用于治疗用途的树突细胞的体外分化和成熟方法
文献发布时间:2023-06-19 09:44:49
发明领域
本发明涉及一种用于生成高产量的加载有1型极化mRNA的树突细胞的加速方法,其用于免疫疗法,尤其是用于癌症疫苗接种。
背景技术
自从40多年前发现树突细胞以来,将这些细胞的独特生物学特性转化为医学应用一直是挑战。大多数努力都集中在以抗癌疫苗的形式将DC引入临床。这是基于DC引发针对肿瘤抗原的T细胞应答的能力,从而导致防止肿瘤发展甚至根除已建立的肿瘤,如无数临床前模型所证明的。
在这种作用的基础上,存在一组独特的生物学特性,这些特性被概括为“4-信号”概念:(1)在主要组织相容性(MHC)分子上呈递大量加工的抗原,(2)上调细胞表面上大量的T细胞共刺激分子,(3)释放驱动T细胞应答正确极化的细胞因子,以及(4)提供额外的信号,这些信号对诱发的T细胞效应子的组织归巢模式进行编程。在抗肿瘤免疫的特定背景下,DC可以通过专门的受体(例如DNGR-1)拾取死细胞,从而导致MHC I交叉呈递并引发抗原特异性细胞毒性T细胞。T细胞共刺激分子CD40的高表达提高了CD4+和CD8+T细胞扩增的幅度,从而导致增强的肿瘤保护以及由耐受性向免疫力的转化,而CD70的上调对于产生强大而持久的记忆细胞毒性T细胞应答至关重要。相反,T细胞抑制性受体或检查点配体(例如程序性死亡配体1(PD-L1))在DC表面上的表达应最小。
接下来,在T细胞接触时分泌足够量的生物活性IL-12的能力对于驱动最佳肿瘤控制所必需的1型极化反应至关重要,同时还支持NK细胞效应子功能。另外,DC释放的趋化因子的模式决定了将募集何种类型的T细胞,即在抗肿瘤免疫的情况下,优先为1型极化效应子,而不是T-helper(Th)2细胞(具有肿瘤支持潜力)或免疫抑制性调节T细胞(T-regs)。
根据这些知识,很明显,设计理想的基于DC的癌症疫苗需要最大程度地控制和优化所有这些关键参数。正确的DC激活或成熟状态在确定T细胞的结果中至关重要,因为不成熟的DC(iDC)在刺激T细胞应答中非常无效,甚至能够提高T细胞耐受性。
因此,在选择强烈的激活刺激物以产生完全有效的成熟的DC时,需要进行深思熟虑的考虑,同时还要避免DC“耗尽”的现象。Toll样受体(TLR)配体是DC成熟的最强的触发因素之一,可以是外源性(即病原体来源的)或内源性的(来自组织损伤或细胞死亡的危险相关分子)。尽管有这些知识,但最常用的成熟策略之一是将单核细胞来源的DC暴露于炎性介质的组合中,这些炎性介质包括肿瘤坏死因子-α(TNF-α),白介素-1β(IL-1β),白介素6(IL-6)和前列腺素E2(PGE2),如Jonuleit等人所首次描述的。添加PGE2的价值在于观察到它能够进一步提高DC产量,成熟度和迁移性(Jonuleit等,1997)。但是,也已经显示PGE2损害DC分泌生物活性IL-12p70以及将T辅助细胞的极化移向Th2而不是Th1发展的能力(Kalinski等,1998)。
从那时起,已经探索了许多替代策略,以最大化DC诱导1型极化反应的能力。Mailliard等人开发了一种方案,其中DC在促炎性细胞因子TNF-α,IL-1β,IFN-γ和干扰素-α(IFN-α)以及TLR3激动剂聚(I:C)的存在下成熟(US7972847;US8691570)。与“标准”TNF-α,IL-1β,IL-6和PGE2成熟的DC相比,这些α1型极化DC(αDC1)产生更高水平的IL-12p70,并诱导更强劲的针对黑色素瘤相关抗原的长寿细胞溶解性T细胞(CTL)的扩增(Maillard等人,2004)。尽管这些αDC1细胞已经在针对复发性恶性神经胶质瘤患者的临床试验中使用(Okada等人2011),但是成熟“混合物(cocktail)”的复杂性对良好生产规范(GMP)兼容性生产过程的实施提出了重大挑战。
一个更简单的替代方案涉及将TLR4配体与干扰素-gamma(IFN-γ)组合。脂多糖(LPS)是DC成熟的最强的先天刺激物之一,可触发免疫刺激性细胞因子(例如IL-12)的大量产生。然而,当LPS刺激的DC随后在体内参与与T细胞的同族相互作用时,对进一步的IL-12释放变得不应。这种“耗尽”现象能够通过将DC与IFN-g共同暴露而抵消,从而在T细胞接触或人工CD40连接触发后能够产生IL-12的“第二次爆发”(Paustian等,2011)。由于其毒性和缺乏GMP规划,LPS的使用在大规模细胞治疗应用中仍然引起问题。然而,由于LPS的酸水解保留了免疫刺激特性但显著减弱毒性水平,因此LPS衍生的单磷酰脂质A(MPLA)已被批准用于临床使用(Boccaccio等,1997)。MPLA是目前大量生产的疫苗的佐剂配方的不可或缺的成分。据报道,在IL-12p70的分泌和趋化因子吸引效应T细胞方面,以及在CD4+和CD8+T细胞引发能力方面,与TNF-α,IL-1β,IL-6和PGE2成熟的DC相比,MPLA/IFN-γDC和α1型极化的DC都同样优越(Flansen等人,2013)。Ten Brinke等人的小组进一步探索了MPLA/IFN-γDC成熟方法,其中在GM-CSF和IL-4存在下培养8天的单核细胞在培养的最后2天中获得了成熟度增强。收获后,获得的DC展示出诱导从头Th1极化以及引发具有高细胞溶解活性的抗原特异性CD8+T细胞的能力(Ten Brinke等人,2007;WO2007/078196;ten Brinke等人,2010),同时保留向CCR7配体迁移的能力。该DC培养方案已就可能的临床实施方式进行了进一步研究,另外的研究表明了在这种情况下人血清对DC成熟和迁移的不利影响(Kolanowski等人,2014)。
在正确的成熟刺激物类型之后,抗原加载方式是临床适用性的重要决定因素。DC被动加载免疫原性肽(如通常用于上述研究中的功能测试的),意味着对所考虑的每种候选抗原都具有免疫显性表位的现有知识,并在符合条件患者的人白细胞抗原(HLA)类型方面施加特异性限制。开发未成熟的DC的高抗原摄取能力的替代方法是与肿瘤裂解物一起温育。然而,这需要足够数量的患者肿瘤材料,这再次限制了在转移性疾病中的可行性,在转移性疾病中,通常只有少量的活检或细胞学样品是可用的。用编码肿瘤抗原的全长mRNA加载DC现已被广泛认为是诱导大量可能表位的呈递的一种简练方法。它还提供了共引入能够优化DC免疫原性力的RNA构建体的机会(Van Lint等人,2014)。这通常是通过细胞电穿孔来实现的,这种方法的另一面是存在大量细胞丢失的风险,从而损害向患者施用足够疫苗剂量的可能性(Tuyaerts等人2003;Ponsaerts等人2003;Bonehill等人2004)。
就疫苗生产而言,一个重要的附加方面是细胞培养的持续时间。对于单核细胞来源的DC,该持续时间传统上为7到8天的范围内,这意味着用新鲜的培养基和细胞因子重复补充培养物。在GMP生产环境的苛刻环境下,这转化为消耗品以及操作员干预方面的成本增加。若干组已经论证了,使用加速培养方案能够将全功能性DC与单核细胞区分开(Jarnjak-Jankovic等,2007;Dauer等,2003;Kvistborg等,2009;Massa等,2013;Truxova等,2014;EP2829600)。
最终的实际考虑是选择使用封闭系统细胞培养:就GMP要求而言,这构成了另一个优势,并允许将生产过程转移到可商购的自动化细胞培养设备上。
考虑到这些考虑,本发明的目的是开发用于生产基于临床级DC的癌症疫苗的方法,从而在一个相同的生产方法中首次将若干种关键资产重新结合:加速培养时间并利用GMP相容的1型极化成熟混合物,并结合通过mRNA电穿孔的抗原加载。此外,已证明可以在无血清条件下,在GMP兼容的细胞培养袋中使用密闭培养系统,并最大程度地使用GMP认证或药物级成分来实现这一目标。
将根据本发明方法的性能与已广泛建立的“标准”8天培养的单核细胞来源的DC进行了比较,该DC是由TNF-α和PGE2的组合成熟的。该成熟混合物是本领域技术人员众所周知的,并且是Jonuleit等人描述的包含TNF-α,PGE2,IL-1b和IL-6的原始经典单DC成熟混合物的简化版本。
重要的是,与许多先前的报道相反,并且为了贴切模拟现实生活中的疫苗接种环境,本发明中用电穿孔DC进行的所有功能测定均在冷冻保存和解冻之后进行,而不是使用新鲜处理的细胞。与标准方案相比,本发明的方法实现在表型和功能上均优越的DC的更高产量。令人惊讶地,我们发现,与经典方案获得的DC相比,在根据本发明生成的DC上的T细胞抑制性检查点配体PD-L1的表达大大降低。而且,在冻存保存的等分试样融化后,经典DC上的表达进一步增加,而用本发明的方法获得的解冻的DC的等分试样则不是这种情况。对于实际上将要注入患者内的细胞而言,这是至关重要的观察,其中这种免疫抑制配体的表达应尽可能低。
本发明涉及在封闭系统(例如具有一个或多个无菌连接的培养物)中生成成熟的,优选自体的,临床级树突细胞的体外方法,并且该方法适于例如癌症患者的疫苗接种。在一个实施方案中,该方法主要包括通过使用临床级细胞因子,优选GM-CSF和IL-4,分化从白细胞去除术获得的单核细胞,并结合DC的进一步成熟,从而生成(例如在封闭系统中)成熟的临床级树突细胞,其中DC的进一步成熟是通过额外暴露于成熟因子优选临床级IFN-g和内毒素解毒衍生物MPLA的组合而获得的。
然后将获得的产物用一种或更多种抗原尤其是肿瘤相关抗原(TAA)加载,所述产物包含在体外培养时间总计约4天内(优选在约3天至约5天培养之内)产生/生成的成熟的树突细胞。根据本发明的这种快速方法能够优选在封闭系统中使用无血清培养基从白细胞去除术产物中生成大量DC。
在一个实施方案中,根据本发明的方法包括以下步骤:
-从患者获得单核细胞白细胞去除术产物,
-从所述白细胞去除术分离单核细胞,
-将所述单核细胞与临床级细胞因子(优选适量的GM-CSF和IL-4)温育,用于分化为树突细胞,
添加成熟因子MPLA和IFN-g,用于单核细胞来源的树突细胞的最终成熟,以及
-回收获得的细胞,并将其用编码一种或若干种抗原或表位的核酸序列(特别是mRNA)转染。
在一个实施方案中,使单核细胞与GM-CSF和IL-4接触约1至4天,优选1至3天,更优选2至3天(以1天计24小时),在此期间DC前体分化为未成熟的树突细胞。在进一步的实施方案中,在IFN-g和MPLA存在下的成熟时间为1至3天,优选为1至2天,更优选为约2天(24小时)。培养条件适合于未成熟的DC的成熟以形成成熟的DC群体。
在进一步的实施方案中,本发明提供可通过本文提供的方法获得的成熟的(和转染的)树突细胞或成熟的(和转染的)树突细胞群体。本发明还提供包含通过本发明的方法获得的树突细胞的组合物,试剂盒,临床级袋或冷冻小瓶。
转染的树突细胞特别可用于制备用于免疫疗法的组合物,特别是其在免疫疗法中的用途,更特别是在治疗癌症中的用途。因此,本发明还提供用于免疫疗法,肿瘤疗法的方法或用于活化T细胞的方法,其包括将通过本文提供的方法获得的转染的树突细胞施用于受试者。
