一种舒肝宁注射液的质量检测方法及其应用
文献发布时间:2024-05-31 01:29:11
技术领域
本发明关于中药制剂分析技术领域,尤其涉及一种舒肝宁注射液的质量检测方法及其应用。
背景技术
舒肝宁注射液以茵陈提取物、栀子提取物、黄芩苷、板蓝根提取物和灵芝提取物为其活性成分,清热解毒,利湿退黄,益气扶正,保肝护肝,用于湿热黄疸,症见面目俱黄,胸肋胀满,恶心呕吐,小便黄赤,乏力,纳差,便溏,以及急、慢性病毒性肝炎见前述症状者。
但舒肝宁注射液在生产过程中需要采用加热、调节pH等工艺步骤,而茵陈、栀子、板蓝根、灵芝作为中药,成分较为复杂,这使得生产舒肝宁注射液过程中可能会产生一些会引起不良反应的物质。因此需要对产品进行严格的质量控制,来保证临床用药安全。
发明内容
针对以上技术问题,本发明提供一种舒肝宁注射液的质量检测方法及其应用。该检测方法通过液质联用的方法能够检测舒肝宁注射液中的有效成分以及可能引起不良反应的物质,还可用于舒肝宁注射液降解产物分析以及差异成分的分析。
为达到上述发明目的,本发明实施例采用了如下的技术方案:
一种舒肝宁注射液的质量检测方法,用液质联用的方法对舒肝宁注射液的成分进行检测;所述液质联用中的液相色谱条件为:
色谱柱:反相色谱柱;
流动相A为含0.05%~0.1%v/v乙酸的水,流动相B为乙腈,进行线性梯度洗脱,梯度洗脱程序为:0min,0%B相;12min,8%B相;13min,8.3%B相;14min,9%B相;17min,16%B相;22min,16.5%B相;24min,20%B相;26min,29%B相;27min,29.5%B相;35min,100%B相;35.1min,0%B相;37min,0%B相;
流速:0.3mL/min;
柱温:25~35℃。
本发明采用的上述色谱条件能够获得峰形好、响应高的色谱峰,并能分离出较多的成分,满足舒肝宁注射液中多种成分的检测,包括有效成分以及可能产生不良反应的成分。上述色谱条件结合质谱分析,能够使舒肝宁注射液的成分检测准确、高效,可用于在生产中对产品进行半成品质量检测和成品质量检测,有助于舒肝宁注射液的质量控制。并且,该检测方法还能够用于舒肝宁注射液降解产物分析以及差异成分的分析。
优选地,所述色谱柱为ACQUITY UPLC HSS T3(2.1×100mm,1.8μm)。采用上述色谱柱得到的色谱峰型更好且响应更高。
优选地,所述柱温为30℃。
优选地,所述流速为0.3mL/min。
优选地,所述液质联用的质谱采用全扫描/数据依赖(Full MS/dd-MS
优选地,所述离子源的参数还包括:鞘气(N
优选地,所述质量检测方法还包括对舒肝宁注射液进行核磁指纹图谱分析。
优选地,所述核磁指纹图谱分析的参数为:谱宽(SW)为20ppm,等待时间(RD)为2s,混合时间(tm)为100ms,t1为4μs,90°脉宽为13.26μs,采样时间为1.36s,采样点数为32K,FID累加次数为128次。
第二方面,本发明还提供上述舒肝宁注射液的质量检测方法在舒肝宁注射液降解产物分析中的应用。
第三方面,本发明还提供上述舒肝宁注射液的质量检测方法在舒肝宁注射液差异成分分析中的应用。
