DNA的标记
文献发布时间:2023-06-19 10:08:35
相关申请
本申请要求2018年6月25日提交的美国临时专利申请系列号62/689,650和2018年7月11日提交的美国临时专利申请系列号62/696,696在35 U.S.C.§119(e)下的利益,这些相关申请的内容为所有目的整体通过参考并入本文。
序列表引用
本申请与电子格式的序列表一起提交。所述序列表作为名为“Sequence_Listing_BNGEN_049WO.txt”的文件提供,所述文件在2019年6月23日生成,大小为1千字节。所述序列表的电子格式中的信息整体通过参考并入本文。
技术领域
本文中的实施方式总的来说涉及DNA分子的标记例如基因组标记,用于纳米通道中的线性化DNA的分析。
背景技术
流体纳米通道中的基因组作图是一种能够在下一代测序(NGS)的检测范围之外的兆碱基长度的DNA分子中寻找基因组结构变异(SV)的鲁棒技术。这些在流体通道中的基因组作图技术例如切口标记物修复染色化学(NLRS)或使用直接标记物的直接标记(非破坏性)和染色化学(DLS)(两者均来自于Bionano Genomics,San Diego,CA),能够为包括30GbpAxolotl基因组在内的大且复杂的植物和动物基因组产生结构准确的基因组组装体。
源自于酿脓链球菌(Streptococcus pyogenes)的II型簇状规则间隔短回文重复序列(CRISPR)-CRISPR相关半胱天冬酶9(Cas9)系统,由于其靶序列可定制性以及高的结合和酶促效率,已变成用于靶向基因组编辑的革命性工具。为了实现位点特异性DNA识别和切割,Cas9蛋白与CRISPR RNA(crRNA)和与所述crRNA部分互补的反式激活crRNA(tracrRNA)的双链体形成核糖核蛋白(RNP)复合体。所述Cas9的HNH和RuvC样核酸酶结构域切割两条DNA链,在由结合的crRNA转录本内大约20个核苷酸的种子序列所定义的位点处产生双链断裂(DSB)。
发明内容
某些实施方式包括一种在靶序列处标记DNA的方法。所述方法可以包括将所述DNA与第一dCas蛋白和包含第一crRNA和含有第一标记物的第一tracrRNA的第一标记的向导RNA(gRNA)相接触,其中所述第一crRNA与所述DNA的第一靶序列或其部分互补。所述方法可以包括将所述DNA、第一dCas蛋白和第一标记的gRNA温育,其中所述第一dCas蛋白、DNA和第一标记的gRNA形成复合体,其中所述第一crRNA与所述第一靶序列或其部分杂交。因此,所述DNA可以在第一靶序列处被标记,以形成标记的DNA。在某些实施方式中,所述方法还包括将所述DNA与第二dCas蛋白和包含第二crRNA和含有不同于所述第一标记物的第二标记物的第二tracrRNA的第二标记的gRNA相接触,其中所述第二crRNA可以与所述DNA的第二靶序列或其部分互补。所述方法还可以包括将所述DNA与所述第二标记的gRNA温育,其中所述第二dCas蛋白、DNA和第二标记的gRNA形成第二复合体,其中所述第二crRNA与所述第二靶序列或其部分杂交。因此,所述DNA可以在所述第二靶序列处用所述不同的标记物标记。在某些实施方式中,将所述DNA与所述第一标记的gRNA和第二标记的gRNA同时相接触,并将所述DNA与所述第一dCas蛋白、第二dCas蛋白、第一标记的gRNA和第二标记的gRNA同时温育。在某些实施方式中,所述DNA在包括接触和温育步骤并至少直至形成标记的DNA的整个方法中保持完整。在某些实施方式中,所述第一靶序列和第二靶序列中的至少一者在包含多个不适合于基于基序的酶促标记例如切口酶标记的重复序列或结构变体的区域中。在某些实施方式中,所述第一靶序列和第二靶序列中的至少一者在据预测在切口平移标记后形成和/或易于形成脆性位点的区域中。在某些实施方式中,所述方法还包括将所述第一靶序列和/或第二靶序列选择成在包含多个不适合于基于基序的酶促标记例如切口酶标记的重复序列或结构变体的区域中。在某些实施方式中,所述第一靶序列和第二靶序列中的至少一者被选择成在据预测在切口平移标记后形成和/或易于形成脆性位点的区域中。在某些实施方式中,所述第一靶序列和第二靶序列中的至少一者由在切口标记后不包含分布不均匀的标记物的基因组区域构成。在某些实施方式中,所述第一靶序列和第二靶序列中的至少一者由重复的序列构成。在某些实施方式中,所述方法不包括切口标记。在某些实施方式中,所述标记的DNA能够在流体纳米通道中线性化后保持完整。在某些实施方式中,所述标记的DNA在流体纳米通道中线性化后保持完整。在某些实施方式中,所述方法还包括将所述标记的DNA在流体纳米通道中线性化,其中所述DNA在所述线性化后保持完整。在某些实施方式中,所述方法还包括在所述流体纳米通道中检测所述线性化的DNA上所述第一标记物与第二标记物之间的相对距离。所述流体纳米通道可以例如具有至少10nm的长度和小于1000nm的横截面直径。在某些实施方式中,所述第一dCas是dCas9。在某些实施方式中,所述第二dCas是dCas9。所述第一dCas和第二dCas中的至少一者可以在HNH结构域和/或RuvC样结构域中包含一个或多个突变或一个或多个缺失。在某些实施方式中,所述第一dCas未被标记。在某些实施方式中,所述第二dCas未被标记。在某些实施方式中,所述第一crRNA和第二crRNA中的至少一者包含与所述第一和第二靶序列或其部分中的一者或多者互补的约10-40个核苷酸的序列,例如约10-30寡核苷酸、约10-20个核苷酸、约15-30个核苷酸、约15-25个核苷酸、约20-30个核苷酸或约20-40个核苷酸。