具体参考附图,应当注意的是,所示出的细节是通过示例的方式并且用于对本发明的不同实施方案的说明性讨论的目的。提出它们是为了提供被认为是对本发明的原理和概念方面最有用和最容易描述的内容。在这一点上,没有试图比基本理解本发明所必需的更详细地示出本发明的结构细节。结合附图的描述使得本领域技术人员清楚如何在实践中实施本发明的几种形式。
图1.收获时4天培养的moDC的特征:(A)排除碎片后,CD11c
图2. 4天moDC和8天moDC之间比较的收获时的DC特征(n=10):(A)成活力和单核细胞到DC的转化率(流式细胞术);(B)表型和成熟标志物的细胞表面表达的比较,计算为相对MFI(阳性荧光信号与背景荧光的几何平均值之比,在活CD11c
图3.收获时4天moDC中MPLA,IFN-γ或二者对诱导成熟特征(profile)的相对贡献(n=3)。DC成熟标志物的相对MFI,以条形图显示。统计学:Kruskal-Wallis与Dunn多重比较检验相结合。
图4.就幼稚T辅助细胞极化潜力而言,MPLA和IFN-γ对4天moDC的组合作用(重复实验合并的n=6至12个重复样品,具有2个不同的DC供体和3个不同的同种异体T细胞供体)。(A)同种异体幼稚T辅助细胞极化测定的实验时间线的示意图。(B)代表性点状图,其显示与未成熟或完全成熟的同种异体DC共培养14天后CD4+T细胞内CD4+T细胞IFN-γ/IL-10细胞因子的产生。(C)MPLA,IFN-γ或其组合对DC介导的幼稚T辅助细胞极化的相对贡献:条形图表示显示IFN-γ、IL-10、IL-4和IL-17的胞内表达的CD4+细胞的百分比。
图5.(A)用载体(MOCK-EP)或eGFP mRNA(1μg mRNA/10e6 DC)EP的4天moDC的代表性点状图,显示在4h电穿孔后的成活的CD11c
图6.(A)在无细胞因子培养基中温育24小时后,冷冻保存的4天(n=5)和8天(n=2)eGFP mRNA-EP DC的细胞因子和趋化因子分泌组,如使用Luminex测定所测量的。统计学:未配对的t检验。(B)与同种异体T辅助细胞共培养的冷冻保存的EP-DC的时间线。(C)冷冻保存和解冻后电穿孔DC的T细胞极化特性(浅灰色条)。没有DC的同种异体幼稚CD4+T细胞用作阴性对照(白色条)。(重复实验合并的n=3到6个重复样品,具有2个不同的DC供体和1个同种异体T细胞供体)。数据显示了表达细胞因子的CD4+T细胞的百分比。统计学:Mann-Whitney检验。
图7.(A)用经指定mRNA电穿孔或用MART-1的AAAGIGILTV A2限制性肽脉冲的自体4d-moDC刺激来自HLA-A2阳性供体的MACS纯化的CD8+T细胞两次。代表性的点状图显示四聚体阳性CD8+T细胞的扩增。将所有测定中使用的DC冷冻保存并解冻。(B)使用不同HLA-A2+供体和不用DC、用MOCK脉冲的DC、用eGFP-mRNA-EP的DC、用MART-1 mRNA-EP的DC和用MART-1肽脉冲的DC刺激的CD8+T细胞获得的数据总结(重复实验合并的n=4至8个重复样品,具有2个不同的HLA-A2阳性供体)。(C)在用指示的DC条件刺激的MART-1特异性CD8+T细胞中细胞内IFN-γ和颗粒酶B的水平。(B-C)统计学:Kruskal-Wallis与Dunn多重比较检验。
图8.(A)使用HLA-A2+供体和MART-1作为模型抗原的自体DC:CD8T细胞共培养后抗原特异性细胞毒性测定的示意图。用自体4d-moDC的每两周一次的轮次的刺激后,将细胞溶解性CD8+T细胞与无T2靶细胞、无关肽脉冲的T2靶细胞(流行性感冒肽)或MART-1肽脉冲的T2靶细胞共培养。DC对应物包括阴性对照DC(MOCK-脉冲的DC(未显示)和eGFP mRNA-EP的DC),MART-1 mRNA-EP的DC和阳性对照DC(被来自MART-1的AAAGIGILTV肽脉冲(未显示))。CD8+T细胞的细胞溶解活性的特征是脱粒标志物CD107a和活化标志物CD137与颗粒酶B和IFN-γ分泌的同时上调。(B)由指定的DC条件先前刺激的CD8+T细胞,代表性的点状图显示与加载有MART-1肽的T2细胞共培养后的CD107a/CD137表达。(C)根据先前的DC刺激和T2靶细胞的类型,自体CD8+T细胞(CD107a/CD137表达)的细胞毒活性(重复实验合并的n=4至8个重复样品,具有2个不同的HLA-A2阳性供体)。统计学:双因素方差分析与Tukey多重比较检验。
图9. 8天TNF-α/PGE2/IL-1β/IL-6成熟的moDC和8天TNF-α/PGE2成熟的moDC之间比较的DC表型(n=3),如在不同时间点所确定的。测定中包括“收获时”,“EP后4小时”,“解冻后立即”和“不存在细胞因子后24小时”的任何相关的时间点。(A)收获时单核细胞到DC的转化率(台盼蓝);(B)随时间变化的生存能力(台盼蓝);(C)表型和成熟标志物的细胞表面表达随时间变化的比较,计算为相对MFI(阳性荧光信号与背景荧光的几何平均值之比,在活CD11c
图10.在4天MPLA/IFN-γ和“经典”moDC方案之间比较的冷冻保存之前和之后的T细胞共抑制分子PD-L1的表达水平。表面PD-L1表达的水平计算为相对MFI(阳性荧光信号相对于背景荧光的几何平均值之比,在活CD11c
图11.在电穿孔后4小时(或在非电穿孔条件下进一步温育),以及冷冻和解冻后评估DC成活力。短DC培养:3天GM-CSF/IL-4;24小时MPLA(2,5μg/mL)和IFN-γ(1000U/mL)。在收获时,DC被分为以下电穿孔组:
·无电穿孔(无-EP);
·指数脉冲(EXP-EP):300V;150μF;200μl;Ω;+/-5×10E6 DCs/比色皿;
方波脉冲(SQW-EP):500V;0,5ms;200μl;1个脉冲;+/-5×10E6 DCs/比色皿。eGFPmRNA以0.5μg/10E6细胞使用。
图12.(A)成活力和eGFP表达的流式细胞术分析;(B)冷冻保存的等分试样解冻后的DC的稳定性,如对活DC相对于预冻的成活力和有效恢复所评估的。单核细胞来源的DC是由2个单独的供体白细胞去除术生成的。在收获时,使用具有以下分组的方波脉冲,用0.5μgeGFP mRNA/10E6细胞电穿孔DC:500V;1.0ms;200μl;1个脉冲;50×10E6 DC/比色皿。
图13.单核细胞来源的树突细胞是根据本发明中所述的方案(“MIDRIX DCs”)或Massa等人,2013中所述的alt-2方案(“Massa DCs”)生成的。DC在各个时间点收获,并用编码eGFP的mRNA电穿孔。数据来自6个不同供体。(A)用两种方案获得的活CD11c+HLA-DR+树突细胞在收获时的成活力和绝对细胞产量。(B)单核细胞标志物CD14相对于DC分化标志物CD83的表达。(C)DC成熟标志物CD40,CD70,CD86和CCR7的表达。(D)T细胞共抑制受体PD-L1的表达。(E)电穿孔效率,以翻译蛋白水平(eGFP信号的相对平均荧光强度)以及电穿孔eGFP-mRNA成功翻译的细胞比例(eGFP+DC百分比)表示,如电穿孔后4小时所测量的。
具体实施方式
现在将进一步描述本发明。在以下段落中,更详细地限定本发明的不同方面。除非明确地相反指示,否则如此限定的每个方面可与任何其它方面组合。如说明书和所附权利要求书中所使用的,单数形式“一个”,“一种”和“所述”包括复数对象,除非上下文另外明确指出。举例来说,“化合物”是指一种化合物或多于一种化合物。在本申请的整个说明书和权利要求书中,单词“包含(comprise)”和该单词的其他形式,例如“包含(comprising)”和“包含(comprises)”,是指包括但不限于,并且不旨在排除例如其他添加剂,组件,整数或步骤。上面描述的术语以及说明书中使用的其他术语对于本领域技术人员来说是众所周知的。本说明书中引用的,所有参考文献,和具体涉及的教导,均通过引用以其整体并入本文。
本发明涉及树突细胞疫苗,特别是自体的单核细胞-树突细胞疫苗的制备。特别感兴趣的是,本发明的方法可以使用临床级的完全封闭的系统来进行。透气的培养袋或容器提供封闭流体路径培养系统的优点,由此细胞悬浮液可以经由无菌连接端口添加到培养袋。理想地,整个细胞收集和预选(如果需要)在封闭的流体路径系统中进行,该系统然后无菌地连接到透气袋以将细胞转移到袋中。然后,培养基可以经由无菌连接端口和无菌管道系统通过袋连续灌注,或定期更新。透气袋内的细胞培养物可以保持在培养箱的气体调节的气氛中,而不暴露于环境危害,例如微生物,否则当细胞最初被引入袋或容器时、当更新培养基时或当添加新的培养基时,微生物可能被引入培养物中。在整个培养期间,培养细胞的样品可以通过无菌连接端口从袋中无菌取出以用于分析。同样,当DC培养物准备收获时,可以无菌地取出细胞用于封闭系统洗涤和/或进一步处理。封闭系统还开辟了在具有粉尘计数方面的较低严格性的洁净室环境(例如C级洁净室环境)中进行细胞培养的可能性。这在操作人员的工作条件方面具有优势,降低了成本,并且还提供将细胞分化过程容易地转移到可商购获得的自动化培养系统的可能性(例如,CliniMACS
因此,如本文所用,术语“封闭系统”是指部件的组合,每个部件对周围环境封闭,并且每个部件设置有用于在部件之间实现无菌连接的装置。在一个实施方案中,封闭系统包含白细胞去除术产物,以及本文提供的分化和成熟组分。GMP认证的透气培养袋系统的实例是
在一个实施方案中,本发明的方法包括分离和/或提供树突细胞前体群体的步骤。通常,本文所用的“树突细胞前体”是(人)外周血单核细胞、单核细胞或另一种骨髓祖细胞。如本文所用,“单核细胞”是指具有分化成树突细胞能力的CD14+单核白细胞。单核细胞可以来自任何哺乳动物,但优选是人单核细胞。可以提供单核细胞,并将其在组合物中温育,所述组合物例如但不限于血液、血液级分(例如,白细胞(WBC)、血沉棕黄层、外周血单核细胞(PBMC)、单核细胞白细胞去除术产物,以及在进一步富集单核细胞的组合物中温育。在优选的实施方案中,单核细胞与其他外周血单核细胞(PBMC)一起提供,例如作为单核细胞单采血液成分分离术(apheresis)产物。从包括血液和骨髓在内的各种来源分离富集树突细胞前体如单核细胞和常规树突细胞的细胞群体的方法是本领域已知的。例如,单核细胞和常规树突细胞可以通过收集肝素化的血液、通过单采血液成分分离术或白细胞去除术、通过制备血沉棕黄层、玫瑰花环(rosetting)、离心、密度梯度离心、细胞的差异裂解、过滤、淘析、荧光激活细胞分选或免疫磁性分离来分离。在优选的实施方案中,单核细胞从单核细胞白细胞去除术中分离。白细胞去除术的方法是本领域已知的。白细胞去除术是从受试者的血液中去除白细胞,然后将其剩余部分输回受试者的程序。白细胞去除术产物通常是富含PBMC的血液级分,具有低水平的污染性红细胞、粒细胞和血小板。用于进行白细胞去除术的方法和设备是本领域众所周知的。单核细胞性树突细胞前体和/或分化的常规树突细胞可以从健康受试者或从需要免疫刺激的受试者分离,所述受试者诸如,例如是癌症患者或其他的细胞免疫刺激对其可以是有益的或期望的受试者(即,具有细菌或病毒感染的受试者等)。树突细胞前体和/或未成熟树突细胞也可以从与HLA匹配的健康个体中获得,以施用于需要免疫刺激的与HLA匹配的受试者。
