与大豆多个产量构成因子相关的QTL共定位位点
文献发布时间:2024-04-18 20:00:50
技术领域
本发明涉及生物技术领域,特别是与大豆多个产量构成因子相关的QTL共定位位点。
背景技术
大豆是一种重要的粮油兼用作物,其种子含有丰富的植物蛋白和油脂,可作为植物油、蛋白食品、饲料等加工工业的原料,具有十分重要的经济价值。因此,培育具有高产基因型的大豆品种是世界各国特别是中国大豆育种者的主要目标。
为了深入了解中国主要大豆种植区大豆品种产量性状的遗传基础,本研究采用全基因组关联分析(GWAS)和单核苷酸多态性分析(SNP)方法,对大豆产量性状构成因子的主要标记-性状关联(MTAs)、候选基因和数量性状基因座(QTL)进行了分析。通过这种深入分析鉴定优良的QTL,对利用基于QTL的育种方法开发具有目标产量特性的优异大豆品种具有深远的意义。
发明内容
为了解决上述技术问题,本发明提供了一种与大豆多个产量构成因子相关的QTL共定位位点。
为达到上述目的,本发明是按照以下技术方案实施的:
与大豆多个产量构成因子相关的QTL共定位位点,该QTL共定位位点位于大豆16号染色体上的3030820bp-3424009bp区域,包含10个与大豆的单株荚数PNpP、单株粒数GNpP、百粒重HGW均相关的候选基因。
进一步地,所述候选基因为Glyma.16g035400、Glyma.16g031800、Glyma.16g035600、Glyma.16g033100、Glyma.16g032700、Glyma.16g034600、Glyma.16g033000、Glyma.16g035000、Glyma.16g032000和Glyma.16g034700。
与现有技术相比,本发明利用全基因组关联分析的方法,对大豆自然群体的产量相关性状和基因型关联性进行了分析,最终找到一个较为特殊的QTL位点,该位点与大豆单株荚数、单株粒数和百粒重三个性状相关,在该位点区域内挖掘出的候选基因所具有的功能均与作物的生长、发育以及代谢等相关,候选基因的功能决定着作物的表现型。由于这三个农艺性状是影响大豆产量的重要因素,因此该QTL位点的发掘对大豆高产育种具有重要意义。
附图说明
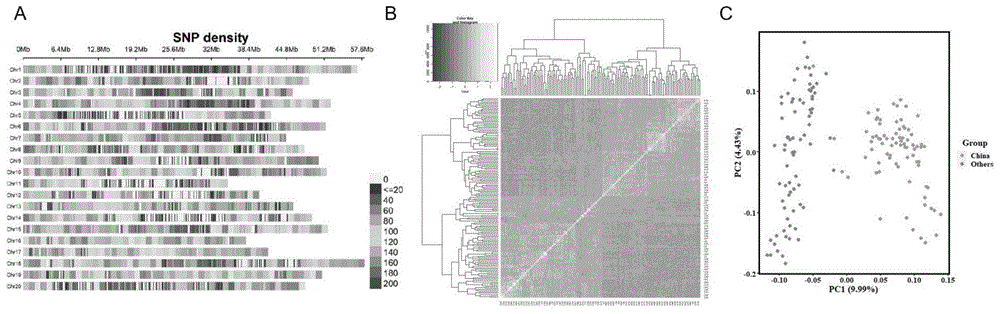
图1为SNP分布和种群结构:(A)用于GWAS的80,163个SNP标记在20个大豆染色体上的分布。(B)158份大豆亲缘关系矩阵的热图。(C)群体结构的主成分分析。在PC1和PC2之下的结合小组中的种质分布。
图2为显示全基因组连锁不平衡(LD)衰变的成对单核苷酸多态性的散点图(r
图3为曼哈顿图和相应的Q-Q图显示在2020年和2021年利用GWAS进行农艺性状分析中检测到的显著相关SNP:(A)单株荚数(GLM);(B)单株荚数(MLM);(C)单株粒数(GLM);(D)单株粒数(MLM);(E)百粒重(GLM);和(F)百粒重(MLM);每个有色点代表一个SNP。
具体实施方式
为使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步的详细说明。此处所描述的具体实施例仅用于解释本发明,并不用于限定发明。
以下实施例使用的材料包括158种不同的大豆品种,其中包括79份中国东北地区的和79份其他国家和地区的。这些资料来自黑龙江省农业科学院齐齐哈尔分院,可代表国内外的遗传多样性。本研究于2020年及2021年在齐齐哈尔(东经117°17′E,北纬33°18′N)种植所有实验材料。田间试验采用单行小区(3米长,间隔0.65米),并采用随机完全区组设计,每个试验环境重复3次。完全成熟后,从每个品系随机选取10个植株及其成熟籽粒进行评价。生长季节的田间管理,包括施肥、杂草管理和杀虫剂的应用是基于一个标准的种植制度。
2020年和2021年秋季,收取正常生长发育的整株包括豆荚,进行性状调查。研究了4个产量相关性状:单株荚数(PNpP)、单株粒数(GNpP)、百粒重(HGW)和小区产量(PY)。