上述舒肝宁注射液的质量检测方法能够准确、高效地检测舒肝宁注射液中的多种成分,从而能够应用于对舒肝宁注射液降解产物和差异成分的分析。
本发明的有益效果在于:本发明通过采用上述液相色谱条件以及质谱条件下的液质联用技术,能够准确、高效地检测舒肝宁注射液中的有效成分以及可能引起不良反应的物质,为质量控制及不良反应物质筛查提供依据,有助于舒肝宁注射液的质量控制。并且,该质量检测方法得到的成分数据还可进一步用于舒肝宁注射液降解产物分析以及差异成分的分析,有利于为舒肝宁注射液的质量控制提供更全面的数据和技术支持。
附图说明
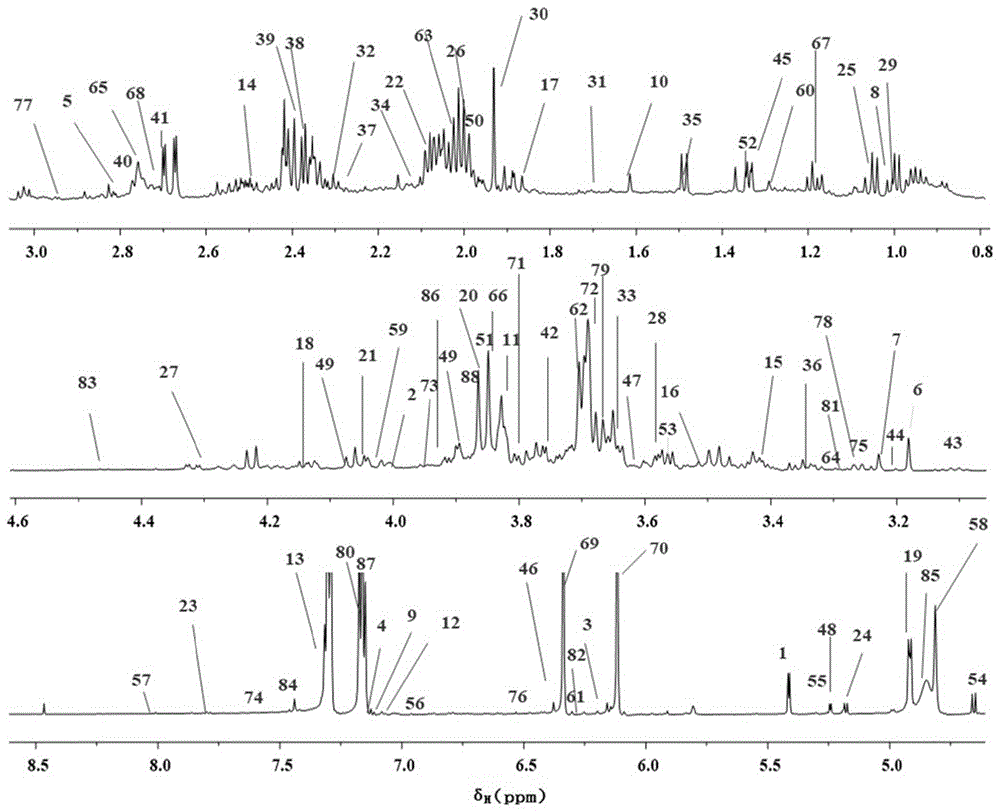
图1是为实施例1中舒肝宁注射液定性分析NMR图谱;
图2为对比例1中舒肝宁样品不同色谱柱总离子流图;
图3为对比例2中舒肝宁注射液在不同水相条件下总离子流图;
图4为对比例2中舒肝宁注射液在不同水相条件下总离子流图;
图5为对比例3中负离子模式下不同喷雾电压(SV)下五种化合物的峰面积;
图6为对比例4中负离子模式下不同毛细管温度(CT)下五种化合物的峰面积;
图7为对比例5中负离子模式下不同辅助气温度(AT)下五种化合物的峰面积;
图8为对比例6中绿原酸在负离子模式下不同归一化碰撞能量下的二级质谱图;
图9为对比例6中咖啡酸在负离子模式下不同归一化碰撞能量下的二级质谱图;
图10为对比例6中新绿原酸在负离子模式下不同归一化碰撞能量下的二级质谱图;
图11为对比例6中隐绿原酸在负离子模式下不同归一化碰撞能量下的二级质谱图;
图12为对比例6中1,3-二咖啡酰奎宁酸在负离子模式下不同归一化碰撞能量下的二级质谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
舒肝宁注射液的活性成分为茵陈提取物、栀子提取物、黄芩苷、板蓝根提取物和灵芝提取物,成分复杂,在包含加热、调节pH等工艺步骤的生产过程中会发生复杂的变化,可能产生一些会引起不良反应的物质,需要进行严格的质量控制来保证临床用药安全。但由于中药提取物中的化合物种类众多,故在检测时既需要考虑检测方法对多种成分的分离能力,还需要兼顾检测方法的准确、高效,才能使之具有助于在实际生产过程中对产品和半成品的质量检测。
针对该问题,本发明提供一种舒肝宁注射液的质量检测方法,该方法采用液质联用技术,首先通过特定的色谱条件实现舒肝宁注射液中多种成分的分离,继而通过质谱分析来进行成分定性,从而能够准确而高效地检测舒肝宁注射液中的有效成分和可能产生不良反应的成分。该方法不仅有助于舒肝宁注射液的质量控制,还能用于舒肝宁注射液降解产物分析以及差异成分的分析。
以下通过具体实施例来对本发明的技术方案进行进一步说明。
以下实施例中所采用的试剂和药品如无特殊说明,均为市售。
以下实施例中所采用的分析仪器:
Ultimate 3000/Q-exative Orbitrap MS高分辨液质联用系统(ThermoFisherScientific,美国);BrukerAVIII 600MHz NMR谱仪(BrukerBiospin,德国)
实施例1
本发明实施例提供了舒肝宁注射液的质量检测方法。
1、舒肝宁注射液的成分检测
1.1待测品溶液制备