在一个实施方案中,在分化步骤之前富集单核细胞。操作可以对单核细胞或PBMC等进行,包括例如离心、淘析、切向流过滤、Ficoll密度梯度、稀释的Ficoll密度梯度离心、稀释的Percoll密度梯度离心、抗体淘选、磁性细胞分选、阳性或阴性免疫磁性选择等。另外,一旦从受试者分离,单核细胞(例如,纯化的单核细胞,富集的单核细胞,包含单核细胞的PBMC等)可以任选地被温育,例如在1℃-34℃的温度下保持一段时间,例如从他们自受试者分离的时间之后的大约1到96小时。
在特定的实施方案中,单核细胞祖细胞是通过免疫磁分离由白细胞去除术获得的。甚至更特别地,成活的单核细胞DC前体的群体是高度纯化的,例如纯度超过90%,95%甚至99%,如使用单核细胞标志物CD14和成活力染色通过流式细胞仪所确定的。
因此,本文公开的方法的第一步包括提供分离的(自体的)单核DC前体,特别是使用本文提供的封闭系统。通常,在细胞培养开始时培养袋或容器中的DC前体细胞密度为0.5×10E6至2×10E6细胞/ml,优选约1×10E6细胞/ml,如通过本领域已知的方法所确定的。
在分离,纯化和/或富集后,诱导DC前体分化为树突细胞。因此,在进一步的实施方案中,本发明的方法包括培养和/或分化步骤以获得未成熟的DC,例如在至少粒细胞-巨噬细胞集落刺激因子(GM-CSF)和白细胞介素-4(IL-4)(称为分化培养基)存在下培养前体细胞,并且这持续约48至96小时,更特别48至84小时,甚至更特别地长达80、75、74、73、72、71、70小时或更短的小时,更特别地长达至少48小时和长达72小时。+/-4小时或+/-2小时的限度是可接受的,并且鉴于实际约束(constraints)可以是必要的。在具体的实施方案中,将分离的DC前体通过封闭系统转移到包含(无血清)分化培养基的透气培养袋中。
GM-CSF和IL-4的使用浓度为每种细胞因子约100U/ml至5000U/ml,优选500U/ml至2500U/ml,更优选500U/ml至1500U/ml或约500至1000U/ml。特别地,GM-CSF的使用浓度可以是500U/ml至2500U/ml,优选地1000至1500U/ml,并且更优选地约1000U/ml。更具体地,IL-4的使用浓度可以是500U/ml至2500U/ml,优选地500至1500U/ml,更优选地500至1000U/ml,甚至更优选地约500U/ml。
在单核细胞分化成未成熟的树突细胞后,未成熟的树突细胞可以成熟为成熟的树突细胞。因此,在一个实施方案中,本发明的方法包括成熟步骤,例如向(分化的)未成熟的DC中加入干扰素gamma(IFN-g)和单磷酰脂质A(MPLA)(称为成熟刺激物或混合物),并且这个过程至少长达30小时,优选至少24小时。特别地,在收获和/或转染前的最后24小时+/-4小时(特别是+/-2小时)的细胞培养期间,将成熟刺激物IFN-g和MPLA加入到培养基中。
IFN-g的使用浓度为500U/ml至2000U/ml,优选为500U/ml至1500U/ml,甚至更优选为500U/ml至1000U/ml,并且在特定的实施方案中为约1000U/ml。MPLA的使用浓度为1至20μg/ml,更特别地为1至10μg/ml,甚至更特别地为1至5μg/ml。在特定的实施方案中,MPLA以约2.5μg/ml的浓度使用。在进一步的实施方案中,IFN-g是药物级或GMP认证的重组人IFN-g。如本文所用,“药物级”化合物是指已经由公认的国家或地区药典为其建立了化学纯度标准的任何活性或非活性药物,生物制剂或试剂。
因此,根据本发明,前体和/或未成熟树突细胞用(至少)以上因子的组合,即分化和/或成熟因子来培育。这可以通过将所述因子添加到培养基中来进行。备选地,其中已经在其中生长了前体细胞和/或未成熟树突细胞的培养基被已经包含所述因子的培养基代替。在进一步的实施方案中,上述物质被添加或可以是添加到所述细胞的培养基中的组合物的一部分。所述培养基可以是任何合适的种类,即可以补充有或没有任何其他补充剂,例如蛋白质、氨基酸或抗生素。在特定的实施方案中,培养基在GMP条件下生产和使用。甚至更特别地,培养基是无血清的,例如无血清GMP
此外,本发明的目的是提供“加速的”体外细胞分化方法,用于产生具有强Th1极化能力并结合核酸编码抗原的有效呈递的临床级树突细胞(DC)。通常,本发明的DC培养方案的持续时间限于约4天,而不是8天的“标准”方案。
当在一系列不同的供体上进行评估时,与标准方案相比,使用本发明的方法的DC成活力和单核细胞到DC的转化率均显著更高(例如,关于转化率:本发明的方法:约45%,标准方法:约25%)。
从表型上看,通过本文提供的方法获得的细胞展示出树突细胞的基本特征,包括:
-通过光学显微镜评估的典型树突细胞形态,
-DC分化标志物CD11c、II类MHC(HLA-DR)和CD83的均匀表达,
单核细胞标志物CD14的均匀下调。
就DC的成熟状态而言,这通过测量具体细胞表面标志物的表达来评估,其中优选通过流式细胞术分析来分析其中的T细胞共刺激分子。在那种情况下,表达水平以相对平均荧光强度(MFI)(阳性荧光信号相对于背景荧光的几何平均值之比)给出,如通过通常已知的方法所确定的。通常将T细胞共刺激分子评估为成熟标志物,该成熟标志物的DC表面上的表达应尽可能高。相反地,努力使T细胞共抑制分子在最终DC产物上的表达保持尽可能低。
根据本发明的方法获得的DC证实:
-T细胞共刺激分子(CD40,CD70,CD86)的均匀上调,以及
-淋巴组织归巢趋化因子受体CCR7的均匀表达。
与使用“经典”方案生成的DC所显示的相比,CD40、CD70和CCR7的中值水平较高,具有统计学显著性(双尾p值<0.05)。
在本发明的一个实施方案中,将本文生成的DC上的细胞表面标志物表达水平与使用“经典”方法生成的DC上的细胞表面标志物表达水平进行比较,所述“经典”方法使用PGE2和TNF-α作为成熟刺激物,由此在8天后获得成熟树突细胞。
例如,对于T细胞共抑制配体PD-L1,用相对平均荧光强度(relMFI)表示的细胞表面水平(对于所用分析方法的描述,请参见“实施例”下的“材料和方法”部分)低于400、350、320、310、300、250、200,尤其是低于150。此外,在根据本发明生产的DC上,在电穿孔、冷冻保存和细胞解冻之后的表面PD-L1的表达(即,代表产品施用至患者的时间)低于500、470、450,特别是低于400。
因此,在细胞收获时(直接在DC成熟后),通过根据本发明方法生成的DC的PD-L1表达水平比通过使用“经典”方法生成的DC的表达低至少3倍、4倍、5倍、6倍、7倍、8倍、9倍,特别是至少10倍,所述经典方法使用PGE2和TNF-α作为成熟刺激物,由此在8天后获得成熟树突细胞(参见实施例的图10)。因此,在细胞解冻时,通过根据本发明方法生成的DC的PD-L1表达水平比通过使用“经典”方法产生的DC的表达低至少4倍、5倍、6倍、7倍、8倍、9倍,特别是至少10倍,所述经典方法使用PGE2和TNF-α作为成熟刺激物,由此在8天后获得成熟树突细胞(参见实施例的图10)。
从功能上讲,冷冻保存和解冻后,在无细胞因子的培养基中延长的温育期间,细胞保持分泌1型极化细胞因子(IL-12,IFN-g)和吸引Th1、CD8和NK细胞的趋化因子的能力。特别地,与通过上述“经典”方法获得的DC相比,根据本发明的方法生产的DC分泌统计学上显著较高水平的CXCR3配体CXCL9(MIG30)和CXCL10(IP-10),以及CCR5配体CCL3(MIP-1α),CCL4(MIR-1β)和CCL5(RANTES),并且CCL17的水平低至不可检测的水平。在一个实施方案中,细胞因子和趋化因子分泌水平以相对术语表示,即当与“经典”方法生成的DC的resp细胞因子或趋化因子的分泌水平比较时,其中所述经典方法使用PGE2和TNF-α作为成熟刺激物并由此在8天后获得成熟树突细胞。分泌水平可以通过使用标准的蛋白质测量方法来确定,例如ELISA或本文提供的
例如,对于原型的1型T细胞极化和支持NK细胞的趋化因子IL-12,在上述条件下解冻并进一步培养后,树突细胞上清液中释放的水平范围为:
·对于根据本发明的方法产生的DC:50至250pg/ml;特别是60至200pg/ml;更特别地为70至150pg/ml;
·对于根据“标准”8天方案产生的DC:0至35pg/ml但小于50pg/ml。
对于趋化因子CXCL10(在募集1型极化的T细胞和NK细胞方面是重要的),在上述条件下解冻并进一步培养后,树突细胞上清液中释放的水平范围是:
对于根据本发明的方法生成的DC:200至2000pg/ml;特别是250至1800pg/ml;更特别地为280至1600pg/ml;
对于根据“标准”8天方案生产的DC:0至5pg/ml但小于10pg/ml。
因此,对于CXCL10,通过根据本发明方法生成的DC的分泌水平是通过使用“经典”方法产生的DC的分泌水平的至少50倍高。用本发明方法获得的DC的类似优势是用另外的细胞因子和趋化因子观察的,所述细胞因子和趋化因子促进抗癌免疫所需的1型极化炎性反应,其中IFN-g、CCL3、CCL4、CCL5和CXCL9。
相反,对于趋化因子CCL17(涉及调节性T细胞和2型极化的T细胞的募集,两者都对抗癌免疫反应有害),通过根据本发明方法生成的DC的释放是通过使用“经典”方法生成的DC的分泌的至少3倍低。
因此,用本发明的方法获得的DC驱动幼稚T辅助细胞向1型极化特征分化,该极化的特征在于高IFN-γ分泌,如例如主动癌症免疫疗法所需的。此外,所述细胞可呈递来源于转染mRNA的免疫原性表位,并随后驱动自体的肿瘤抗原特异性CD8+T细胞的扩增,所述CD8+T细胞表达IFN-γ和细胞毒性分子粒酶B,如已经提到的。
在进一步的实施方案中,本发明的方法包括用抗原编码核酸,特别是RNA,更特别是mRNA加载或转染成熟的DC。如本文所用,“抗原”不限于本发明。在一个实施方案中,抗原选自由以下各项组成的组:肿瘤抗原,肿瘤相关抗原,睾丸癌抗原,突变组来源的抗原,(致癌的)病毒抗原,细菌抗原,酵母抗原,寄生虫抗原和真菌抗原。抗原对受试者可以是自体的,并且可以用于制备加载有抗原的自体DC疫苗以施用于受试者。对受试者自体是指抗原(或其序列)获自或源自相同受试者。作为非限制性实例,抗原可以来自从受试者获得的癌细胞或肿瘤组织。癌抗原可以作为癌细胞、癌细胞或组织裂解物、癌细胞或组织提取物、癌细胞或组织的纯化或克隆组分、总RNA或总mRNA、或来自这些细胞或组织的选定RNA或mRNA(无论是存在于提取物中的、纯化的、扩增的、体外翻译的等)加载到树突细胞中。备选地,抗原可以获自或源自受试者中存在的病原体或感染病原体的细胞。术语“核酸”是指单链,双链和三螺旋分子,基因或基因片段,外显子,内含子,mRNA,tRNA,rRNA,核酶,cDNA,重组多核苷酸,分支多核苷酸,质粒,载体,任何序列的分离的DNA,任何序列的分离的RNA,核酸探针和引物。更具体地,将树突细胞在体外用一种或更多种编码抗原的mRNA转染。任选地并且在替代实施方案中,在成熟期完成之后,DC可以在进一步处理(例如转染)之前首先收获,由此收集、离心细胞和/或洗出细胞因子。