在某些实施方式中,所述温育包含所述第一dCas蛋白、第二dCas蛋白、第一标记的gRNA和第二标记的gRNA中的一者或多者,其浓度为每150ng所述用于杂交的DNA约120nM(每ng DNA约0.8nM),例如每150ng DNA不超过或不超过约5nM、10nM、15nM、20nM、50nM、80nM、100nM、110nM、120nM、130nM、140nM、150nM、200nM、250nM、300nM、400nM、450nM或500nM,包括所列出的任两个值之间的范围,例如5nM至25nM、5nM至20nM、5nM至15nM、5nM至500nM、10nM至25nM、10nM至20nM、10nM至15nM、10nM至500nM、50nM至500nM、100nM至500nM、120nM至500nM、5nM至300nM、10nM至300nM、50nM至300nM、100nM至300nM、120nM至300nM、5nM至200nM、10nM至200nM、50nM至200nM、100nM至200nM、120nM至200nM、5nM至120nM、10nM至120nM、50nM至120nM或100nM至120nM。在某些实施方式中,所述浓度为10nM至20nM,例如15nM。不受理论限制,已观察到如果这些物质的浓度过高,则可能存在高背景,其可以干扰真阳性标记物的检测。在某些实施方式中,所述温育包含所述dCas蛋白和标记的gRNA中的一者或多者,其浓度为每150ng用于杂交的DNA约120nM。在某些实施方式中,所述方法还包括通过另外的化学方法标记所述DNA,例如使用酶例如DLE-1的直接酶促标记并且任选地除了所述酶促标记之外还包括染色剂(例如BIONANO GENOMICS的“DLS”技术)或产生切口并随后进行切口标记和修复(例如BIONANO GENOMICS的“NLRS”技术),以产生具有两个或更多个带有不同标记物(例如不同颜色)的不同特异性基序的DNA。在某些实施方式中,所述切口标记包含切口平移,例如将所述切口的DNA与聚合酶和标记的核苷酸温育,其中所述核苷酸用与所述标记物相同或不同的核苷酸标记物标记,由此所述聚合酶将所述标记的核苷酸以5’->3’的方向掺入到所述DNA中。在某些实施方式中,所述方法不包括通过包含切口平移的切口标记来标记所述DNA。在某些实施方式中,所述方法还包括用直接标记酶例如DLE-1进行标记(例如使用DLS化学)。所述第一标记物可以是例如荧光团、量子点、树枝状聚合物、纳米丝、珠子、半抗原、链霉亲和素、亲和素、中性亲和素、生物素和反应性基团、肽、蛋白质、磁珠、放射性标记物、非光学标记物或所列出的物品中的两者或更多者的组合。在某些实施方式中,所述第二标记物选自:荧光团、量子点、树枝状聚合物、纳米丝、珠子、半抗原、链霉亲和素、亲和素、中性亲和素、生物素和反应性基团、肽、蛋白质、磁珠、放射性标记物或非光学标记物或所列出的物品中的两者或更多者的组合。在某些实施方式中,将所述DNA进一步用非特异性标记物例如骨架标记物如YOYO标记物标记(所述非特异性标记物在本文中也可以被称为“染色剂”)。所述非特异性标记物可以在所述CRISPR-dCas标记后添加。在某些实施方式中,所述方法的标记在单一步骤中进行。
某些实施方式包括一种DNA组合物。所述DNA组合物可以包含DNA分子、第一dCas蛋白(例如dCas9)和第一标记的向导RNA(gRNA)。所述第一标记的gRNA可以包含第一crRNA和包含第一标记物的第一tracrRNA。所述第一dCas、第一标记的gRNA和DNA可以构成第一复合体,所述第一复合体包含与所述DNA分子的第一靶序列或其部分杂交的第一crRNA。在某些实施方式中,所述DNA组合物还包含第二dCas蛋白和包含第二cRNA和用不同于所述第一标记物的第二标记物标记的第二tracrRNA的第二标记的gRNA。所述第二dCas蛋白、第二标记的gRNA和DNA可以构成第二复合体,所述第二复合体包含与所述DNA的第二靶序列杂交的第二crRNA。在某些实施方式中,将所述DNA组合物在流体纳米通道中线性化。在某些实施方式中,所述DNA在所述流体纳米通道中是完整的。在某些实施方式中,所述流体纳米通道具有至少10nm的长度和小于1000nm的横截面直径。在某些实施方式的DNA组合物中,所述第一dCas是dCas9。在某些实施方式的DNA组合物中,所述第二dCas是dCas9。在某些实施方式的DNA组合物中,所述第一dCas在HNH结构域和/或RuvC样结构域中包含一个或多个突变或一个或多个缺失。在某些实施方式的DNA组合物中,所述第二dCas在HNH结构域和/或RuvC样结构域中包含一个或多个突变或一个或多个缺失。在某些实施方式的DNA组合物中,所述第一dCas未被标记。在某些实施方式的DNA组合物中,所述第二dCas未被标记。在某些实施方式的DNA组合物中,所述第一和第二crRNA中的至少一者包含与所述第一和/或第二靶序列或其部分互补的约10-40个核苷酸的序列,基本上由所述序列构成或由所述序列构成。在某些实施方式的DNA组合物中,所述DNA包含掺入到所述DNA中的标记的核苷酸,其中所述核苷酸用与所述第一标记物和/或第二标记物相同或不同的核苷酸标记物标记。在某些实施方式中,所述DNA未被切口标记。在某些实施方式中,所述第一标记物选自:荧光团、量子点、树枝状聚合物、纳米丝、珠子、半抗原、链霉亲和素、亲和素、中性亲和素、生物素和反应性基团、肽、蛋白质、磁珠、放射性标记物或非光学标记物或所列出的物品中的两者或更多者的组合。在某些实施方式中,所述第二标记物选自:荧光团、量子点、树枝状聚合物、纳米丝、珠子、半抗原、链霉亲和素、亲和素、中性亲和素、生物素和反应性基团、肽、蛋白质、磁珠、放射性标记物或非光学标记物或所列出的物品中的两者或更多者的组合。在某些实施方式中,所述DNA进一步用非特异性标记物例如骨架标记物如YOYO标记物标记。在某些实施方式中,所述DNA已用直接标记酶例如DLE-1标记。
某些实施方式包括一种用于执行本文中描述的任何标记方法的试剂盒。所述试剂盒可以包含本文中所描述的dCas蛋白(例如dCas9)。在某些实施方式中,所述试剂盒还包含标记物。在某些实施方式中,所述标记物未被附连到所述dCas蛋白。在某些实施方式中,所述试剂盒还包含含有crRNA和tracrRNA的gRNA。在某些实施方式中,所述gRNA包含标记物,例如被所述tracrRNA包含的标记物。在某些实施方式中,所述试剂盒还包含切口酶。在某些实施方式中,所述试剂盒还包含直接标记酶例如DLE-1。
附图说明
