,然而这些软件只能模拟单碱基变异(SNVs)和小片段的插入缺失(InDels)。另外,肿瘤中常见的变异(EGFR L858R、BRAF V600E等)习惯性的以氨基酸的变化进行表示,而目前这些软件都需要输入变异在基因组水平上的改变形式,均不能直接读取变异在RNA或氨基酸水平的改变形式并输出相应的模拟数据。
综上所述,如何开发新型构建模拟变异数据的方法,以满足评估生物信息学流程的需求,是目前仅诊断领域亟需解决的问题之一。
发明内容
针对现有技术的不足和实际需求,本发明提供一种制作模拟变异数据的方法、装置、计算机可读存储介质及应用,以满足目前评估生物信息学流程需求。
为达上述目的,本发明采用以下技术方案:
第一方面,本发明提供一种制作模拟变异数据的方法,所述方法包括以下步骤:
(1)读取输入的bed文件,提取bed文件覆盖的基因列表和基因的基因组坐标;
(2)读取输入的变异位点,获取基因组水平的变异信息,包括染色体、开始和结束坐标,突变序列;
(3)根据bed文件覆盖的基因列表和基因的基因组坐标、突变位点在基因组水平的信息,制作野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件;
(4)从野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件截取FASTQ文件;
(5)利用比对软件将野生型的FASTQ文件和突变型的FASTQ文件与参考基因组进行比对,得到初始的野生型的BAM文件和突变型的BAM文件,提取初始的野生型的BAM文件和突变型的BAM文件中bed文件区域的部分,得到野生型的BAM文件和突变型的BAM文件;
(6)读取突变位点目标的突变频率,按照突变频率将野生型的BAM文件和突变型的BAM文件进行混合,生成模拟变异数据的BAM文件。
本发明中,针对目前模拟方法中无法读取变异在RNA或氨基酸水平的改变形式并输出相应的模拟数据,以及无法模拟基因组变异的多碱基变异和复杂变异等问题,采取从头合成策略,设计制作模拟变异数据的方法,可以直接读取变异在基因组、RNA或氨基酸上的HGVS格式的改变形式,模拟所有基因组变异形式,并输出相应的BAM文件。
本发明中,bed文件指一种文本文件的格式,文件中至少包含3列,染色体、开始坐标和结束坐标;HGVS格式注释规则:人类基因组变异协会(HGVS:Human Genome VariationSociety)制定的目前学术界所公认的突变命名规则;FASTA格式:在生物信息学中,FASTA格式(又称为Pearson格式),是一种基于文本用于表示核苷酸序列或氨基酸序列的格式。序列文件的第一行是由大于号">"或分号";"打头的任意文字说明(习惯常用">"作为起始),用于序列标记。从第二行开始为序列本身,只允许使用既定的核苷酸或氨基酸编码符号。通常核苷酸符号大小写均可,而氨基酸常用大写字母;FASTQ格式:FASTQ是一种存储了生物序列(通常是核酸序列)以及相应的质量评价的文本格式。FASTQ格式是序列格式中常见的一种,FASTQ格式的序列一般都包含有四行,第一行由'@'开始,后面跟着序列的描述信息,这点跟FASTA格式是一样的,第二行是序列,第三行由'+'开始,后面也可以跟着序列的描述信息。第四行是第二行序列的质量评价(quality values,注:应该是测序的质量评价),字符数跟第二行的序列是相等的;BAM文件指SAM文件的二进制格式,SAM文件是短序列比对默认的标准格式,是以TAB为分割符的文本格式,主要应用于测序序列mapping到基因组上的结果表示,另外也可以表示其他的多重比对结果。一般常见的比对软件把FASTQ文件往参考基因组上比对后,得到的就是BAM文件。
优选地,步骤(1)具体包括:
利用第一数据库对bed文件进行注释,提取bed文件覆盖到的基因列表,及基因列表中每个基因在基因组上对应的所有转录本的染色体、开始和结束坐标;在第二数据库中提取每个基因优选的唯一的转录本,并由此得到每个基因唯一对应的在基因组对应的染色体、开始和结束坐标。
优选地,所述第一数据库包括refGene数据库或ensemble数据库。
优选地,所述第二数据库包括HGNC数据库。
本发明中,HGNC数据库:全称为HUGO Gene Nomenclature Committee,叫做HUGO基因命名委员会,负责对人类基因组上包括蛋白编码基因、ncRNA基因、假基因和其他基因在内的所有基因提供唯一的、标准的、可以广泛传播的symbol,该数据库为NCBI和EMBL联合为每个基因选择了一个最具有代表性的转录本,并收录进该数据库。
优选地,步骤(2)具体包括:
(2-1)读取输入的变异位点,判断输入的变异位点的注释形式,利用Transvar软件将输入的变异位点进行转化,提取其中基因组水平的注释;
(2-2)根据Transvar注释结果中基因组水平的注释,得到基因组水平的变异信息,并从基因组水平的变异信息中获取染色体、突变开始坐标、突变终止坐标和突变序列;
(2-2-1)基因组水平注释信息中包含的“chr”字符串后的数字,即为突变位点所在的染色体;
(2-2-2)若基因组水平注释信息中包含1个数字,则判断该数字同时为突变初始和结束坐标,继续读取数字后跟着的字符串,若字符串中存在“delins”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“delins”后的序列串;若字符串中存在“del”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“”(表示突变序列为空);若字符串中存在“dup”,则突变开始坐标即为突变初始坐标,突变结束坐标在突变初始结束坐标加1,突变序列为“dup”后的序列串;若字符串中存在“>”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“>”后的序列串;
(2-2-3)若基因组水平注释信息中包含2个数字,且数字间用“_”分隔开,则判断第一个数字为突变初始开始坐标,第二个数字为突变初始结束坐标;继续读取数字后跟着的字符串,若字符串中存在“delins”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“delins”后的序列串;若字符串中存在“del”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“”(表示突变序列为空);若字符串中存在“dup”,则突变开始坐标即为突变初始坐标,突变结束坐标在突变初始结束坐标加1,突变序列为“dup”后的序列串;若字符串中存在“ins”,则突变开始坐标即为突变初始坐标,突变结束坐标即为突变初始结束坐标,突变序列为“ins”后的序列串。
优选地,步骤(3)具体包括:
(3-1)遍历bed文件覆盖到的基因列表,若基因中不包含需要模拟的突变位点,则根据基因的染色体坐标,利用截取软件从基因组上截取该基因的序列信息,分别输出到野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件中;
(3-2)若基因中包含需要模拟的突变位点,首先根据基因的染色体坐标,利用截取软件从基因组上截取该基因的序列信息,输出到野生型的模拟基因组FASTA文件;其次根据基因在基因组上的开始坐标和突变位点的开始坐标,从基因组上截取第一段序列,把突变位点的突变序列作为第二段序列,根据基因突变位点的结束坐标和在基因组上的结束坐标,从基因组上截取第三段序列,将第一段、第二段和第三段序列合并在一起,输出到突变型的模拟基因组FASTA文件中;