精密吸取待测舒肝宁注射液样品100μL于1.5mL离心管中,加50%甲醇(v/v)稀释至1000μL,涡旋混匀20min后,14000rpm离心10min,取上清液作进样准备。
1.2液相与质谱条件
1.2.1液相色谱条件:
色谱柱为ACQUITY UPLC HSS T3(2.1×100mm Column,1.8μm);
流动相A:含0.1%v/v乙酸的水,流动相B:乙腈;进行线性梯度洗脱,梯度洗脱程序0min,0%B相;12min,8%B相;13min,8.3%B相;14min,9%B相;17min,16%B相;22min,16.5%B相;24min,20%B相;26min,29%B相;27min,29.5%B相;35min,100%B相;35.1min,0%B相;37min,0%B相;
流速:0.3mL/min;
进样量:2μL;柱温:30℃。
1.2.2质谱条件:
质谱条件:采用Full MS/dd-MS
2、指纹图谱分析
2.1实验仪器:
所有样品的1D
2.2检测方法:
样品配制:吸取450μL注射液和50μL磷酸盐缓冲液(0.1M,100%D
样品检测:使用NOESYGPPR1D序列[RD-90°-t1-90°-tm-90°-ACQ]采集谱图。
参数设置如下:谱宽(SW)为20ppm,等待时间(RD)为2s,混合时间(tm)为100ms,t1为4μs,90°脉宽为13.26μs,采样时间为1.36s,采样点数为32K,FID累加次数为128次。
0.1M Na
2.3实验结果
2.3.1样品相似度
测定100个批次舒肝宁注射液的NMR指纹图谱,分别采用相关系数及夹角余旋方法计算相似度。各批次的化学位移和峰型相似度很高,相似度均在0.9以上。根据上述100个批次舒肝宁注射液的NMR指纹图谱生成标准对照指纹图谱,标示出共有特征峰,以化学位移表示,分别为:2.01±0.01;2.07±0.01;2.94±0.01;3.13±0.01;3.51±0.02;3.70±0.01;3.86±0.02;3.89±0.01;3.91±0.01,5.17±0.01;5.92±0.02;6.10±0.01;6.12±0.02;6.34±0.01;6.35±0.01;7.06±0.02;7.16±0.01;7.45±0.02。
2.3.2方法学考察
本实验主要考察稳定性、精密度以及重复性等。以样品中一些特征峰对应的化学位移以及峰面积,计算其相对标准偏差(RSD)值。
稳定性试验:随机选择一个批次的舒肝宁注射液(20200921),按“2.2”项下方法制备供试品溶液,分别于0,2,4,10,12,24h进行
精密度试验:随机选择一个批次的舒肝宁注射液(20200921),按“2.2”项下方法制备供试品,连续采集六次
重复性试验:随机选择一个批次的舒肝宁注射液(20200921),按“2.2”项下方法重复制备6份供试品,分别采集
实验结果如表1~表3所示:在不同时间内测定的舒肝宁注射液NMR指纹图谱中特征峰的峰面积和化学位移值的RSD值均小于5%,表明样品在24h内稳定性良好;在实验条件下连续测定得到的舒肝宁注射液20200921 6次得到的NMR指纹图谱中特征峰的峰面积和化学位移值的RSD值均小于5%,表明仪器的精密度良好;重复制备的6份舒肝宁注射液S71样品测定得到的NMR指纹图谱中特征峰的峰面积和化学位移值的RSD值均小于5%,表明方法的重现性良好。