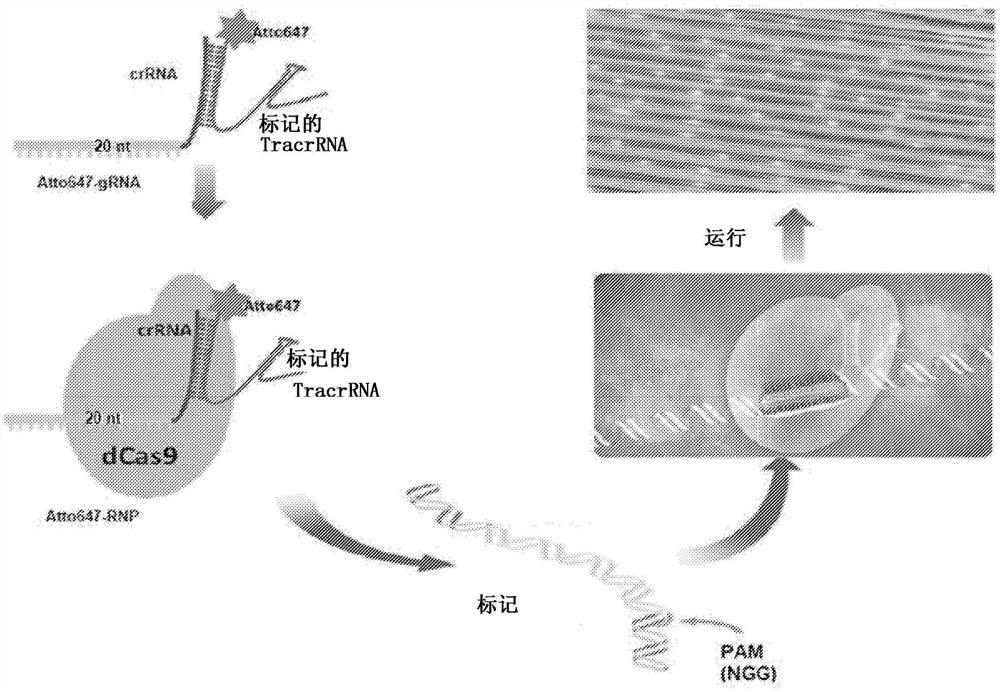
图1A-1E是一系列示意图,示出了根据本文中的某些实施方式用于标记DNA的方法。
图2A是根据本文中的某些实施方式的纳米通道中的标记的DNA的荧光显微照片。
图2B是根据本文中的某些实施方式通过标记物图案对齐的分子的图。
图3A-3B分别是描绘了根据本文中的某些实施方式在切口标记和标记的CRISPR-dCAS9复合体结合后的标记的人类基因组DNA的图。
图3A示出了在CHM1上通过dCas9/Alu-Atto647进行的基因组作图。
图3B描绘了在CHM1上通过dCas9/Alu进行的Alu重复序列作图。
图4A-4B是示出了作为切口产生的结果可能在脆性位点处发生的片段化的示意图,其中切口彼此更近(图4A),并且移动得更加远离的切口抑制了脆性位点形成的风险(图4B)。
图5A-5D是一系列示意图,示出了根据本文中的某些实施方式用于标记DNA的方法。
详细描述
根据本文中的某些实施方式的标记方法、DNA组合物和试剂盒,使用修饰的核酸酶缺陷型簇状规则间隔短回文重复序列(CRISPR)/CRISPR相关半胱天冬酶(Cas;例如CRISPR相关半胱天冬酶9“Cas9”)复合体作为探针来标记DNA,所述复合体荧光标记序列特异性基因组位点而不产生DNA切口。所述Cas可以是核酸酶缺陷型Cas(dCas)蛋白例如dCas9。所述DNA-CRISPR-dCas复合体可以在体外组装。有利情况下,所述dCas/gRNA复合体(例如dCas9/gRNA)稳定地结合它的靶,并适合于使用不同颜色的dCas/gRNA复合体(例如dCas9/gRNA)同时探测多个靶,允许靶的多色标记。例如,第一gRNA可以包含第一标记物和与所述DNA上的第一靶序列或其部分互补的第一crRNA。第二gRNA可以包含第二标记物(其不同于所述第一标记物)和与所述DNA上的第二靶序列(其不同于所述第一靶序列)或其部分互补的第二crRNA。所述第一gRNA可以在所述DNA的第一靶序列处与dCas(例如dCas9)形成第一复合体,并且所述第二gRNA可以在所述DNA的第二靶序列处与dCas(例如dCas9)形成第二复合体。在某些实施方式中,可以将所述用第一复合体(包含所述第一标记物)和第二复合体(包含所述第二标记物)标记的DNA在流体纳米通道中线性化。可以测量所述线性化的DNA上所述第一标记物与第二标记物之间的相对距离。由于所述gRNA包含标记物,因此在某些实施方式中所述dCas(例如dCas9)可以不被标记。
在本文中公开的某些实施方式的标记方法、DNA组合物和试剂盒中,用本文中所描述的荧光标记的CRISPR-dCas复合体(例如CRISPR-dCas9复合体)直接标记DNA。所述标记方法可以包括对DNA进行标记,其包括使用没有核酸酶活性但在被靶RNA指导时维持靶向特异性和效率的dCas(例如dCas9)将标记物靶向到所述DNA上选定的靶序列。通过用标记物例如Atto647标记tracrRNA并产生靶向基因组区域的复合体,标记可以通过直接结合来实现。在本文中,使用组装有与各种不同crRNA相组合的荧光标记的tracrRNA的核酸酶缺陷的Cas9(dCas9)蛋白,演示了重复的Duff1220元件和Alu位点的快速标记(分别参见实施例1和2)。在某些实施方式中,所述标记方法除了用CRISPR-dCas标记之外,还包括通过另外的化学方法标记所述DNA,例如使用酶例如DLE-1的直接酶促标记并且任选地除了所述酶促标记之外还包括染色剂(例如BIONANO GENOMICS的“DLS”技术)或产生切口并随后进行切口标记和修复(例如BIONANO GENOMICS的“NLRS”技术),以产生包含两个或更多个带有不同标记物(例如不同颜色)的特异性基序(例如靶序列)的DNA。设想了使用多种颜色标记多个特异性基序与标记较少数目的基序相比可以产生更高的信息密度。有利的是,本文中的标记方法可以使用只需温育的简单流程来实现,并且它不损坏DNA。还设想了与在流体纳米通道中线性化和移动DNA有关的力可以引起受损DNA的双链断裂,使标记图案的分析变得复杂。因此,在本文的某些实施方式中,所述CRISPR-dCas(例如CRISPR-dCas9)标记的DNA是未受损的(因此也是完整的),可以适合于如本文中所述通过在流体纳米通道中线性化来分析所述标记的DNA。不受理论限制,设想了本文中的标记方法与切口标记相比可以更快地实现标记,并且可用于靶向更多种类的靶序列。在某些实施方式中,所述标记方法不包含切口标记。
本文中描述的某些实施方式的标记DNA的方法可用于例如靶向重复序列、为基因组区域和结构变体添加条形码,这些应用不适合基于基序的酶促标记例如产生切口然后使用酶例如DLE-1进行直接酶促标记(例如BIONANO GENOMICS的“DLS”技术)或基于切口酶的方法例如产生切口然后进行切口标记和修复(例如BIONANO GENOMICS的“NLRS”技术),后者可能涉及基序例如切口酶基序在所述DNA中的不均匀分布。这种快速、方便、无损和成本效益高的技术为自动高通量基因组广度作图以及含有重复和结构变体DNA的复杂区域的靶向分析两者,提供了有价值的工具。因此,在某些实施方式中,一种标记DNA的方法包括在不适合用基于基序的标记例如需要限制性位点基序在DNA中不均匀分布的切口平移来标记的基因组区域中选择靶序列。所述方法可以包括使用本文中所描述的包含crRNA的gRNA,所述crRNA与所述不适合用切口平移标记的基因组区域中的靶序列或其部分互补。在某些实施方式中,所述标记DNA的方法包括在包含多个重复序列(例如Alu或Duff1220)的基因组区域中选择靶序列,为不适合于基于基序的酶促标记例如基于切口酶的标记方法(例如切口平移)的基因组区域和/或结构变体添加条形码。在某些实施方式中,所述标记DNA的方法包括将DNA与特异性针对SNP的一组标记的gRNA相接触。在某些实施方式中,所述标记DNA的方法包括对定制的靶进行标记。在某些实施方式中,所述标记DNA的方法包括对条形码序列进行标记。