(3-3)对野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件进行处理,使得文件中的序列部分每行的碱基数相等。
优选地,所述截取软件包括samtools软件或twoBitToFa软件;
优选地,调整FASTA文件每行碱基数的软件包括Picard软件。
优选地,步骤(4)具体包括:
(4-1)读取输入的模拟数据的目标平均深度,根据bed文件覆盖到的基因列表中所有基因的起止坐标,计算所有基因起止坐标的差值的总和,平均深度与差值总和相乘得到需要的数据量;
(4-2)分别从野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件截取需要数据量的野生型的FASTQ文件和突变型的FASTQ文件,优选地,可采用wgsim软件进行该步骤。
优选地,步骤(5)具体包括:
(5-1)利用比对软件将野生型的FASTQ文件和突变型的FASTQ文件与参考基因组进行比对,得到初始的野生型的BAM文件和突变型的BAM文件,提取初始的野生型的BAM文件和突变型的BAM文件中bed文件区域的部分,得到野生型的BAM文件和突变型的BAM文件(可采用bedtools软件的intersect模块);
(5-2)读取突变位点目标的突变频率,按照比例将野生型的BAM文件和突变型的BAM文件进行混合,生成模拟变异数据的BAM文件。
第二方面,本发明提供一种制作模拟变异数据的装置,所述装置用于执行第一方面所述的制作模拟变异数据的方法中的步骤,所述装置包括:
读取bed文件模块,用于执行包括:
读取输入的bed文件,提取bed文件覆盖的基因列表和基因的基因组坐标;
读取变异位点模块,用于执行包括:
读取输入的变异位点,获取基因组水平的变异信息,包括染色体、开始和结束坐标,突变序列;
制作FASTA文件模块,用于执行包括:
根据bed文件覆盖的基因列表和基因的基因组坐标、突变位点在基因组水平的信息,制作野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件;
制作FASTQ文件模块,用于执行包括:
从野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件截取FASTQ文件;
制作模拟变异数据模块,用于执行包括:
读取突变位点目标的突变频率,利用野生型的FASTQ文件和突变型的FASTQ文件、基因组文件和bed文件制作模拟变异数据的BAM文件。
优选地,所述读取bed文件模块用于执行包括:
利用第一数据库对bed文件进行注释,提取bed文件覆盖到的基因列表,及基因列表中每个基因在基因组上对应的所有转录本的染色体、开始和结束坐标;在第二数据库中提取每个基因优选的唯一的转录本,并由此得到每个基因唯一对应的在基因组对应的染色体、开始和结束坐标。
所述读取变异位点模块,用于执行包括:
(2-1)读取输入的变异位点,判断输入的变异位点的注释形式,用Transvar软件将输入的变异位点进行转化,提取其中基因组水平的注释;
(2-2)根据transvar软件注释结果中基因组水平的注释,得到基因组水平的变异信息,从基因组水平的变异信息中获取染色体、突变开始坐标、突变终止坐标、突变序列;
(2-2-1)基因组水平注释信息中包含的“chr”字符串后的数字,即为突变位点所在的染色体;
(2-2-2)若基因组水平注释信息中包含1个数字,则判断该数字同时为突变初始和结束坐标,继续读取数字后跟着的字符串,若字符串中存在“delins”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“delins”后的序列串;若字符串中存在“del”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“”;若字符串中存在“dup”,则突变开始坐标即为突变初始坐标,突变结束坐标在突变初始结束坐标加1,突变序列为“dup”后的序列串;若字符串中存在“>”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“>”后的序列串;