2.3.3舒肝宁注射液NMR图谱定性分析
通过
/>
对比例1
本对比例考察了不同色谱柱得到的色谱图。
精密吸取舒肝宁注射液(批次:20191112)100μL于1.5mL离心管中,加50%甲醇(v/v)稀释至1000μL后,涡旋混匀10min后,14000rpm离心10min,取上清液作进样准备。
色谱柱分别为ACQUITY UPLC HSS T3(2.1×100mm Column,1.8μm)、Waters BEHC
结果如图2所示,ACQUITY UPLC HSS T3色谱柱在当前条件下色谱峰型最好且响应最高。
对比例2
本对比例考察了不同水相得到的色谱图。
选择纯乙腈作为有机相,水相分别为纯水、0.05%v/v乙酸水、0.1%v/v乙酸水和0.1%v/v甲酸水,结果如图3和图4所示,水相为0.1%v/v乙酸水时对舒肝宁样品的分离效果最好。
对比例3
本对比例考察了不同喷雾电压下的离子色谱峰面积。
在实施例1的质谱条件的基础上,在负离子模式下,分别考察喷雾电压为2.5kV、3.0kV和3.5kV时绿原酸、京尼平苷、木樨草苷、腺苷和咖啡酸五种化学成分的离子色谱峰面积。结果如图5所示,随着喷雾电压的增加,离子色谱峰面积呈现逐渐增加的趋势,但过高的喷雾电压可能导致放电,同时考虑到高分辨质谱仪Q-Orbitrap MS喷雾电压的常用参数范围,3.0kV为最佳喷雾电压。
对比例4
本对比例考察了不同毛细管温度下的离子色谱峰面积。
在实施例1的质谱条件的基础上,在负离子模式下,分别考察毛细管温度为250℃、300℃和350℃时绿原酸、京尼平苷、木樨草苷、腺苷和咖啡酸五种化学成分的离子色谱峰面积。结果如图6所示,随着毛细管温度的增加,绿原酸、京尼平苷、腺苷和咖啡酸离子色谱峰面积呈现逐渐降低的趋势,而木樨草苷在毛细管温度为300℃时离子色谱峰面积更高。综合考虑各化学成分的离子色谱峰面积,毛细管温度为300℃时各成分的离子色谱峰面积更为理想。
对比例5
本对比例考察了不同辅助气温度下的离子色谱峰面积。
在实施例1的质谱条件的基础上,在负离子模式下,分别考察辅助气温度为300℃、350℃和400℃时绿原酸、京尼平苷、木樨草苷、腺苷和咖啡酸五种化学成分的离子色谱峰面积。结果如图7所示,当辅助气温度为400℃时,以上五种化学成分具有更高的离子色谱峰面积。
对比例6
本对比例考察了不同碰撞能量下的二级碎片。
在实施例1的质谱条件的基础上,在负离子模式下,分别考察10/20/30V,10/20/40V,20/30/40V和20/40/60V四个归一化碰撞能量时绿原酸、隐绿原酸、新绿原酸、咖啡酸和1,3-二咖啡酰奎宁酸五种化学成分的二级质谱图。结果如图8~图12所示,20/40/60V的碰撞能量使准分子离子峰丰度低且存在,同时二级碎片丰度更高。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 一种补肾舒脊的中药组合物的质量检测方法及其应用
- 一种补肾舒脊的中药组合物的质量检测方法及其应用