在某些实施方式中,除了用CRISPR-dCas标记之外,所述标记方法还包括通过另外的化学方法标记所述DNA,例如使用酶例如DLE-1的直接酶促标记并且任选地除了所述酶促标记之外还包括染色剂(例如BIONANO GENOMICS的“DLS”技术)或产生切口并随后进行切口标记和修复(例如BIONANO GENOMICS的“NLRS”技术),以产生具有两个或更多个带有不同标记物(例如不同颜色)的特异性基序(例如靶序列)的DNA。在某些实施方式中,所述切口标记包括使用切割单链(切口酶)而不是两条链的修饰的限制性酶在所述DNA上产生切口。然后可以将标记的核苷酸直接地(任选地随后进行修复)或通过切口平移掺入到所述切口的DNA中。任选地,所述DNA可以在所述切口平移后用连接酶修复。任选地,所述DNA也可以用非特异性骨架标记物例如YOYO标记物染色。所述非特异性标记物可以在所述CRISPR-dCas标记后添加。这种切口标记方法可能极其高效,并产生高分辨率全基因组组装体。然而,基于基序的酶标记方法例如直接酶促标记(例如DLS)和切口标记受到可用酶中的靶序列数目的限制。因此,在某些实施方式中,切口标记与本文中所描述的CRISPR-dCas(例如dCas9)标记联合进行,这可以提供更多种类的靶序列。
还设想了作为包含切口平移的切口标记方法的一部分,当在同一DNA的相反链上存在切口时,所述切口可以移动得更加靠近在一起(如图4A中所示)或更加远离(如图4B中所示)。不受理论限制,已观察到当两个切口在相反的DNA链上相隔<1Kb时,出现脆性位点。由于例如下述因素,在脆性位点处可能发生片段化:(1)机械操作,(2)标记所需的热,(3)与标记和某些种类的修复(例如使用聚合酶的外切核酸酶活性)有关的链延伸,和/或(4)与DNA分子的线性化有关的剪切力。因此,根据本文中的某些实施方式,使用CRISPR-dCas的标记可用于在据预期在切口平移期间产生脆性位点的区域中维持链完整性。
当在本文中使用时,“dCas蛋白”具有本领域技术人员鉴于本公开而理解的惯常含义。它是指一类没有核酸酶活性的Cas蛋白(例如一类Cas9)。所述dCas蛋白(例如dCas9)可以缺少核酸酶活性,但以高特异性和效率与向导RNA(gRNA)-DNA复合体结合。应该指出,存在多种类型的Cas蛋白,包括例如Cas1直至Cas9。根据某些实施方式的标记方法、DNA组合物和试剂盒,所述dCas蛋白是dCas9蛋白。在某些实施方式的标记方法、DNA组合物和试剂盒中,所述dCas蛋白(例如dCas9蛋白)不在所述DNA上产生切口。设想了dCas蛋白中的核酸酶活性可以通过所述dCas蛋白的HNH和/或RuvC样结构域中的一个或多个突变和/或一个或多个缺失来抑制或阻止。根据某些实施方式的标记方法、DNA组合物和试剂盒,所述dCas(例如dCas9)蛋白在HNH和RuvC样结构域中的一者或两者中包含一个或多个突变。在某些实施方式中,所述dCas(例如dCas9)蛋白在HNH结构域中包含突变,例如点突变、移码、插入、所述结构域的一部分或全部的缺失或缺失了所述结构域的一部分或全部的插入-缺失。在某些实施方式中,所述dCas(例如dCas9)蛋白在RuvC样结构域中包含一个或多个突变,例如点突变、移码、插入、所述结构域的一部分或全部的缺失或缺失了所述结构域的一部分或全部的插入-缺失。在某些实施方式中,所述dCas(例如dCas9)蛋白在HNH结构域中包含一个或多个突变,例如点突变、移码、插入、所述结构域的一部分或全部的缺失或缺失了所述结构域的一部分或全部的插入-缺失,并在RuvC样结构域中包含突变例如点突变。在某些实施方式中,所述dCas(例如dCas9)蛋白在HNH结构域中包含突变,例如点突变、移码、插入、所述结构域的一部分或全部的缺失或缺失了所述结构域的一部分或全部的插入-缺失,并在RuvC样结构域中包含突变例如移码。在某些实施方式中,所述dCas(例如dCas9)蛋白在HNH结构域中包含突变,例如点突变、移码、插入、所述结构域的一部分或全部的缺失或缺失了所述结构域的一部分或全部的插入-缺失,并在RuvC样结构域中包含突变例如插入。在某些实施方式中,所述dCas(例如dCas9)蛋白在HNH结构域中包含突变,例如点突变、移码、插入、所述结构域的一部分或全部的缺失或缺失了所述结构域的一部分或全部的插入-缺失,并在RuvC样结构域中包含突变例如所述RuvC样结构域的一部分或全部的缺失。在某些实施方式中,所述dCas9蛋白在HNH结构域中包含突变,例如点突变、移码、插入、所述结构域的一部分或全部的缺失或缺失了所述结构域的一部分或全部的插入-缺失,并在RuvC样结构域中包含突变例如缺失了所述RuvC样结构域的一部分或全部的插入-缺失。在某些实施方式的方法和DNA组合物中,所述dCas9蛋白未被标记。应该指出,Cas9 D10A突变体的切割位点之一已被失活,可以将CRISPR-Cas9复合体转变成具有可定制的靶序列的切口酶。尽管Cas9D10A可用于与基于限制性酶的切口标记相似的标记(例如涉及作为独立步骤的切口形成、标记和通过连接的修复),因为Cas9 D10A仍具有核酸酶(切口)活性,但应该理解,Cas9 D10A不代表本文中使用的dCAS。因此,在某些实施方式中,所述dCAS不包含Cas9D10A。