考虑到加速培养方案,在单核细胞分离或向前体DC中加入分化刺激物后,成熟的DC的转染可能在约72至96小时,特别是86小时+/-4小时后。
在本发明的上下文中,转染方法包括但不限于电穿孔、光穿孔、脂转染、病毒载体系统、裸核酸的温育或DC与感染细胞或肿瘤细胞的融合。这些标准方法是本领域公知的,并且是可行的,并且将核酸,例如抗原编码质粒、它们的RNA或DNA,引入DC中。也可能存在与原始MHC分子的其它抗原组合,例如可想到的用作任何种类的抗原来源的膜片段或外来体。在具体的实施方案中,通过电穿孔转染成熟的DC。三种不同类型的脉冲可用于电穿孔,所述三种不同类型的脉冲为例如指数衰减脉冲,方波脉冲和时间常数。在本发明的特定实施方案中,电穿孔由方波脉冲组成。通常,诱导1或2个脉冲以完成转染。
在本发明的一个实施方案中,通过用核酸,优选mRNA对树突细胞进行电穿孔来加载抗原。优选地,树突细胞是用每10E6树突细胞约0.25至4μg RNA转染的,最优选地是用每10E6树突细胞约1至3μg RNA转染的。在一个实施方案中,每次转染使用每百万DC 1-2μg抗原RNA。
本文论证了通过本发明的方法获得的细胞均匀且稳定地表达源自转染的mRNA的蛋白质(参见实施例)。此外,所述细胞可呈递来源于转染的mRNA的免疫原性表位,并随后驱动具有细胞毒性特征的自体肿瘤抗原特异性CD8+T细胞的扩增,如例如主动癌症免疫疗法所需的。
在本发明的上下文中,已经发现,与通过8天的“经典”方案制备的mDC相比,在缩短的温育时间(例如约3天内)下,如本文所提供的未成熟树突细胞的刺激可生成具有改善的成活力,功能和/或免疫刺激活性的成熟树突细胞。
如本文所用,术语“免疫刺激活性”是指成熟树突细胞或成熟树突细胞群体产生和/或分泌足够量的特定细胞因子和趋化因子(特别是IL-12和CXCL10)的能力,所述细胞因子和趋化因子介导1型-极化效应T细胞和NK细胞的正确分化和动员,如针对癌症和特定病原体的免疫所需的。
在本发明的一个实施方案中,可以将加载/转染的树突细胞冷冻在包含冷冻保护剂的组合物中。许多冷冻保护剂和用于冷冻DC的方法是本领域技术人员已知的。例如,使用可控速率的冷冻机将树突细胞冷却,并转移至液氮容器的气相中用于冷冻保存和存储。特别地,将树突细胞以20-70×10E6活细胞/mL,更具体地约40-60×10E6活细胞/mL,甚至更具体地约50×10E6活细胞/mL以100μL的体积等分试样重悬浮于合适的冷冻保存介质中。在进一步的步骤中,解冻的树突细胞疫苗准备好在解冻后的任何时间(通常多达约4小时)施用于受试者。
在一个实施方案中,本发明提供冷冻小瓶,其包含如本文提供的冷冻保存的,成熟的,转染的,特别是电穿孔的DC,特别是以每100μL约5×10E6个细胞的量,如在冷冻保存之前所测量的。
本发明进一步提供用于加载有抗原的树突细胞疫苗的施用方法,该方法包括解冻根据本文提供的方法制备的冷冻保存的活树突细胞并将其施用于受试者。
本发明还提供通过本文公开的方法获得的加载有抗原的树突细胞作为药物的用途,特别是用于制备药物或药物组合物的用途。本发明提供本文所述的DC或组合物,其用于免疫疗法,特别是用于治疗或预防癌症或病原体感染。
在进一步的方面,本发明涵盖药物组合物,其包含根据本发明的成熟树突细胞和药学上可接受的载体和/或赋形剂。此外,本发明还涉及本发明的成熟树突细胞或成熟树突细胞群体,其用于治疗选自由以下各项组成的组的疾病的方法中:恶性病症(癌症)、特定非恶性病症(例如LAM肺病(淋巴管肌瘤病)、和感染性疾病(例如由病毒、细菌、胞内细菌或真菌引起的)。此外,本发明涉及用于治疗患有肿瘤疾病(例如癌症)或感染性疾病的患者的方法,其中将有效量的本发明的成熟树突细胞施用至所述患者。
本发明的加载有抗原的树突细胞可作为疫苗用于治疗或预防疾病或用于T细胞的活化。例如,加载有抗原的树突细胞可用于引发针对抗原的免疫应答。它们可以用作疫苗,以预防未来的感染或疾病(“预防性疫苗接种”),或激活免疫系统以治疗正在进行的疾病(“治疗性疫苗接种”),例如但不限于病原体感染或癌症。如本文制备的加载有抗原的树突细胞可以被配制以用作具有合适的载体例如生理缓冲液或其他可注射液体的疫苗或药物组合物。疫苗或药物组合物以足以引起免疫反应的治疗有效量施用。
本文所用的术语“治疗(treatment)”和“治疗(treating)”通常是指获得所需的药理和/或生理效果,并且涵盖哺乳动物尤其是人中的任何疾病治疗,包括:
(1)防止疾病或症状发生于可能易患该疾病或症状但尚未被诊断为患有该疾病或症状的受试者中;
(2)抑制疾病症状,即阻止其发展;或
(3)减轻疾病症状,即引起疾病或症状的消退。
就完全或部分预防疾病或其症状而言,该效果可以是预防性的,和/或就部分或完全稳定或治愈疾病和/或归因于该疾病的副作用而言,该效果可以是治疗性的。此外,除了主要疗法或初始疗法外,还可将疫苗用作“辅助疗法”以使其在治疗环境中的有效性最大化,或在初始疗法后用作“维持”或“巩固(consolidative)”疗法以使疾病控制最大化并延迟疾病复发。
在本发明的上下文中,术语“癌症”是指由恶性肿瘤引起的任何种类的疾病。如本文所用,术语“感染性疾病”是指由病原微生物剂(包括病原病毒,病原细菌,真菌,原生动物或多细胞寄生虫)的存在引起的任何种类的临床上明显的疾病。
用于配制树突细胞疫苗的方法是本领域技术人员已知的。用于施用的合适的制剂可以包括水性等渗无菌注射溶液,该溶液可以含有抗氧化剂,缓冲液,抑菌剂和使制剂与预期接受者血液等渗的溶质,以及水性和非水性无菌悬浮液,其中所述悬浮液可以包括悬浮剂,增溶剂,增稠剂,稳定剂,防腐剂,免疫刺激剂,细胞因子和佐剂。
树突细胞组合物/疫苗可以通过多种方法施用,例如但不限于注射(例如皮下,皮内,静脉内,淋巴管内,关节内,肌肉内,腹膜内),通过连续输注,从植入物中缓释等。DC疫苗能够以特定的间隔施用。在一个实施方案中,以2至4周的间隔,特别是两周的间隔施用DC。树突细胞疫苗能够与生理上可接受的载体,缓冲液,稀释剂,佐剂,免疫调节剂等一起施用。优选地,树突细胞疫苗对于其所施用的患者是自体的,或者是最大程度的HLA匹配的。
向受试者施用的细胞剂量为有效量,可有朝一日在受试者中有效地实现所需的有益治疗反应,或抑制癌细胞的生长,或抑制感染,同时保持良好的耐受性特征(最小毒性)。足以完成此效果的量定义为“治疗有效剂量”。该剂量将由所产生的树突细胞的生物学和/或临床活性以及任选地患者的病况决定。该剂量的大小也将由在特定患者中施用特定细胞所伴随的任何不良副作用的存在,性质和程度来确定。在确定治疗或预防疾病诸如癌症(例如,转移性黑色素瘤,前列腺癌等)的细胞有效量时,医师(或研究者)需要评价针对包含在疫苗中的靶标的免疫反应(即免疫监测),以及使用可测量参数(通过常规或与免疫相关的RECIST标准的放射学肿瘤负荷,肿瘤标志物,循环肿瘤细胞,血浆循环肿瘤DNA,或疾病负荷或疾病活性的其他替代标志物)评价肿瘤临床进展。
本领域技术人员众所周知,没有证据表明待施用的DC的优选剂量实现特定水平的生物学和/或临床效果。同样,没有观察到明确的剂量限制毒性(DLT),因此也没有观察到最大耐受剂量(MTD)。最经常施用的剂量取决于从一轮白细胞去除术获得的DC的产量和后续疫苗接种的所需次数。在一个实施方案中,剂量落入每轮疫苗接种5-100×10E6 DC之内,重复2至8次,特别是2至6次,更特别是2至4次。同样地,注射的细胞数量与毒性之间也没有关系。DC疫苗接种的毒性通常较低,并且与施用途径有关(与皮内途径相比,静脉内途径的急性副作用更大)。注射可以是例如以1、2或3周的间隔重复2、3、4、5或6次,并且应通过以下方式给予:静脉内或在淋巴结附近通过皮内或皮下注射或直接注射到淋巴结中。可以在暂停例如1到若干个月之后进行加强注射。
任选地添加生物反应调节剂,以通过本发明的DC或活化的T细胞进行治疗。例如,任选地细胞与佐剂或诸如GM-CSF,IL-12,IFN-α或IL-2的细胞因子一起施用。
本文描述的所有特征(包括任何所附权利要求,摘要和附图)和/或如此公开的任何方法或过程的所有步骤,可以以任何组合与上述任何方面进行组合,除非存在至少一些此类特征和/或步骤互斥的组合。
通过以下附图,表格和实施例将进一步描述本发明,这些附图,表格和实施例不旨在限制权利要求中限定的保护范围。实施例中描述的方法和实验主要涉及使用匿名供体血沉棕黄层作为起始材料的临床前开发。
单核细胞来源的树突细胞培养
从地方输血中心获得血沉棕黄层,并通过Ficoll-paque密度梯度离心(GEHealthcare Life Science,Chicago,Illinois,USA)分离外周血单核细胞(PBMC)。根据制造商的规程,使用人抗CD14免疫磁性微珠(Miltenyi Biotec,Bergisch Gladbach,德国)将单核细胞进行免疫磁纯化。如通过流式细胞术所评估的,始终获得>90%的纯度(数据未显示)。
将单核细胞耗尽的级分(外周血淋巴细胞(PBL))在RPMI-GlutaMAX培养基(Invitrogen by Life Technologies,California,USA)中冷冻,所述培养基具有10%胎牛血清(FBS)(Sigma-Aldrich,Missouri,USA),100U/ml青霉素/链霉素(P/S)(Gibco,LifeTechnologies,California,USA)和10%二甲基亚砜(DMSO)(Sigma-Aldrich,Missouri,USA)。
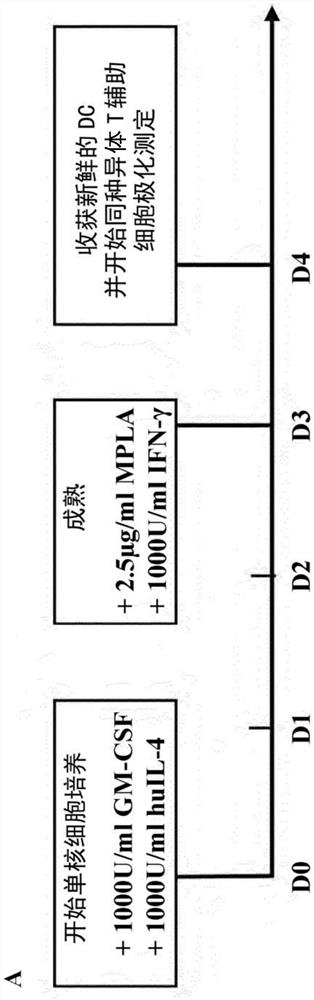
对于我们加快的(即4天)DC培养方案,将单核细胞在无血清GMP CellGro(CG)培养基(CellGenix GmBH,Freiburg,Germany)中以2×10E6细胞/ml的密度培养在30ml GMP细胞分化袋(Miltenyi Biotec,Bergisch Gladbach,Germany)中,其中所述培养基含有1000U/ml药物级粒细胞巨噬细胞集落刺激因子(GM-CSF)(Leukine(Berlex),Bayer HealthcarePharmaceuticals,New Jersey,USA),1000U/ml GMP认证的重组人白细胞介素4(huIL-4)(Miltenyi Biotec,Bergisch Gladbach,德国)和100U/ml P/S(Gibco,LifeTechnologies,California,USA)。在第3天,将2.5μg/ml合成的MPLA(Invivogen,California,USA)和1000U/ml药物级IFN-γ(Immukine,Boehringer Ingelheim BV,Ingelheim,德国)添加到培养基中在持续另外的24小时。第4天收获成熟的DC(mDC)。