(2-2-3)若基因组水平注释信息中包含2个数字,且数字间用“_”分隔开,则判断第一个数字为突变初始开始坐标,第二个数字为突变初始结束坐标;继续读取数字后跟着的字符串,若字符串中存在“delins”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“delins”后的序列串;若字符串中存在“del”,则突变开始坐标在突变初始坐标基础上减1,突变结束坐标在突变初始结束坐标加1,突变序列为“”;若字符串中存在“dup”,则突变开始坐标即为突变初始坐标,突变结束坐标在突变初始结束坐标加1,突变序列为“dup”后的序列串;若字符串中存在“ins”,则突变开始坐标即为突变初始坐标,突变结束坐标即为突变初始结束坐标,突变序列为“ins”后的序列串。
所述制作FASTA文件模块,用于执行包括:
(3-1)遍历bed文件覆盖到的基因列表,若基因中不包含需要模拟的突变位点,则根据基因的染色体坐标,利用截取软件从基因组上截取该基因的序列信息,分别输出到野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件中;
(3-2)若基因中包含需要模拟的突变位点,首先根据基因的染色体坐标,利用截取软件从基因组上截取该基因的序列信息,输出到野生型的模拟基因组FASTA文件;其次根据基因在基因组上的开始坐标和突变位点的开始坐标,从基因组上截取第一段序列,把突变位点的突变序列作为第二段序列,根据基因突变位点的结束坐标和在基因组上的结束坐标,从基因组上截取第三段序列,将第一段、第二段和第三段序列合并在一起,输出到突变型的模拟基因组FASTA文件中;
(3-3)对野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件进行处理,使得文件中的序列部分每行的碱基数相等,优选地,可使用Picard软件进行该步骤。
所述制作FASTQ文件模块,用于执行包括:
(4-1)读取输入的模拟数据的目标平均深度,根据bed文件覆盖到的基因列表中所有基因的起止坐标,计算所有基因起止坐标的差值的总和,平均深度与差值总和相乘得到需要的数据量;
(4-2)分别从野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件截取需要数据量的野生型的FASTQ文件和突变型的FASTQ文件,优选地,可采用wgsim软件进行该步骤。
所述制作模拟变异数据模块,用于执行包括:
(5-1)利用比对软件将野生型的FASTQ文件和突变型的FASTQ文件与参考基因组进行比对,得到初始的野生型的BAM文件和突变型的BAM文件,提取初始的野生型的BAM文件和突变型的BAM文件中bed文件区域的部分,得到野生型的BAM文件和突变型的BAM文件(可采用bedtools软件的intersect模块);
(5-2)读取突变位点目标的突变频率,按照比例将野生型的BAM文件和突变型的BAM文件进行混合,生成模拟变异数据的BAM文件。
第三方面,本发明提供一种存储有计算机程序的计算机可读存储介质,所述计算机程序使计算机建立和/或运行如第一方面所述制作模拟变异数据的方法的步骤,或,所述计算机程序使计算机建立和/或运行如第二方面所述制作模拟变异数据的装置的模块。
第四方面,本发明提供第一方面所述制作模拟变异数据的方法、第二方面所述制作模拟变异数据的装置或第三方面所述的存储有计算机程序的计算机可读存储介质在制备生物测序数据的生物信息学分析标准参考数据中的应用或在测试生物信息学分析软件或流程中的应用。