当在本文中使用时,“向导RNA(gRNA)”具有本领域技术人员鉴于本公开而理解的惯常含义。它是指一类包含反式激活crRNA(tracrRNA)和crRNA、基本上由它们构成或由它们构成的RNA。所述crRNA可以包含与本文中所描述的DNA上的靶序列或其部分互补的序列、基本上由所述序列构成或由所述序列构成。例如但没有任何限制,所述crRNA可以包含与所述靶序列或其部分互补的序列或其至少10、15、20、25、30、35或40个核苷酸的序列,基本上由所述序列构成或由所述序列构成,包括在所列出的任两个值之间的范围,例如与所述靶序列或其部分互补的10–15个核苷酸、10–20个核苷酸、10–25个核苷酸、10–30个核苷酸、10–35个核苷酸、10–40个核苷酸、15–20个核苷酸、15–25个核苷酸、15–30个核苷酸、15–35个核苷酸、15–40个核苷酸、20–25个核苷酸、20–30个核苷酸、20–35个核苷酸、20–40个核苷酸、25–30个核苷酸、25–35个核苷酸、25–40个核苷酸、30–35个核苷酸、30–40个核苷酸或35–40个核苷酸。所述tracrRNA可以包含本文中描述的任何标记物。所述tracrRNA可以至少部分与所述crRNA互补。不受理论限制,所述tracrRNA在所述DNA的靶序列或其部分处与所述crRNA的结合,可以导致在所述靶序列处与Cas9形成复合体。在某些实施方式中,所述tracrRNA和crRNA是单一分子的部分(采取顺式)。在某些实施方式中,所述tracrRNA和crRNA是分开的分子的部分。设想了根据某些实施方式的标记方法、DNA组合物和试剂盒,所述crRNA与所述靶序列的一部分互补(例如不与所述感兴趣的靶序列的全长互补)。在某些实施方式中,所述crRNA与定制的靶序列互补。在某些实施方式中,所述crRNA与条形码序列互补。
设想了在某些实施方式的标记方法、DNA组合物和试剂盒中,两种或更多种不同的标记的gRNA可用于标记DNA的不同的靶序列。因此,设想了在某些实施方式的标记方法、DNA组合物和试剂盒中,两种或更多种gRNA可以各自包含本文中所描述的不同标记物。因此,在某些实施方式的标记方法、DNA组合物和试剂盒中,所述DNA标记是多重的。
流体纳米通道可用于分析长的(例如千碱基或兆碱基长度)DNA分子以及短的DNA分子的结构特点。关于适合的流体纳米通道的详细信息,可以在例如美国专利号8722327、8628919和9533879中找到,每个所述专利整体通过参考并入本文。适合用于某些实施方式的标记方法、DNA组合物和试剂盒的纳米通道可以具有例如小于所述大分子在伸展形式下的回转半径的约2倍的直径。这样的纳米通道可以执行自由伸展的波动的DNA卷曲的熵约束,以便伸展并拉伸所述DNA。
某些实施方式的方法和DNA组合物和试剂盒的适合的流体纳米通道区段可以例如具有小于约1000nm、或小于约500nm、或小于约200nm、或小于约100nm、或小于约50nm、约10nm、约5nm、约2nm或小于约0.5nm的特征性横截面维度。在某些实施方式中,流体纳米通道区段具有小于所述DNA分子的回转半径的约两倍的特征性横截面维度。在某些实施方式中,所述纳米通道具有至少大约所述DNA分子的维持长度的特征性横截面维度。适合于本文中的某些实施方式的方法、试剂盒和DNA组合物的流体纳米通道区段可以具有例如至少约100nm、至少约500nm、至少约1000nm、至少约2微米、至少约5微米、至少约10微米、至少约1mm或至少约10mm的长度。在某些实施方式中,流体纳米通道区段以每立方厘米至少1个流体纳米通道区段的密度存在。
因此,在某些实施方式的标记方法、DNA组合物和试剂盒中,所述流体纳米通道能够将所述DNA分子线性化(以便对所述DNA卷曲进行熵约束,以伸展并拉伸所述DNA分子)。在流体纳米通道中线性化后,所述DNA分子维持线性化的拉伸构象,允许确定标记物沿着所述DNA长度的相对位置。这些标记物可用于研究DNA结构变异例如复杂的重排、单体型分析、长的(千碱基或兆碱基规模)DNA上重复元件拷贝数的定量,定量短的DNA、解析多个重复序列、插入和/或在支架上组装指示DNA结构的序列或标记图案。
在某些实施方式中,描述了一种在靶序列处标记DNA分子的方法(这些方法在本文中也被描述为“标记方法”)。所述标记方法可以包括将所述DNA与本文中所描述的dCas蛋白(例如dCas9)相接触。所述标记方法可以包括将所述DNA与本文中所描述的标记的gRNA相接触。所述标记的gRNA可以包含本文中所描述的与靶序列或其部分互补的crRNA和包含标记物的tracrRNA。所述dCas蛋白、DNA和标记的gRNA形成复合体,其中所述crRNA与所述靶序列杂交。因此,所述DNA可以通过所述标记方法标记。在某些实施方式中,将所述DNA与所述gRNA和dCas蛋白(例如dCas9)同时相接触。在某些实施方式中,将所述DNA与单一组合物中的所述gRNA和dCas蛋白(例如dCas9)相接触。在某些实施方式中,将所述DNA与分开的组合物中的所述gRNA和dCas蛋白(例如dCas9)同时或在不同时间相接触。在某些实施方式中,所述方法的标记在单一步骤中进行。在某些实施方式中,所述标记的DNA具有千碱基或兆碱基范围内的长度,例如至少1kb、2kb、3kb、5kb、10kb、20kb、30kb、40kb、50kb、100kb、150kb、250kb、500kb、1Mb、1.5Mb或2Mb,包括所列出的任两个值之间的范围(例如1kb–2Mb、5kb–2Mb、10kb–2Mb、20kb–2Mb、100kb–2Mb、500kb–2Mb、1kb–1Mb、5kb–1Mb、10kb–1Mb、100kb–1Mb、200kb–1Mb、500kb–1Mb、1kb–500kb、5kb–500kb、10kb–500kb、20kb–500kb、100kb–500kb、1kb–100kb、5kb–100kb、10kb–100kb、20kb–100kb、50kb–100kb、1kb–50kb、5kb–50kb、10kb–50kb、1kb–10kb、5kb–10kb或1kb–5kb)。
在某些实施方式中,所述标记方法包括在两个或更多个不同靶序列处使用对于每个靶序列来说不同的标记物来标记所述DNA。因此,某些实施方式的标记方法或复合体还包含两种或更多种gRNA,其各自包含与所述DNA的不同靶序列或其部分互补的crRNA以及用不同标记物标记的tracRNA,以便用不同标记物标记所述DNA上的不同靶序列。在某些实施方式中,每个靶序列用独特的标记物标记。例如,所述标记方法可以包括将所述DNA与包含与所述DNA上的第一靶序列(或其部分)互补的crRNA和含有第一标记物的tracrRNA的第一gRNA、包含与所述DNA上的不同于所述第一靶序列的第二靶序列(或其部分)互补的crRNA和含有不同于所述第一标记物的第二标记物的tracrRNA的第二gRNA和/或包含与所述DNA上的不同于所述第一靶序列和/或第二靶序列的第三靶序列(或其部分)互补的crRNA和含有不同于所述第一标记物和/或第二标记物的第三标记物的tracrRNA的第三gRNA相接触。