对于“经典”(8天)方案,将单核细胞在聚苯乙烯培养瓶(Nunc,Thermo FisherScientific,Massachusetts,USA)中以1×10E6的密度在相同的完全培养基中培养,不同之处是重组huIL-4(250U/ml;Miltenyi Biotec,Bergisch Gladbach,Germany)的浓度较低,并加入1%合并的人AB血清(huAB血清)(Sigma-Aldrich,Missouri,USA)。在第3或4天,加入新鲜的含有GM-CSF和IL-4的培养基。在第6天,将20ng/ml重组人TNF-α(Miltenyi Biotec,Bergisch Gladbach,Germany)和2.5μg/ml药物级PGE2(Prostin E2,Pfizer,纽约,美国)添加到培养基中,额外持续48小时。第8天收获成熟的DC。
DC表型分析
对于表面染色,首先将细胞洗涤,然后重悬于磷酸盐缓冲盐水(PBS)(Invitrogenby Life Technologies,California,USA)中,然后与FcR阻断剂(Miltenyi Biotec,Bergisch Gladbach,Germany)和可固定成活力染料eFIuor 506(eBioscience by ThermoFisher Scientific,Massachusetts,USA)的组合在4℃温育20分钟,以对死细胞进行染色。
接下来,用FACS缓冲液洗涤细胞,该缓冲液由以下组成:补充有0.5mM乙二胺四乙酸(EDTA)的PBS(Invitrogen by Life Technologies,California,USA);0.25%牛血清白蛋白(BSA);和0.05%NaN3(全部来自Sigma-Aldrich,Missouri,USA),然后在4℃下添加表面抗体(Ab)30分钟。使用了以下荧光染料缀合的单克隆抗体:抗CD40 FITC;抗HLA-ABCFITC;抗CCR7 APC;抗CD1 1c Alexa Fluor 700;抗HLA-DR APC-Cy7(eBioscience byThermo Fisher Scientific,Massachusetts,USA);抗HLA-A2 FITC;抗DNGR-1PE;抗CD86PE德克萨斯红;抗CD83 PE-Cy7;抗PD-L1太平洋蓝(BD Biosciences,New Jersey,USA);抗CD70 PE;和抗CD14太平洋蓝(Miltenyi Biotec,Bergisch Gladbach,Germany)。
在LSR Fortessa分析型流式细胞仪(BD Biosciences,美国新泽西州)上采集样品,并使用FlowJo软件(版本9.9.4;BD Biosciences,美国新泽西州)进行分析。表型和成熟标志物的表达水平显示为相对平均荧光强度(MFI)(阳性荧光信号相对于背景荧光的几何平均值之比,在活CD11c
Luminex测定
将4天和8天单核细胞来源的DC(moDC)的冷冻保存的等分试样解冻,并在补充有100U/ml P/S(Gibco,Life Technologies,California,USA)的无血清和无细胞因子的CG培养基(CellGenix GmBH,Freiburg,Germany)中培养。收集DC培养上清液,并使用Luminex测定法(R&D Systems,Minneapolis,美国)进行分析,并对其进行定制以包括以下人细胞因子和趋化因子:IL-12p70;IFN-γ;IL-10;CCL3;CCL4;CCL5;CXCL9;CXCL10;CCL17;CCL20;和CXCL12。Luminex测定是在Bio-Plex(Bio-Rad,California,USA)读数器上分析的。
DC的mRNA电穿孔
收获后,分别在第4天或第8天,将DC电穿孔,然后冷冻保存在Plasma-Lyte A(Baxter,Illinois,USA)中,所述Plasma-Lyte A富含3.5%人血清白蛋白(Sanquin,Amsterdam,The Netherlands);6.25%羟乙基淀粉(HES)(Grifols,Barcelona,Spain);和6.25%DMSO(Sigma-Aldrich,Missouri,USA)。eGFP mRNA源自pST1-eGFP2质粒,该质粒由布鲁塞尔自由大学(Free University of Brussels)分子与细胞治疗实验室(Laboratory ofMolecular and Cellular Therapy)(LMCT)K.Thielemans教授惠赠。首先使用Sapl限制性内切酶(New England Biolab,Massachusetts,USA)将质粒线性化,然后使用mMESSAGEmMACHINE T17Ultra试剂盒(Ambion by Thermo Fisher Scientific,Massachusetts,USA)将其体外转录为mRNA。MART-1mRNA也由LMCT捐赠。如Bonehill等人(2004年)所述,将MART-1的开放阅读框与溶酶体蛋白DC-LAMP1的HLA II类靶向序列融合。将4至16×10E6个DC重悬于170μl无血清CG培养基(CellGenix GmBH,Freiburg,Germany)中,所述培养基补充有1μgmRNA/10E6 DC的剂量的溶解于无核酸酶水中的30μl mRNA(Applied Biosystems by LifeTechnologies,California,USA),并转移至4mm缺口的比色杯中(Bio-Rad,California,USA)。使用指数波脉冲的电穿孔是使用Gene Pulser Xcell电穿孔系统(Bio-Rad,California,USA)用以下参数进行的:电容150μF;电压300V;电阻。EP后,立即将DC在超低附着平板(Corning,New York,USA)上于37℃和5%CO
同种异体T辅助细胞极化测定
将电穿孔的和冷冻保存的DC解冻,使其在温暖RPMI-GlutaMAX培养基(Invitrogenby Life Technologies,California,USA)中于37℃和5%CO
同种异体共培养结束时,加入50ng/ml佛波醇12-肉豆蔻酸酯13醋酸盐(PMA);1μg/ml离子霉素(iono)和10μg/ml的布雷菲德菌素A(BFA)(均来自Sigma-Aldrich,Missouri,USA)在37℃和5%CO
抗原特异性自体CTL的扩增
来自HLA-A2+供体的血沉棕黄层用于生成4天MPLA/IFN-γ成熟的DC,将其收获后4小时冷冻(即非 EP Dc),或者在冷冻保存前先用任何一种载体(即eGFP mRNA-EP DC)或抗原MART-1 mRNA(即MART-1 mRNA-EP DC)电穿孔。有关DC培养和操作的更多详细信息,请参阅上述材料和方法部分“单核细胞来源的树突细胞培养”和“DC的mRNA电穿孔”。
解冻后,将非EP DC;eGFP mRNA-EP DC;和MART-1 mRNA-EP DC,在RPMI-GlutaMAX培养基(Invitrogen by Life Technologies,California,USA)中于37℃和5%CO
使用阳性免疫磁性选择试剂盒(Miltenyi Biotec,Bergisch Gladbach,Germany)从冷冻保存的自体CD14阴性级分中纯化CD8+T细胞。将DC和T细胞在RPMI-GlutaMAX培养基(Invitrogen by Life Technologies,Califotnia,USA)中以1∶10比率共培养14天,所述培养基补充有10%huAB血清(Invitrogen by Life Technologies,California,USA)和100U/ml P/S(Gibco,Life Technologies,California,USA)。在第3天和第10天加入20ng/ml重组人IL-2(R&D Systems,Minneapolis,USA)。包括具有自体CD8+T细胞不具有DC的培养孔作为附加对照。在共培养的第7天,用相应的DC(即MOCK-脉冲的DC,eGFP mRNA-EP DC,MART-1mRNADC和MART-1肽脉冲的DC)再次刺激自体CD8+T细胞。在共培养结束时,如上所述,将细胞与PMA/iono/brefA温育5小时并收获,用于使用PE缀合的A*02:01/人MART-1 MHC四聚体(Sanquin,Amsterdam,The Netherlands)的表面染色,以及使用以下标志物:抗IFN-γFITC(BioLegend,California,USA);和抗粒酶B太平洋蓝(BD Biosciences,New Jersey,USA)的细胞内染色。
DC诱导的抗原特异性细胞溶解活性的评价
如上所述,在自体DC:T细胞共培养设置的第14天收获效应T细胞。靶细胞由TAP2缺陷的T2细胞组成,该细胞加载有如上所述的来自MART-1的相同肽(Genscript,New Jersey,USA),或具有序列GILGFVFTL的来自流感基质蛋白的无关A2限制性肽(AnaSpec,California,USA;SEQ ID NO 2)作为对照,两者均以10μg/ml的量使用。将T2细胞脉冲3小时,并彻底洗涤以除去未结合的肽。在莫能菌素(Golgistop,BD Biosciences,New Jersey,USA)和抗CD107a太平洋蓝抗体(Miltenyi Biotec,Bergisch Gladbach,Germany)的存在下,以10∶1的E∶T比例共培养14小时。共培养结束时,将细胞用表面抗CD3,抗CD8和抗CD137(eBioscience,Thermo Fisher Scientific,Massachusetts,USA)染色。
统计
使用GraphPad Prism(7.02版,GraphPad Software,California,USA)进行统计分析。正态分布首先使用D'Agostino-Pearson综合正态检验进行测试。使用针对2组的未配对或配对t检验或针对3组或更多组的ANOVA检验与Tukey多重比较检验相结合来分析正态分布的数据。对于非正态分布的数据,使用非参数检验,即Mann-Witney检验用于不配对的数据集,Wilcoxon匹配对符号秩检验用于两组的配对数据集。对于超过2组,将非参数Kruskal-Wallis检验与Dunn多重比较检验结合使用。统计显著性水平用星号符号编码如下:p值0.01-0.05(*),p值0.001-0.01(**),p值<0.001(***)和p值<0.0001(****)。