与现有技术相比,本发明具有以下有益效果:
采取从头合成策略,设计制作模拟变异数据的方法,能够读取基于HGVS格式注释规则的不同水平(基因组、RNA、氨基酸)的变异位点的注释形式,并将其转化为基因组水平的突变起止坐标以及突变序列;能够模拟除了结构变异之外所有形式的基因组变异,包括单碱基变异(SNVs)、多碱基变异(MNVs)、插入缺失(InDels)以及复杂变异(Complexdeletion-insertion);能够根据使用者所需要的模拟数据的平均深度、突变频率制作对应的模拟数据,方便使用。
附图说明
图1为单碱基变异(MET:c.3028+3A>C)实际突变频率与预期突变频率的相关性图;
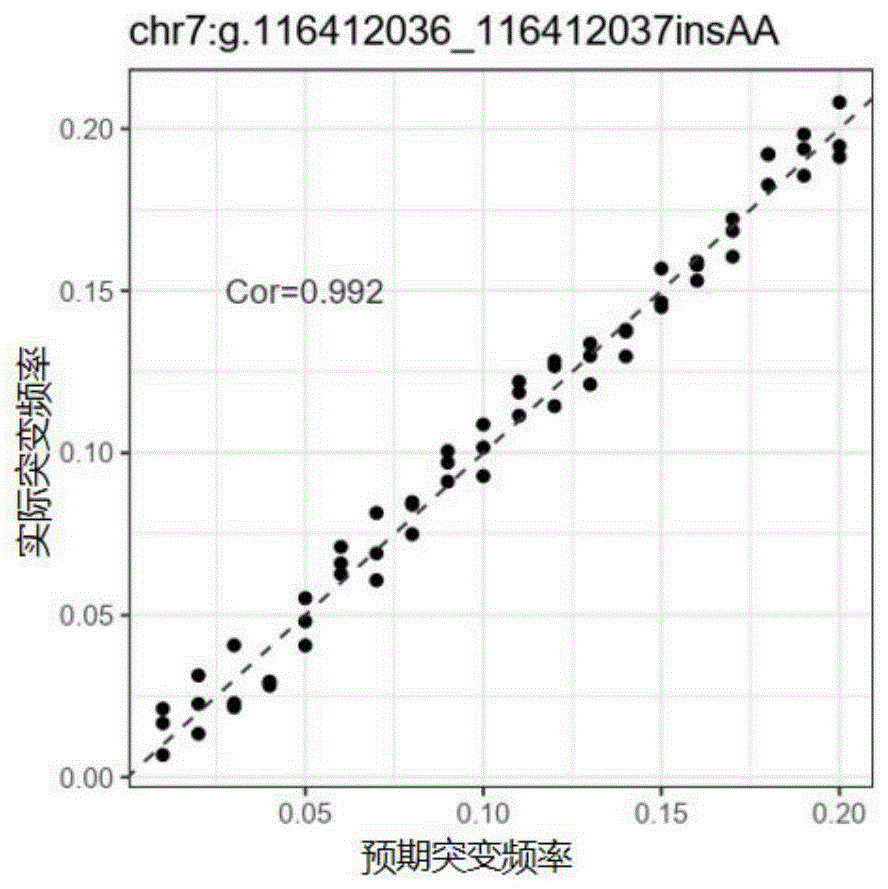
图2为插入变异(chr7:g.116412036_116412037insAA)实际突变频率与预期突变频率的相关性图;
图3为缺失变异(MET:c.2888-18_2888-2del)实际突变频率与预期突变频率的相关性图;
图4为单碱基变异(MET:c.3028+3A>C)、插入变异(chr7:g.116412036_116412037insAA)和缺失变异(MET:c.2888-18_2888-2del)实际有效平均深度与预期有效平均深度的箱线图。
具体实施方式
为进一步阐述本发明所采取的技术手段及其效果,以下结合实施例和附图对本发明作进一步地说明。可以理解的是,此处所描述的具体实施方式仅仅用于解释本发明,而非对本发明的限定。
实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道购买获得的常规产品。
实施例1
本实施例制作模拟变异数据。
验证本发明方法对基因组不同类型变异的模拟效果,分别选取了单碱基变异(MET:c.3028+3A>C)、多碱基变异(chr7:g.116412045_116412046delinsCC)、插入突变(chr7:g.116412036_116412037insAA)、缺失突变(MET:c.2888-18_2888-2del)和复杂变异(MET:c.3028+2_3028+4delinsACC)。
制作模拟变异数据的方法包括以下步骤:
(1)读取输入的bed文件,提取bed文件覆盖的基因列表和基因的基因组坐标;(2)读取输入的变异位点,获取基因组水平的变异信息,包括染色体、开始和结束坐标,突变序列;(3)根据bed文件覆盖的基因列表和基因的基因组坐标、突变位点在基因组水平的信息,制作野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件;(4)从野生型的模拟基因组FASTA文件和突变型的模拟基因组FASTA文件截取FASTQ文件;(5)利用比对软件将野生型的FASTQ文件和突变型的FASTQ文件与参考基因组进行比对,得到初始的野生型的BAM文件和突变型的BAM文件,提取初始的野生型的BAM文件和突变型的BAM文件中bed文件区域的部分,得到野生型的BAM文件和突变型的BAM文件;(6)读取突变位点目标的突变频率,按照比例将野生型的BAM文件和突变型的BAM文件进行混合,生成模拟变异数据的BAM文件。
将制作模拟数据的流程用python代码串行起来,封装到名字为simulate.py的脚本。可以通过命令行“python simulate.py-I<突变位点>-p<生成模拟数据的前缀>-d<有效平均深度>-f<突变频率>”来生成模拟数据。上述挑选的不同类型的变异位点,其模拟数据可采用下述命令行生成:
a.python3 simulate.py-i“MET:c.3028+3A>C”-p a-d 1000-f0.05;