在某些实施方式中,所述标记方法还包括将所述DNA至少与包含第二crRNA和含有第二种不同标记物的第二tracrRNA的第二标记的gRNA相接触,其中所述第二crRNA与第二种不同的靶序列或其部分互补。所述标记方法可以包括将所述DNA与所述第二标记的gRNA温育,其中dCas蛋白(例如dCas9)、DNA和第二标记的gRNA形成第二复合体,其中所述第二crRNA(以及因此所述第二种不同的标记物)与所述DNA上的所述第二种不同的靶序列或其部分杂交。因此,可以在至少两个不同的靶序列处用至少两种不同的标记物对所述DNA进行标记。在某些实施方式中,将所述DNA与所述第一标记的gRNA和第二标记的gRNA同时,例如在单一组合物中相接触。因此,可以将所述第一标记的gRNA和第二标记的gRNA与所述DNA和dCas蛋白(例如dCAS9)同时温育。在某些实施方式中,将所述DNA与所述第一标记的gRNA和第二标记的gRNA分开地相接触。例如,可以将所述DNA与包含所述第一标记的gRNA的第一组合物和包含所述第二标记的gRNA的分开的第二组合物相接触。在某些实施方式中,将所述DNA与所述第一和第二组合物同时相接触,并将所述第一标记的gRNA和第二标记的gRNA与所述DNA和dCas蛋白(例如dCSA9)同时温育。在某些实施方式中,将所述DNA与所述第一和第二组合物在不同时间相接触,并将所述第一标记的gRNA和第二标记的gRNA与所述DNA和dCas蛋白(例如dCAS9)在不同时间温育。在某些实施方式中,将所述DNA与所述第一和第二组合物在不同时间相接触,并将所述第一标记的gRNA和第二标记的gRNA与所述DNA和dCas蛋白(例如dCAS9)温育不同但交叠的时间段(例如如果所述第一和第二组合物被顺序添加的话)。
设想了所述dCAS(例如dCAS9)不在所述DNA中产生双链断裂,并因此可用于在所述标记期间和之后维持DNA链的完整性。在某些实施方式的标记方法中,所述DNA在整个所述方法中保持完整(例如没有断裂的双链)。在某些实施方式的标记方法中,所述DNA在如本文中所述通过流体纳米通道线性化和/或运输后保持完整。在某些实施方式的DNA组合物中,所述DNA在被标记后保持完整(例如没有断裂的双链)。在某些实施方式的DNA组合物中,所述DNA当如本文中所述在流体纳米通道中线性化时保持完整。在某些实施方式中,对所述DNA的靶序列进行选择以避免可能由切口标记(带有或不带有切口平移)引起的断裂。例如,在某些实施方式的标记方法、DNA组合物和试剂盒中,所述靶序列被选择成在据预测在切口平移标记后形成或易于形成脆性位点的区域中(参见例如图4A)。在某些实施方式的第一标记方法、DNA组合物和试剂盒中,所述靶序列在据预测在切口平移标记后形成脆性位点的区域中。在某些实施方式的标记方法、DNA组合物和试剂盒中,所述标记的DNA能够在流体纳米通道中线性化后保持完整。在某些实施方式的标记方法、DNA组合物和试剂盒中,所述标记的DNA在流体纳米通道中线性化后保持完整。
根据某些实施方式的标记方法,所述靶序列被选择成由在基于基序的酶促标记例如切口标记后不包含不均匀分布的标记物的基因组区域、例如包含多个重复序列的基因组区域构成。在某些实施方式的标记方法、DNA组合物和试剂盒中,所述靶序列由在基于基序的酶促标记例如切口标记后不包含不均匀分布的标记物的基因组区域、例如包含多个重复序列的基因组区域构成。根据某些实施方式的标记方法,所述靶序列被选择成由一个或多个重复序列(例如重复的元件例如Alu元件或Duff1220元件)构成。在某些实施方式的标记方法、DNA组合物和试剂盒中,所述靶序列由一个或多个重复序列构成。根据某些实施方式的标记方法、DNA组合物和试剂盒,可以构成所述靶序列的重复元件的实例包括但不限于Alu元件和Duff1220元件。根据某些实施方式的标记方法,所述靶序列被选择成包含SNP。在某些实施方式中,所述方法包括将所述DNA与特异性针对SNP的一组gdRNA相接触。所述组中的每个gDRNA可以特异性针对不同的SNP并包含不同的标记物。在某些实施方式中,所述标记方法包括对定制的靶进行标记。在某些实施方式中,所述标记方法包括对条形码序列进行标记。
在某些实施方式的标记方法中,所述方法还包括将所述标记的DNA在流体纳米通道中线性化。所述DNA可以例如在线性化后保持完整。例如但没有任何限制,所述流体纳米通道可以具有至少10nm的长度和小于1000nm例如小于900nm、800nm、700nm、600nm、500nm、400nm、300nm、200nm或100nm的横截面直径。在某些实施方式中,所述方法还包括在所述纳米通道中检测所述线性化的DNA上如本文中所描述的两个标记物之间的相对距离。在某些实施方式中,所述方法包括在所述纳米通道中检测所述线性化的DNA上如本文中所描述的两个标记的CRISPR-dCas复合体之间的相对距离。
在某些实施方式的标记方法中,所述dCas蛋白包含本文中所描述的dCas9蛋白、基本上由其构成或由其构成。在某些实施方式的涉及形成两个或更多个标记的CRISPR-dCas复合体的标记方法中,所有的所述dCas蛋白可以包含本文中所描述的dCas9、基本上由其构成或由其构成。在某些实施方式中,所述dCas(例如dCas9)如本文中所述在HNH结构域和/或RuvC样结构域中包含突变或缺失。在某些实施方式的标记方法中,所述dCas蛋白(例如dCas9)未被标记。在某些实施方式的包含多个CRISPR-dCas复合体的标记方法中,所述dCas蛋白(例如dCas9)均未被标记。
在某些实施方式的标记方法中,所述crRNA包含与本文中所描述的靶序列或其部分互补的约10-40个核苷酸的序列。