可以通过缩短单核细胞培养方案(包括用TLR4-配体和IFN-γ成熟)来获得高产量的全分化成熟树突细胞
通过使用大规模的一系列从血沉棕黄层开始的小规模培养,评估了通过组合大大减少的单核细胞培养持续时间以及使用已建立的1型极化因子组合进行成熟来生成DC的可行性。选择了细胞培养基,细胞因子和封闭系统容器,以便直接翻译到我们的GMP生产环境中。
为了减少操作员干预的需要,我们的目标是将标准的8天DC培养持续时间缩短为4天的总时间。这由以下组成:在补充GM-CSF/IL-4的符合GMP的无血清培养基中培养3天,然后在收获前暴露于MPLA和IFN-γ的组合中持续另外24小时。该方案产生了CD11c
细胞的表型与完全分化的成熟的DC的表型一致,具有单核细胞标志物CD14的深度下调,同时CD83的上调与T细胞共刺激标志物CD40,CD70和CD86的高表面表达以及高水平的HLA I类和II类抗原呈递分子。此外,可以以高水平检测到分子DNGR-1的观察结果表明了捕获并交叉呈递外源细胞结合抗原的潜力。CCR7在成熟的DC上被诱导,表明迁移至次级淋巴器官的能力。T细胞检查点分子PD-L1也被上调,反映moDC的整体活化状态(图1D)。
然后,在与疫苗生产相关的几个关键参数的方面,我们将这种4天的moDC分化方案与已建立的“经典”临床级8天DC培养进行了比较。8天moDC在补充有GM-CSF/IL-4的培养基中生成,并通过添加TNF-α和PGE2使其在最后两天成熟。尽管由Jonuleit等人(1997年)首次描述的原始成熟混合物由TNF-α,PGE2,IL-1β和IL-6组成,但我们和其他人已经观察到IL-1β和IL-6的缺失对由此产生的DC的成活力,分化和成熟度没有不利影响(图9),对DC功能也没有负面影响(Van Driessche等人2009年)。
首先,我们始终观察到,收获时,4天moDC比8天MODC的成活力明显更高(p值0.0010),4天moDC的中值成活力(流式细胞仪)为96.3%[95%Cl:92.7-98],而8天MODC的中值成活力为58%[95%Cl:45.1-69.1]。4天moDC还产生最高中值单核细胞到DC转化率(46.9%[95%Cl:27.2-63.2]vs 26.8%[95%Cl:14.1-36.2]),达到统计学显著性(p值0.0195)(图2A)。
接下来,我们看了收获时表型特征的差异。4天moDC比标准的8天moDC显示出显著更高的CD40、CD70和HLA-ABC水平(MFI)。出乎意料的是,尽管没有暴露于PGE2,CCR7在MPLA/IFN-γ成熟的4天moDC上也以更高的水平表达。相比之下,CD86的表达在8天moDC中更高(图2B和表1)。在两种DC培养方案中,CD83,HLA-DR和DNGR-1的表达均不显示统计学显著差异。出乎意料的是,在标准的8天moDC中,PD-L1的表达始终比在4天moDC中的更高(平均四倍),并且在冷冻保存的DC等分试样解冻后甚至进一步增加(图10)。
表1.
*背景信号:活的CD11c
从这些数据,我们总结出将单核细胞培养持续时间减少一半并结合活化因子MPLA和IFN-γ产生完全分化的成熟的DC,具有更高的转化产量,更高的细胞成活力,并且对共刺激分子表达水平没有有害影响。
据我们所知,仅有一份报道描述了在具有仅24-36小时的单核细胞至DC分化时期的加速DC分化方案中MPLA+IFN-γ作为成熟混合物的整合(Massa等人,2013)。然而,对于所述替代DC,并没有评估DC疫苗活性的结果。作为比较,单核细胞来源的树突细胞是根据本文之前所述的方案(“MIDRIX DC”)或根据Massa等人描述的alt-2方案(“Massa DC”)生成的。如前所述,从血沉棕黄层中分离CD14+单核细胞。在各个时间点收获DC,并用编码eGFP的mRNA电穿孔。数据源自6个不同的供体,并评价了以下方面:
(A)用两种方法获得的活CD11c+HLA-DR+树突细胞在收获时的成活力和绝对细胞产量。
(B)单核细胞标志物CD14相对于DC分化标志物CD83的表达;
(C)DC成熟标志物CD40、CD70、CD86和CCR7的表达;
(D)T细胞共抑制受体PD-L1的表达;
(E)电穿孔效率,表示为被翻译的蛋白的水平(eGFP信号的相对平均荧光强度)以及经电穿孔的eGFP-mRNA的成功翻译的细胞比例(eGFP+DC百分比),如电穿孔后4小时所测量的。
如图13所示,分化步骤持续24到36小时的时间不足以实现单核细胞向DC的充分分化,并产生与T细胞刺激能力相关的成熟表型特征。重要的是,与本发明的方法相比,使用Massa等人描述的方案的成活的DC的绝对产量显著更低,这被认为显著削弱了用电穿孔和冷冻保存进一步处理这些细胞的可能性,从而对DC疫苗产生了影响。“Massa DC”不易被编码全长蛋白的mRNA电穿孔,而根据本发明产生的DC显示出高的电穿孔效率。
另外,“Massa DC”显示出单核细胞标志物CD14的更少的下调,DC分化标志物CD83的更少的上调以及DC成熟/T细胞共刺激受体CD40、CD70和CD86的更低的水平。迁移到淋巴组织的T细胞区所需的CCR7水平在D1 DC上也较少上调。更多地,PD-L1水平显示出在“MassaDC”上更高表达的趋势。
对于赋予短期培养的DC完全成熟的表型和诱导从头T辅助细胞1极化的能力,MPLA和IFN-γ都是必要的
接下来,我们剖析了MPLA、IFN-γ或它们的组合对表型成熟状态的相对贡献,以及就4天培养的moDC的T辅助细胞极化能力而言对功能影响的相对贡献。
我们发现,两个成熟刺激都是需要的以使T细胞共刺激分子CD40、CD70、CD86以及CD83和CCR7的表面表达水平最大化(图3)。关于HLA-DR或DNGR-1的表达,未观察到这种作用,后者相对于未成熟的DC保持稳定。值得注意的是,观察到moDC上的PD-L1诱导主要是由MPLA而不是IFN-γ暴露引起的。
在功能水平上,仅通过将DC事先暴露于MPLA和IFN-γ才能实现幼稚同种异体CD4+T细胞的IFN-γ分泌的最大诱导。幼稚的T辅助细胞中不成熟的DC诱导了有限量的IL-10产生,在预先暴露于MPLA的DC的存在下这可被进一步抑制,无论之前是否有IFN-γ暴露。T辅助细胞IL-4的产生仅在低水平下诱导,如IL-17一样,IL-17在MPLA/IFN-γ成熟的DC存在下显示出少量增加(图4)。
因此,将短期分化的moDCs暴露于MPLA和IFN-γ两者是必要的,以获得完全成熟的表型,并使这些细胞具有诱导稳健的从头1-型极化的T辅助细胞反应的能力。
短期培养的DC表现出对电穿孔的优异弹性以及高mRNA翻译效率
除了表型成熟和1型免疫极化潜力外,应在电穿孔和冷冻保存的应激后恢复足够的DC量,以便在临床实践中实施。
我们首先评估了MPLA/IFN-γ成熟的、短期培养的DC成功且稳定地表达源自电穿孔抗原编码mRNA的蛋白抗原的能力。使用编码eGFP的mRNA作为电穿孔效率的标志物,我们在电穿孔后4小时/冷冻保存前,细胞解冻后立即和细胞解冻后24小时(在无细胞因子的培养基中进一步温育)观察了eGFP的表达。
通过指数脉冲电穿孔的eGFP阳性DC的中值百分比从冷冻保存前的64.8[95%Cl:55.2-87.7]演变为解冻后立即为80.2[95%Cl:73.1-87.7],并在接下来的24小时内保持稳定(86.3[95%Cl:75.2-86.4),在该时间段内表达强度(MFI)没有明显变化(图5A,B)。
指数脉冲电穿孔导致在4天moDC中的成活力(台盼蓝)平均降低17.3%。结合电穿孔诱导的净细胞损失,这转化为51.4%[95%Cl:36-67%]的活DC恢复中值百分比(电穿孔后相对于电穿孔前的活细胞恢复)(图5C)。使用单独的一系列供体,我们比较了4天MPLA/IFN-γmoDC与标准8天moDC的电穿孔弹性。我们观察到,EP后4天DC的成活力(台盼蓝)比8天DC显著提高,中值成活力分别为67.3%[95%Cl:18.2-93.5]和16.5%[95%Cl:2.8-57.8]。eGFP mRNA EP后,8天moDC也更容易受到净细胞丢失的影响,平均活细胞恢复率为24.6%[95%Cl:3.7-47.5],而4天moDC的平均活细胞恢复率为41.5%[95%Cl:12.8-83.8](图5D)。
在进一步的测试中,我们评估了方波脉冲电穿孔后的结果。使用小规模的运行,使用在一系列细胞因子浓度产生的DC,我们发现,与指数脉冲程序相比,使用方波脉冲的电穿孔后和冷冻保存后的细胞成活力始终较高(图11)。
方波脉冲的进一步评估是在GMP环境下的全面DC生产轮次上进行的。就%eGFP阳性细胞而言,与指数脉冲相比,使用方波脉冲电穿孔的DC的eGFP表达的流式细胞术分析是不劣的(代表性数据如图12A所示)。通过方波脉冲的电穿孔可导致在解冻后立即>80%的DC恢复,冷冻保存的DC具有>75%的成活力(图12B)。
方波脉冲电穿孔后未观察到肉眼可见的细胞聚集体的形成,这极大地促进了进一步的细胞处理并改善了整体细胞的恢复率(结果未显示)。
电穿孔和冷冻保存不会损害短期培养的DC选择性促进1型极化的T细胞应答的能力
在电穿孔和冷冻保存应激之后,应保持完整的关键DC特性是当施用于患者时选择性动员1型极化和细胞溶解性T细胞的潜力。为了提供该功能的评估,在无细胞因子培养基中温育24小时后,我们分析了电穿孔的和冷冻保存的4天moDC与标准8天DC的细胞因子和趋化因子分泌组(图6A)。我们发现4天moDC仍然能够分泌生物活性的IL-12和IFN-γ,而通过8天的moDC分泌的这些细胞因子的产量却低于检测限。在两种DC类型之间未观察到IL-10产量的差异。更令人惊讶的是,我们发现只有MPLA/IFN-γ成熟的4天moDC才能产生大量的趋化因子,这些因子参与吸引1型极化的T辅助细胞,细胞溶解性T细胞和NK细胞(Colantonio等,2002),从标准的8天MoDC没有可检测到的分泌。这包括高水平的CXCR3配体CXCL9(MIG30)和CXCL10(IP-10)(Groom等2011),以及CCR5配体CCL3(MIP-1α),CCL4(MIR-1β)和CCL5(RANTES)(Samson等,1997)。CXCR3配体CXCL11(Groom等2011)的分泌低于检测限。相反,在标准的8天moDC中,T-reg和Th2动员的趋化因子CCL17(TARC)的分泌(Yoshie等人,2015)是五倍高。存在着通过4天moDCs高释放Th17和T-reg吸引的趋化因子CCL20(Yamazaki等人2008)的趋势,而T-reg吸引的CXCR4配体CXCL12(SDF-1α)(Colantonio等人2002)的产量在两种DC培养方案之间没有差异(数据未显示)。
我们还研究了电穿孔和冷冻保存是否影响4天moDC诱导从头T辅助细胞1极化反应的能力(图6C)。同种异体幼稚CD4 T细胞与解冻的4天moDC的共培养可导致高的IFN-γ产量水平,该水平可与与新鲜收获的未经电穿孔的4天moDC的共培养所获得的水平相当(图4)。在这种情况下,IL-10产量的诱导非常低(图6C),与用新鲜DC获得结果一致(图4)。