通过CTAB方法从每个样本材料的大量新鲜叶组织中提取基因组DNA,然后基于特定基因座扩增片段测序(SLAF-seq)方法进行基因分型(Sun等,2013)。通过短寡核苷酸比对程序软件(SOAP2)进行原始配对末端读数和大豆参考基因组Williams 82(版本:Glyma.Wm82.a2)之间的比对。在SNP调用中,次要等位基因频率(MAF)被设置为0.05。当次要等位基因的深度大于样品总深度的五分之一时,基因型被定义为杂合子,并且总共选择80163个高质量SNP用于关联作图。进行全基因组LD分析,筛选出3026个R2>0.2为阈值的未连锁位点,然后进行群体结构分析,生成结果并调整阈值。
使用TASSEL 3.0版的两个不同模型:混合线性模型(MLM)和一般线性模型(GLM)进行分析。使用QQ和曼哈顿图用qqman软件作图分析结果。利用Glyma.Wm82.a2.v1(Williams82)的基因模型在10kb区域内显示候选基因;TAIR的拟南芥基因同源序列比对;PANTER或GO数据库进行候选基因分析。
方差分析(ANOVA)结果显示,年份对单株荚数、单株粒数和百粒重没有显著影响。F值和显著性水平表明,年份和这些变量之间没有显著的关系,具体如表1所示。
表1产量构成因子与年份的方差分析
利用特异位点扩增片段测序技术对158份大豆品种的总DNA进行了测序。测序后,从1.53亿对末端读数中鉴定出一组高质量的标签,每个大豆样品的平均标签数为80,163。读取长度和测序深度分别为45bp和6.51倍。随后,去除了等位基因频率低于0.05的标记,保留了80,163个高质量的SNP标记,拥有属性的等位基因频率≥0.05,缺失的数据≤0.03,定位在20条大豆染色体上。如图1所示,筛选的SNP标记被用于进行GWAS分析。作为了解大豆遗传组成的一部分,进行了全面的分析,产生了一组覆盖广阔基因组空间的单核苷酸多态性(SNP)标记。这些标记跨越大约9.4701亿个碱基对,覆盖了大豆基因组的86.09%。在20条染色体上鉴定出的SNP数量不同,其中18号染色体的SNP数量最多(5,162),11号染色体的SNP数量最少(2,037)。每条染色体的平均SNP约为4,008。随着标记密度也显示出显着的变化(从Chr.18上每个SNP 33.44kb到Chr.11上每个SNP 50.48kb),发现全基因组平均约为每1Mb一个SNP(图1A)。这些发现为大豆基因组研究提供了重要的见解,对于指导今后通过分子标记辅助选择开发改良大豆品种的研究工作具有重要价值。
我们采用连锁不平衡分析,阈值为R2>0.2,共筛选出3,026个SNP标记。群体的结构亲缘关系概况显示在图1B。基因型评估显示,GWAS小组由二至四个亚群组成。系统发育和主成分分析(PCA)结果表明,样品中有两个不同的组,组间有一些杂合材料。基于这些发现,我们选择K=2作为对所讨论的GWAS数据进行结构分析的最佳值,得到如图2所示。
进一步地,通过图2中的图表检查158个大豆基因型的连锁不平衡特征。在标记距离为5Mb时,得到的R2值小于0.1,发现LD衰变以0.7的R2值开始。此外,半衰变在0.35时达到,LD衰变曲线与半衰变在200Kbp处相交,代表全基因组检测连锁的临界距离。最后,应注意的是LD衰减曲线锚定在200kb,其中包括100kb的上游或下游区域。这个分析强调了基于GWAS模块阈值选择(如K=2)的适当值作为决策过程的一部分的相关性和重要性。
使用GWAS结合表型和基因型性状的分析在2020年没有检测到任何关联位点,而2021年的数据在GLM模式中具有较高的假阳性,同时选择MLM模式来检测显著相关的位点。在GLM模式中共检测到3275个SNP,1118个与PNpP相关,1043个与GNpP相关和1114个与HGW相关;在MLM模式中共检测到179个SNP,123个与PNpP相关,46个与GNpP相关,10个与HGW相关。