在某些实施方式的标记方法中,将所述DNA、dCas蛋白和标记的gRNA温育,以便在单次温育中标记所述DNA。在某些实施方式的标记方法中,所述DNA、dCas蛋白和/或标记的gRNA以至少约1ng/μl的浓度进行标记,例如至少约1、2、3、4、5、6、7、8、9、10、15、20、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450或500ng/μl,包括在所列出的任两个值之间的范围例如约1ng/μl至约10ng/μl、约1ng/μl至约50ng/μl、约1ng/μl至约100ng/μl、约1ng/μl至约200ng/μl、约1ng/μl至约300ng/μl、约1ng/μl至约500ng/μl、约5ng/μl至约10ng/μl、约5ng/μl至约50ng/μl、约5ng/μl至约100ng/μl、约5ng/μl至约200ng/μl、约5ng/μl至约300ng/μl、约5ng/μl至约500ng/μl、约10ng/μl至约50ng/μl、约10ng/μl至约100ng/μl、约10ng/μl至约200ng/μl、约10ng/μl至约300ng/μl、约10ng/μl至约500ng/μl、约50ng/μl至约100ng/μl、约50ng/μl至约200ng/μl、约0ng/μl至约300ng/μl或约50ng/μl至约500ng/μl。
在某些实施方式中,除了用CRISPR-dCas标记之外,所述标记方法还包括通过另外的化学方法标记所述DNA,例如使用酶例如DLE-1的直接酶促标记并且任选地除了DNA的酶促标记之外还包括染色剂(例如BIONANO GENOMICS的“DLS”技术)或产生切口并随后进行切口标记和修复(例如BIONANO GENOMICS的“NLRS”技术),以产生包含两个或更多个带有不同标记物(例如不同颜色)的特异性基序(例如靶序列)的DNA。在某些实施方式中,所述标记方法还包括通过包含切口平移的切口标记来标记所述DNA,以便掺入本文中所描述的标记的核苷酸。在某些实施方式中,所述切口平移包括用切口酶在所述DNA上产生切口,并将所述切口的DNA与聚合酶和标记的核苷酸温育。所述核苷酸可以用与所述标记物相同或不同的核苷酸标记物标记。所述聚合酶可以将所述标记的核苷酸以5’->3’的方向掺入到所述DNA中。
正如本文中所公开的,非限制性的示例性标记物包括:荧光团、量子点、树枝状聚合物、纳米丝、珠子、半抗原、链霉亲和素、亲和素、中性亲和素、生物素、反应性基团、肽、蛋白质、磁珠、放射性标记物、非光学标记物以及所列出的物品中的两者或更多者的组合。在某些实施方式中,所述标记物是光学标记物。如果所述标记方法包含两种或更多种不同标记物,则所述标记物中的两者或更多者可以是相同类型的(例如两种不同的荧光团),或者所述不同标记物中的两者或更多者可以是两种或更多种不同类型的(例如荧光团和量子点)或所列出的物品中的两者或更多者的组合。在某些实施方式的标记方法、DNA组合物和试剂盒中,所述DNA还用非特异性标记物例如骨架标记物如YOYO标记物标记。
某些实施方式包括用于执行本文中描述的任何标记方法的试剂盒。所述试剂盒可以包含本文中描述的dCas蛋白,例如dCas9蛋白。在某些实施方式中,所述试剂盒包含标记物。在某些实施方式中,所述标记物未被附连到所述dCas蛋白。在某些实施方式中,所述试剂盒包含gRNA。所述gRNA可以包含标记物,例如本文中描述的任何标记物。在某些实施方式中,所述gRNA包含本文中所描述的crRNA和tracrRNA。所述crRNA可以与指定的DNA靶序列或其部分例如重复序列如Alu或Duff1220互补。在某些实施方式中,所述crRNA与SNP互补。在某些实施方式的试剂盒中,所述试剂盒还包含本文中所描述的切口酶,例如Nt.BspQI。在某些实施方式中,所述切口酶被修饰成仅在DNA的一条链上产生切口。在某些实施方式中,所述试剂盒还包含本文中所描述的流体纳米通道。在某些实施方式中,所述试剂盒还包含直接标记酶。所述直接标记酶可以是非切口酶例如DLE-1。在某些实施方式中,所述试剂盒还包含DLE-1。
某些实施方式包括DNA组合物。所述DNA组合物可以包含DNA、本文中所描述的dCas蛋白(例如dCas9)和本文中所描述的包含crRNA和tracrRNA的标记的gRNA,基本上由它们构成或由它们构成。所述dCas(例如dCas9)、标记的gRNA和DNA可以构成复合体,所述复合体包含与所述DNA的靶序列或其部分杂交的crRNA。
在某些实施方式中,所述DNA组合物的DNA在本文中所描述的流体纳米通道中线性化。由于设想了本文中的某些实施方式的CRISPR-dCas标记不切口或切割所述DNA,因此在某些实施方式中,所述DNA组合物的DNA在所述纳米通道中可能是完整的(例如与标记前的DNA相比没有任何双链断裂)。在某些实施方式中,所述流体纳米通道具有至少10nm的长度和小于1000nm的横截面直径。在某些实施方式中,所述DNA组合物的DNA具有千碱基或兆碱基范围内的长度,例如至少1kb、2kb、3kb、5kb、10kb、20kb、30kb、40kb、50kb、100kb、150kb、250kb、500kb、1Mb、1.5Mb或2Mb,包括在所列出的任两个值之间的范围(例如1kb–2Mb、5kb–2Mb、10kb–2Mb、20kb–2Mb、100kb–2Mb、500kb–2Mb、1kb–1Mb、5kb–1Mb、10kb–1Mb、100kb–1Mb、200kb–1Mb、500kb–1Mb、1kb–500kb、5kb–500kb、10kb–500kb、20kb–500kb、100kb–500kb、1kb–100kb、5kb–100kb、10kb–100kb、20kb–100kb、50kb–100kb、1kb–50kb、5kb–50kb、10kb–50kb、1kb–10kb、5kb–10kb或1kb–5kb)。在某些实施方式中,所述DNA组合物的DNA在本文中所描述的流体纳米通道中线性化后维持其完整性(和长度)。