短期培养的DC可有效引发和扩增具有细胞溶解活性的肿瘤抗原特异性CD8+T细胞
在电穿孔/低温保存后的产量、表型、恢复以及促进1型极化的T细胞应答的能力方面建立了短期培养的moDC的优越性,接下来我们测试了这些细胞呈递来自电穿孔的肿瘤抗原编码mRNA的免疫原性表位的能力。同样,为了反映DC疫苗在现实生活中的临床设置的实现,我们用冷冻保存的而不是新鲜的mRNA-EP DC进行了所有测定。考虑到在HLA-A2阳性健康供体中使用四聚体检测MART-1特异性CD8+T细胞的可能性,将MART-1/Melan-A用作模型肿瘤相关抗原。
我们观察到,与用加载有不相关抗原(eGFP)的DC刺激相比,用MART-1-mRNA-EP DC进行的总共的每两周一次的刺激轮次足以诱导抗原特异性(四聚体阳性)CD8+T细胞扩增30倍以上(中值为0.43%[95%Cl:0.22-0.53])vs 13.2%[95%Cl:1.21-37.6])。无论用MART-1 mRNA还是eGFP mRNA电穿孔4天moDC,在电穿孔后的成活力和恢复率方面均未观察到差异(数据未显示)。与使用MART-1肽脉冲的DC(阳性对照)获得的相比,MART-1特异性CD8+T细胞的扩增处于相同的数量级(中值为18.9%[95%Cl:5.75-28.8])。这些结果表明,MPLA/IFN-γ成熟的4天moDC能够从电穿孔的编码MARTI的mRNA中提取免疫原性表位,以有效地呈递给Ag特异性的自体CD8+T细胞(图7A-B)。
为了评估受刺激的CD8+T细胞的效应潜能,对于IFN-γ和粒酶B,我们将四聚体检测与IC染色相结合。与阴性对照条件(即用MOCK脉冲或eGFP mRNA-EP-DC刺激,或不使用DC刺激)相比,我们发现MART-1-mRNA-EP 4天moDCs诱导了最高数量的产生IFN-γ和粒酶B的抗原特异性CD8+T细胞(图7C)。
为了进一步评估4天受moDC刺激的CD8+T细胞的细胞溶解能力,我们使用TAP缺陷型HLA-A2+T2细胞作为被动加载有A2限制性MART-1肽与无关(Flu)肽的靶标(图8A所示的实验安排)。如前所述(Bonehill等人2009),着眼于T细胞活化标志物CD137/4-1BB与细胞溶解性脱粒标志物CD107a的双重表达的流式细胞术分析,用于检测靶标接合和杀伤活性。我们观察到,在2周期间内,只有用MART-1-mRNA-加载的和MART-1肽脉冲的4天moDC刺激的CD8+T细胞在与MART-1-肽加载的T2细胞接触后上调CD137/CD107a(代表性点图绘制在图8B中)。在大多数供体中都检测到了该信号,并且该信号是特异性的,因为不相关的靶标(Flu肽加载的T2细胞)的接合不会诱导细胞溶解性标志物的表达,而先前用MOCK脉冲的或eGFP-mRNA电穿孔的DC刺激效应CD8+T细胞也不会诱导这种表达(图8C)。
讨论
据我们所知,这是对加速的体外细胞分化方法的首次描述,该方法允许产生具有较强Th1极化能力的临床级DC,并结合电穿孔引入的mRNA编码的肿瘤抗原的有效呈递。
之前其他小组已经描述了缩短经典的7-8天体外培养以产生完全成熟的DC的可行性。通常称为“快速DC”,是通过以下获得的细胞:在GM-CSF和IL-4的存在下24(Dauer等人2003;Kvistborg等人2009;Jarnjak-Jankovic等人2007)至72小时(Truxova等人2014)的单核细胞到DC分化时间,然后使用标准的炎性细胞因子混合物TNF-α,IL-1β,IL-6,PGE2或TLR配体(Truxova等2014年)进行24小时的成熟期,在成熟度特征和功能方面,与经典的长期DC培养相比表现相当。只有一项报告描述了MPLA+IFN-γ作为成熟混合物在加速DC分化方案中的整合,作为比较性研究的一部分,该比较性研究在24-36小时的单核细胞到DC分化期后使用了4种不同的成熟策略(Massa等,2013)。与用TNF-α+IL-1β+IL-6+PGE2的经典混合物或备选方案TNF-α+IL-1β+IFN-α+IFN-γ+poly(I:C)或TNF-α+IL-1β+IFN-γ+CL097成熟的DC相比,MPLA+IFN-γ成熟的DC表达出最高水平的共刺激分子表达,并产生最佳的IL-12p70/IL-10释放比率。
Ten Brinke等人(2007;2010)进行的研究还记录了MPLA/IFN-γ在1-型极化效力方面的使用,尽管使用了6-7天的培养时间。本发明证明了当应用于加速培养方案时,MPLA/IFN-γ也可以驱动DC的完全成熟(图1D;2B)。此外,我们证明了两种试剂的组合对于诱导关键T细胞共刺激分子(例如CD86,CD40和CD70以及淋巴结归巢趋化因子受体CCR7)的最大表达是必要的(图3)。其中,CD40和CD70的上调始终高于使用复杂炎性混合物成熟的8天DC获得的上调。两种分子的足够水平对于抗肿瘤免疫反应至关重要:CD40在促进T辅助细胞-DC活化中是重要的,允许CD8+细胞毒性T淋巴细胞的下游最佳刺激,而CD70在驱动Thl而不是T-reg或Th17 T细胞分化中是关键的,并且用于赋予CD8+T细胞效应子和记忆特性。因此,探究CD40/CD40L和CD70/CD27轴的潜力已被成功开发为增加用于临床癌症疫苗应用的DC免疫原性的策略。
我们惊奇地检测到MPLA/IFN-γ成熟的DC相对于TNF-α/PGE2 DC上的T细胞共抑制受体PD-L1的水平较低(图2B)。引人注目的是,在冷冻保存/解冻后,收获时的PD-L1表达水平的差异进一步增加(图10),即,将有效施用于患者的生物制剂,其中该免疫抑制配体的表达应尽可能低。尽管2型干扰素是PD-L1在许多细胞类型上表达的原型诱导物(Gato-Canas等人2017),但PGE2已被描述为骨髓细胞上PD-L1上调的有力驱动物,如在具有免疫抑制能力的肿瘤相关骨髓细胞中所显示的情况(Prima等人2017)。在DC培养方案中使用PGE2通常是由其诱导DC上CCR7最佳表达的能力产生动机的,从而使向淋巴组织的T细胞依赖性区域迁移的效率最大化。然而,在本发明中,4天MPLA/IFN-γDC表达的CCR7至少与TNF-α/PGE2成熟的DC表达的一样多。结合在我们的TNF-α/PGE2成熟的DC中看到的IL-12抑制作用(图6A),总之,这些发现强烈支持了从经典DC成熟混合物向下一代基于DC的癌症疫苗的转移。
冷冻保存,解冻并在无细胞因子培养基中进一步培养24小时后释放的趋化因子特征进一步证实了4天MPLA/IFN-γ成熟的DC支持1型极化免疫反应的能力(图6A)。与8天TNF-α/PGE2成熟的DC相比,只有4天MPLA/IFN-γ成熟的DC分泌高水平的Th1吸引趋化因子CCL3,CCL4,CCL5,CXCL9和CXCL10。在体内,DC分泌的CXCL10和CXCR3受体在CD4+T细胞上的表达之间的相互作用显示确保在淋巴结中这些细胞类型之间形成稳定接触。这种稳定的细胞接触与将这些CD4+T细胞置于高IFN-γ产生的潜在生态位(nich)中组合,可进一步促进Th1-分化。另外,我们的实验显示T-reg和Th2-动员趋化因子CCL17主要由8天TNF-α/PGE2成熟的DC释放,可能是PGE2预处理的结果。虽然统计学意义尚未达到,但4天MPLA/IFN-γ成熟的DCs仍倾向于释放更多的Th17-和T-reg-吸引的趋化因子CCL20。选择DC成熟刺激物定义Th1-或Th2-T细胞动员特征的事实已经通过Lebre等人(2005年)用文件证实。在他们的试验中,评估了新鲜收获的成熟的DC响应于CD40连接的趋化因子产生。在LPS和IFN-γ存在下成熟的DCs显示主要释放Th1-吸引趋化因子,而当PGE2存在于成熟混合物中时,Th2-相关的趋化因子CCL22的表达水平显著增加。与我们的发现相反,在Lebre等的论文中CCL17的表达模式不依赖于DC类型。
潜在影响所实现的DC成熟水平的额外因素是所用培养容器的物理性质。通过本发明的方法,在构成与临床认证的免疫磁性分离系统相容的封闭系统的透气袋中分化和活化细胞是可行的。我们的结果与早期研究相矛盾,早期研究表明在临床级袋中产生的DC具有成熟程序受损,共刺激分子表达、趋化因子和IL-12分泌下调(Rouas等2010)。令人惊讶的是,我们显示所有这些特征在我们的DC中被诱导,并且在冷冻保存、解冻和在无细胞因子的基础培养基中进一步培养后甚至是完整的。G.Gaudernack等人/和G.Kvalheim等人(Kyte等人2005;Mu等人2003)的小组进行了类似的研究,加强了具有完整免疫原性特性的临床级DC确实可以在袋中生成的想法。在细胞分化袋中的培养也将允许我们容易地将我们的方法转移到可商购获得的、完全自动化的封闭细胞培养系统。这种选择将能够进一步减少操作者干预,降低污染风险,并提高总体再现性。
本发明通过选择mRNA电穿孔作为用抗原加载DC的方式,进一步将其与集中于备选培养持续时间和/或成熟方案的早期报道相区分。该技术的优点是在合成编码肿瘤相关抗原的定制序列或含有突变来源的新表位的定制序列方面的灵活性,以及选择掺入序列以优化MHCI和MHCII呈递两者。此外,与其中DC被动加载了选择的HLA限制性肽的先前的研究相反,用全长mRNA电穿孔确保了广泛的表位的加工和潜在呈递,而不需要在HLA类型方面施加任何患者预选择。此外,DC中翻译的蛋白质的半衰期确保了MHC I-表位复合物的延长生成,而被动加载的外源肽仅瞬时结合至表面HLA分子或通过内化耗尽。用mRNA电穿孔的DC诱导T细胞应答的能力与加载肽的DC一样稳健,这已经在之前得到证实。在此我们显示,用模型肿瘤相关抗原电穿孔的4天培养的MPLA/IFN-γ成熟的DC可诱导配备有必需的抗肿瘤工具包的罕见抗原特异性CD8+T细胞的剧烈扩增(例如IFN-γ和穿孔素的高表达),这通过有效和高度特异性细胞毒活性反映。重要的是,我们在冷冻保存和解冻后评价了这种基本的DC特性,这反映了现实的疫苗接种情况。
总之,本发明证实了4天MPLA/IFN-γ成熟的单核细胞来源的DC在细胞产量、表型和1型极化特征方面优于“经典的”8天TNF-α/PGE2成熟的DC。在封闭系统中使用符合GMP的材料和无血清培养基缩短培养时间,电穿孔和冷冻保存不会损害短期培养的MPLA/IFN-γ-DC诱导细胞溶解性肿瘤来源的抗原特异性CD8+T细胞应答的能力,这进一步强调了这种生产方法在临床实施中的稳健性。
参考文献
H.Jonuleit,U.Kuhn,G.Muller,K.Steinbrink,L.Paragnik,et al,Pro-inflammatory cytokines and prostaglandins induce maturation of potentimmunostimulatory dendritic cells under fetal calf serum-free conditions,European journal of immunology27(12)(1997)3135-42.