b.python3 simulate.py-i“chr7:g.116412045_116412046delinsCC”-p b-d1000-f0.05;
c.python3 simulate.py-i“chr7:g.116412036_116412037insAA”-p c-d 1000-f0.05;
d.python3 simulate.py-i“MET:c.2888-18_2888-2del”-p d-d 1000-f0.05;
e.python3 simulate.py-i“MET:c.3028+2_3028+4delinsACC”-p e-d 1000-f0.05。
利用VarDict软件对生成的BAM文件进行检测,利用transvar软件对VarScan的检测结果进行注释,查看检测结果与预期结果是否一致,结果如表1所示。
表1
由表1结果可知,本发明方法能够有效的对单碱基变异、多碱基变异、插入突变、缺失突变和复杂变异进行模拟。
实施例2
测试实施例1制作的模拟数据,包括数据的有效平均深度、突变频率与输入参数相比的准确度。
(1)选取单碱基变异(MET:c.3028+3A>C)、插入变异(chr7:g.116412036_116412037insAA)、缺失变异(MET:c.2888-18_2888-2del)分别制作BAM文件,深度分别设置500X,1000X和2000X三个梯度,突变频率从0.01到0.2,每0.01为1个步长,各制作60个BAM文件;
(2)利用bamdst软件计算不同BAM文件的有效平均深度,统计不同BAM文件的实际平均深度和预期平均深度的相关性、利用VarDict软件对上述BAM文件进行检测,统计不同变异位点的实际突变频率与预期突变频率的相关性,从而评估制作的模拟数据的有效平均深度、突变频率与输入参数相比的准确度;
(3)统计3种变异类型实际突变频率与预期突变频率的相关性,结果如图1-图3所示,相关系数均达到0.95以上。
(4)统计3种变异类型实际有效平均深度与预期有效平均深度的关系,绘制箱线图,结果如图4所示,实际有效平均深度均在预期有效平均深度上下浮动。
根据上述结果可知,本发明方法制作的模拟数据的有效平均深度、突变频率与输入参数相比的准确度均较高。
实施例3
利用模拟数据软件分别按照表2制作携带17个变异位点的模拟数据,有效平均深度1400X,突变频率0.3%,每个位点重复10次,制作模拟数据,共计17x10=170个。利用北京优迅的《人EGFR/KRAS/BRAF/NRAS/PIK3CA基因突变联合检测试剂盒分析软件》对模拟数据进行分析,统计检出率,每个位点检出率均为100%。因此,通过本发明,验证了《人EGFR/KRAS/BRAF/NRAS/PIK3CA基因突变联合检测试剂盒分析软件》是可靠的。
表2
综上所述,本发明采取从头合成策略,设计制作模拟变异数据的方法,能够读取基于HGVS格式注释规则的不同水平(基因组、RNA、氨基酸)的变异位点的注释形式,并将其转化为基因组水平的突变起止坐标以及突变序列;能够模拟除了结构变异之外所有形式的基因组变异,包括单碱基变异、多碱基变异、插入缺失以及复杂变异;能够根据使用者所需要的模拟数据的平均深度、突变频率制作对应的模拟数据,方便使用。
申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 一种元数据存储方法、装置、设备及计算机可读存储介质
- 一种数据存储方法、装置、设备及计算机可读存储介质
- 一种可回滚档案采集数据的导入方法、计算机装置及计算机可读存储介质
- 一种数据处理方法、数据处理装置、计算机设备及可读存储介质
- 一种数据采集系统定位方法、装置和计算机可读存储介质
- 一种可保留分子标签信息的在测序数据中模拟小变异的方法、装置及计算机可读存储介质
- 一种可保留分子标签信息的在测序数据中模拟小变异的方法、装置及计算机可读存储介质