所有与PNpP、GNpP和HGW相关的SNP中,在16号染色体上发现了一个区域3030820-3424009bp,区间内包括10个标记位点,其中与单株荚数相关的7个标记位点是三个性状共有的。这7个标记位点分别是Gm16_3130820、Gm16_3203580、Gm16_3260654、Gm16_3271292、Gm16_3283717、Gm16_3304635和Gm16_3324009。同时发现,在所有标记中,Gm16_3130820、Gm16_3195848和Gm16_3260654分别位于Glyma.16G033100、Glyma.16g034100和Glyma.16g034600这三个候选基因上,参见表2。
表2产量构成因子相关的标记
在共定位区域发现了10个候选基因,编码5个激酶(蛋白激酶,糖基水解酶,m6A甲基转移酶,蛋白酪氨酸激酶和泛素结合酶),两个结构域(细胞周期蛋白样结构域、锌指和环指蛋白)和两个重复。Glyma.16G034600和Glyma.16g034700相距2300bp,分别编码蛋白酪氨酸激酶、锌指蛋白和环指蛋白(见表3)。
表3候选基因的相关信息
荚数、粒数和百粒重是决定大豆品种产量高低的重要性状,利用GWAS方法在该大豆群体中共找到很多个与荚数、粒数和百粒重相关的SNPs,在第16号染色体上找到了一个共定位位点,与这三个性状均相关联,该位点包含10个与作物生长发育相关的候选基因,是未报道过的。
这些功能性的候选基因与作物的生长发育息息相关,有的参与蛋白质编码基因的转录起始,有的转录调节参与DNA修复、细胞周期和细胞凋亡的基因,还有的调节RNA选择性剪接。候选基因Glyma.16g035400代表一个细胞周期蛋白样结构域,它在各种真核TFIIB及其古细菌对应物的C末端区域中重复出现。在真核生物中,所有蛋白质编码基因的转录起始都涉及聚合酶II系统。该系统受一般和特定转录因子的调节,其中一般因子包括TFIIB等通过常见的启动子元件(例如TATA盒)运行。
候选基因Glyma.16g031800和Glyma.16g035600被称为PPR重复。这种重复与预测的植物蛋白有关O49549具有类似于人类BRCA1蛋白的结构域结构。研究表明,BRCA蛋白是维持染色体稳定性所必需的,从而保护基因组免受损害。此外,BRCA转录调节一些参与DNA修复、细胞周期和细胞凋亡的基因。
候选基因Glyma.16g033100编码MT-A70,是人mRNA:m
在大多数细胞活动中起关键作用的蛋白质磷酸化是由蛋白激酶和磷蛋白磷酸酶介导的可逆过程。蛋白激酶催化γ磷酸从三磷酸核苷酸(通常是ATP)转移到蛋白质底物侧链中的一个或多个氨基酸残基,从而导致影响蛋白质功能的构象变化。以结构域为表现形式的候选基因Glyma.16g032700和Glyma.16g034600就存在于蛋白激酶中。此外,有些酶在细胞壁代谢中很重要。植物的原始细胞壁是由果胶物质、纤维素和半纤维素组成的复杂网络。果胶物质由长多糖链组成,骨架由同型半乳糖醛酸和鼠李糖半乳糖醛酸组成。编码糖基水解酶家族28的候选基因Glyma.16g033000,包括多聚半乳糖醛酸酶以及鼠李糖半乳糖醛酸酶A(RGaseA)均参与细胞壁的降解过程。
泛素-蛋白酶体系统(UPS)/泛素-蛋白酶体途径是一个普遍存在的蛋白质转换系统,可以降解或修饰真核细胞中的蛋白质。该系统可以调节许多细胞过程,几乎参与生长发育的各个方面,因此泛素化的修饰可能比磷酸化的修饰更重要。该系统由泛素(Ub),泛素活化酶(E1),泛素缀合酶(E2),泛素连接酶(E3),26S蛋白酶体,DUB和靶蛋白组成。其中,候选基因Glyma.16g035000编码泛素结合酶(E2),候选基因Glyma.16g032000和Glyma.16g034700代表锌指和环指蛋白,被认为是E3泛素连接酶。
本发明是以中国大豆品种和外国大豆品种作为研究群体,利用GWAS进行表型性状相关位点的鉴定,最终获得一个与单株荚数、单株粒数和百粒重相关的共定位位点。在该共定位位点区域内,找到10个候选基因和7个共有标记,其中3个标记定位了3个候选基因。这些与产量性状相关的候选基因紧密的分布,可能代表了该位点的重要性,我们将进一步研究这些候选基因在特定阶段的表达模式,以此对品种进行改良、突破产量瓶颈。
本发明的技术方案不限于上述具体实施例的限制,凡是根据本发明的技术方案做出的技术变形,均落入本发明的保护范围之内。
- 一种电厂脱硫设备用烟气处理方法
- 一种电厂脱硫设备用烟气系统