P.Kalinski,J.H.Schuitemaker,C.M.Hilkens,M.L.Kapsenberg,ProstaglandinE2 induces the final maturation of IL-12-deficient CD1a+CD83+dendritic cells:the levels of IL-12 are determined during the final dendritic Cell maturationand are resistant to further modulation,Journal of immunology(Baltimore,Md,;1950)161(6)(1998)2804-9.
R.B.Mailiard,A.Wankowicz-Kalinska,Q.Cai,A.Wesa,C.M.Hilkens,M.L.Kapsenberg,et al,alpha-type-1 polarized dendritic cells:a novelimmunization tool with optimized CTL-inducing activity,Cancer research 64(17)(2004)5934-7.
H.Okada.P.Kalinski,R.Ueda,A,Hoji,G,Kohanbash,T.E.Donegan,et al,Induction of CD8+T-cell responses against novel glioma associated antigenpeptides and clinical activity by vaccinations with{alpha}-type 1polarizeddendritic cells and polyinosinic-polycytidylic acid stabilized by lysine andcarboxymethylcellulose in patients with recurrent malignant glioma,Journal ofclinical oncology:official journal of the Amencan Society of ClinicalOncology 29(3)(2011)330-6.
C.Paustian,R.Caspell,T.Johnson,P.A.Cohen,S.Shu,S.Xu,et al,Effect ofmultiple activation stimuli on the generation of Th1-polarizirg dendriticcells,Human immunology 72(1)(2011)24-31.
C.Boccaccio,S.Jacod,A.Kaiser,A.Boyer.J.P.Abasuado,A.Nardin,Identification of a clinical-grade maturation factor for dendritic cells,Journal of immunotherapy(Hagerstown,Md.1997)25(1)(2002)88-96.
A.G.Johnson,M.Tomai,L.Solem,L.Beck,E.Ribi,Characterization of anontoxicmonophosphoryl lipid A,Reviews of infectious diseases 9 Suppl 5(1987)S512-6
K.A.Gregg,E.Harberts,F.M.Gardner,M.R.Pelletier,C.Cayatte,et alRationally Designed TLR4 Ligands for Vaccine Adjuvant Discovery,mBio 8(3)(2017).
M.Hansen,G.M.Hjorto,M.Donia,O.Met,N.B.Larsen,M.H.Andersen,et al,Comparison of clinical grade type 1polarized and standard matured dendriticcells for cancer immunotherapy,Vaccine 31(4)(2013)639-46.
A.Ten Brinke,M.L.Karsten,M.C,Dieker,J.J.Zwaginga,S.M.van Ham,Theclinical grade maturation cocktail monophosphoryl lipid A plus IFNgammagenerares monocyte-derived dendritic cells with the capacity to migrate andinduce Th1 polarization,Vaccine 25(41)(2007)7145-52.
A.ten Brinke,G.van Schijndel,R.Visser,T.D.de Gruijl,J.J.Zwaginga,S.M.van Ham,Monophosphoryl lipid A plus IFNgamma maturation of dendriticcells induces antigen-specific CD8+cytotoxic T cells with high cytolyticpotential,Cancer Immunol Immunother 59(8)(2010)1185-95.
S.T.Kolanowski,L.Sritharan,S.N.Lissenberg-Thunnissen,G.M.VanSchijndel,S.M.Van Ham,A.ten Brinke,Comparison of media and serumsupplementation for generation of monophosphoryl lipid A/interferon-gamma-matured type I dendritic cells for immunotherapy,Cytotherapy 16(6)(2014)826-34.
S.Van Lint,S.Wilgenhof,C.Heirman,J.Corthals,K.Breckpot,A.Bonehill,etal,Optimized dendritic cell-based immunotherapy for melanoma:the TriMix-formula,Cancer Immunol Immunother 63(9)(2014)959-67.
S.Tuyaerts,A.Michiels,J.Corthals,A,Bonehill,C.Heirman.C.de Greef,etal,Induction of Influenza Matrix Protein 1and MelanA-specific T lymphocytesin vitro using mRNA-electroporated dendritic cells,Cancer gene therapy 10(9)(2003)696-706.
P.Ponsaerts,V.F.Van Tendeloo,Z.N.Berneman.Cancer immunotherapy usingRNA-loaded dendritic cells,Clinical and experimental immunology 134(3)(2003)378-84.
A.Bonehill,C.Heirman,S.Tuyaerts.A.Michiels,K.Breckpot,F.Brasseur,etal,Messenger RNA-electroporated dendritic cells presentingMAGE-A3simultaneously in HLA classland class II molecules,Journal of immunology(Baltimore,Md.:1950)172(11)(2004)6649-57.
S.Jarnjak-Jankovic,H.Hammerstad,S.Saeboe-Larssen,G.Kvalheim,G.Gaudernack,A full scale comparative study of methods for generation offunctional Dendritic cells for use as cancer vaccines,BMC cancer 7(2007),119.
M.Dauer,B.Obermaier,J.Herten,C.Haerle,K.Pohl,S.Rothenfusser,et al,Mature dendritic cells derived from human monocytes within 48hours:a novelstrategy for dandritic call differentiation from blood precursors,Journal ofimmunology(Baltimore,Md.:1950)170(8)(2003)4069-76.
P.Kvistborg,M.Boegh,A.W.Pedersen,M.H.Claesson,M.B.Zocca,Fastgeneration of dendritic cells,Cellular immunology 260(1)(2009)56-62.
C.Massa,B.Seliger,Fast dendritic cells stimulated with alternativematuration mixtures induce polyfunctional and Iong-lasting activation ofinnate and adaptive effector cells with tumor-killing capabilities,Journal ofimmunology(Baltimore,Md.:1950)190(7)(2013)3328-37.
I.Truxova,K.Pokorna,K.Kloudova,S.Partlova,R.Spisek,J.Fucikova,Day3Poly(I:C)-activated dendritic cells generated in CellGro for use in cancerimmunotherapy trials are fully comparable to standard Day 5DCs,Immunol Lett160(1)(2014)39-49.
A.Van Driessche,A.L.Van de Velde,G.Nijs,T.Braeckman,B.Stein,J.M.DeVries,et al,Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemiapatients in a phase l dose-escalation clinical trial,Cytotherapy 11(5)(2009)653-68.
L.Colantonio,H.Recalde,F.Sinigaglia,D.D′Ambrosio,Modulation ofchemokine receptor expression and chemotactic responsiveness duringdifferentiation of human naive T cells into Th1 or Th2 cells,European journalof immunology 32(5)(2002)1264-73.
J.R.Groom,A.D.Luster,CXCR3 ligands:redundant,Collaborative andantagonistic functions,lmmunology and cell biology 89(2)(2011)207-15.
M.Samson,G.LaRosa,F.Libert,P.Paindavoine,M.Detheux,G.Vassart,et al,The second extracellular loop of CCR5 is the major determinant of ligandspecificity,The Journal of biological chemistry 272(40)(1997)24934-41.
O.Yoshie,K.Matsushima,CCR4 and its ligands:from bench to bedside,International immunology 27(1)(2015)11-20.
T.Yamazaki,X.O.Yang,Y.Chung,A.Fukunaga,R.Nurieva,B.Pappu,et al,CCR6regulates the migration of inflammatory and regulatory T cells,Journal ofimmunology(Baltimore,Md.:1950)181(12)(2008)8391-401.
A.Bonehill,A.M.Van Nuffel,J.Corthals,S.Tuyaerts,C,Heirman,V.Francois,et al,Single-step antigen loading and activation of dendritic cells by mRNAelectroporation for the purpose of therapeutic vaccination in melanomapatients,Clinical cancer research:an official journal of the AmericanAssociation for Cancer Research 15(10)(2009)3366-75.
M.Gato-Canas,M,Zuazo,H,Arasanz,M.Ibenez-Vea,L.Lorenzo,G.Fernandez-Hinojal,et al,PDL1 Signals through Conserved Sequence Motifs to Overcomelnterferon-Medated Cytotoxicity,Cell reports 20(8)(2017)1818-1829.
V.Prima,L.N.Kaliberova.S.Kaliberov,D.T.Curiel,S.Kusmartsev,COX2/mPGES1/PGE(2)pathway regulates PD-L1 expression in tumor-associatedmacrophages and myeloid-derived suppressor cells,P Natl Acad Sci USA 114(5)(2017)1117-1122.
M.C.Lebre,T.Burwell,P.L.Vieira,J.Lora,A.J.Coyle,M.L.Kapsenberg,et al,Differential expression of inflammatory chemokines by Th1-and Th2-cellpromoting dendritic cells:a role for different mature dendritic cellpopulations in attracting appropriate effector cells to peripheral sites ofinflammation,Immunology and cell biology 83(5)(2005)525-35.
R.Rouas,H.Akl,H.Fayyad-Kazan,N.EI Zein,B.Badran,B.Nowak,et al,Dendritic cells generated in clinical grade bags strongly differ in immunefunctionality when compared with classical DCs generated in plates,Journal ofimmunotherapy(Hagerstown,Md.:1997)33(4)(2010)352-63.
J.A.Kyte,G.Kvalheim,S.Aamdal,S.Saeboe-Larssen,G.Gaudernack,Preclinical full-scale evaluation of dendritic cells transfected withautologous tumor-mRNA for melanoma vaccinetion,Cancer gene theraPy 12(6)(2005)579-591.
L.J.Mu,G.Gaudernack,S.Saeboe-Larssen,H.Hammerstad,A.Tierens,G.Kvalheim,A protocol for generation of clinical grade mRNA-transfectedmonocyte-derived dendritic cells for cancer vaccines,Scandinavian joumal ofimmunology 58(5)(2003)578-86.
Valmori D,Gervois N,Rimoldi D,Fonteneau JF,Bonelo A,Liēnard D,Rivoltini L,Jotereau F,Cerottini JC,Romero P.Diversity of the finespecificity displayed by HLA-A*0201-restricted CTL specific for theimmunodominant Melan-A/MART-1antigenic peptide.J Immunol.1998Dec 15;161(12):6956-62.
- 用于治疗用途的树突细胞的体外分化和成熟方法
- 给予体外部分成熟的树突细胞治疗肿瘤的制药用途