在某些实施方式中,所述DNA组合物的dCas如本文中所述在HNH结构域和/或RuvC样结构域中包含一个或多个突变和/或一个或多个缺失。在某些实施方式中,所述DNA组合物包含本文中所描述的dCas9。在某些实施方式中,对于包含两种或更多种dCas-CRIPSR标记物的DNA组合物来说,每个dCas是本文中所描述的dCas9。在某些实施方式中,对于包含两种或更多种dCas-CRIPSR标记物的DNA组合物来说,每个dCas如本文中所述在HNH结构域和/或RuvC样结构域中包含突变或缺失。在某些实施方式中,所述DNA组合物的dCas本身未被标记。在某些实施方式中,所述DNA组合物的dCas均未被标记。
在某些实施方式中,所述DNA组合物的gRNA如本文中所述包含与DNA上的靶序列或其部分互补的crRNA。所述crRNA如本文中所述可以包含与所述靶序列或其部分互补的约10-40个核苷酸的序列。设想了所述靶序列可以包含适合于CRISPR靶向的任何序列(例如通过替换所述crRNA探针,可以靶向所需序列)、基本上由所述序列构成或由所述序列构成。
在某些实施方式中,所述DNA组合物的DNA如本文中所述被进一步切口标记。因此,在某些实施方式中,所述DNA组合物的DNA包含标记的核苷酸。所述核苷酸可以用与所述gRNA上也标记所述DNA的标记物相同或不同的核苷酸标记物来标记。在某些实施方式的DNA组合物中,所述DNA的每种标记物(gRNA和/或核苷酸标记物)选自:荧光团、量子点、树枝状聚合物、纳米丝、珠子、半抗原、链霉亲和素、亲和素、中性亲和素、生物素、反应性基团、肽、蛋白质、磁珠、放射性标记物或非光学标记物以及所列出的物品中的两者或更多者的组合。在某些实施方式的DNA组合物中,所述标记物包含光学标记物。在某些实施方式例如包含两种或更多种标记物的DNA组合物中,所述DNA组合物的DNA包含彼此不同但各自是相同类型的两种或更多种标记物(例如两种不同颜色的荧光团)。在某些实施方式例如包含两种或更多种标记物的DNA组合物中,所述DNA组合物的DNA包含彼此不同并且属于不同类型的两种或更多种标记物(例如荧光团和量子点)。在某些实施方式中,所述DNA还用非特异性标记物例如骨架标记物例如YOYO标记物标记。
本文中的某些实施方式的方法和DNA组合物的dCas(例如dCas9)结合标记系统使用向导RNA(gRNA)将所述dCas(例如dCas9)引导到靶位点。所述gRNA包含荧光偶联的反式激活crRNA(tracrRNA)和含有与感兴趣的位点互补的~20个核苷酸的序列的crRNA,基本上由它们构成或由它们构成。HNH和RuvC样结构域核酸酶中的双重突变将所述Cas9酶改变成以非常高的特异性和效率结合,但没有催化活性。在本文的某些实施方式中,通过合成带有标记物(例如Atto647)的TracrRNA代替所述dCas9蛋白的标记,对复合体进行标记。使用dCas9、标记的TracrRNA和定制的crRNA探针,可以通过替换所述crRNA探针来靶向适合于CRISPR靶向的任何序列。这种DNA标记方法CRISPR结合体,已被应用于标记BAC DNA和兆碱基长度的人类基因组DNA和使用SAPHYR或IRYS系统对这种DNA在纳米通道阵列中进行成像(Bionano Genomics,San Diego,CA,参见图1A-1E和5A-5D)。
Cas9核酸酶缺陷型衍生物(dCas9)通过分别与转录-调控结构域或荧光蛋白融合,也可用于活细胞中基因表达的控制和基因组位点的可视化。CRISPR系统的体外研究表明,dCas9/sgRNA对其靶DNA具有强且稳定的亲和性。
根据本文中某些实施方式的方法、DNA组合物和试剂盒,描述了使用dCas9蛋白的序列特异性标记。所述方法是快速、方便、成本效益高且无损的。特定序列的灵活且高效的荧光标记允许能够在SAPHYR纳米通道阵列(Bionano Genomics,San Diego,CA)中获得沿着长的线性DNA分子的背景特异性序列信息。这种集成的荧光DNA双链标记不仅可以使全基因组作图更加准确并提供更多信息,而且可以特异性靶向用于临床测试、包括SNP的检测的某些位点。另外,它可以使所述标记的双链DNA以长的完整的拉伸形式获得,用于纳米通道阵列中的高通量分析,以及使用用于标记的大DNA分子的拉伸和成像的可选方法用于标记的DNA区域的较低通量的靶向分析。因此,某些实施方式的标记方法极大改进了自动高通量基因组广度作图以及含有重复和结构变体DNA的复杂区域的靶向分析两者。dCas9荧光结合系统允许开发组合的、多重的多色成像系统,因此可以为结构变异的快速遗传诊断提供优势。
根据本文中描述的某些实施方式的方法和试剂盒,使用dCas蛋白的序列特异性标记方法可能在光学作图中有用。某些实施方式的该单步骤标记不损坏所述DNA,并且当在纳米通道阵列例如Bionano Genomics IRYS或SAPHYR系统中进行单分子光学作图时,所述特定序列的灵活且高效的荧光标记能够获取背景特异性序列信息。某些实施方式的方法和试剂盒不仅可以通过添加第二种颜色和提高信息密度为全基因组结构变异分析产生优越的品质和灵敏度,而且能够靶向广泛种类的序列例如长串联重复序列、病毒整合位点、转入基因,并且甚至可用于单核苷酸变体的基因分型。
下述参考文献整体通过参考并入本文:
McCaffrey,J.,Sibert,J.,Zhang,B.,Zhang,Y.,Hu,W.,Riethman,H.和Xiao,M.(2016),“用于全基因组作图和结构变异分析的双链DNA的CRISPR-CAS9 D10A切口酶靶特异性荧光标记”(CRISPR-CAS9 D10A nickase target-specific fluorescent labeling ofdouble strand DNA for whole genome mapping and structural variationanalysis),Nucleic Acids Res.Jan 29;44(2):e11
Bittel DC,Z.X.(2014),“非综合征性先天性心脏缺陷法洛四联症的超高分辨率基因中心基因组结构分析(Ultra high-resolution gene centric genomic structuralanalysis of a non-syndromic congenital heart defect,Tetralogy of Fallot),PLoSOne,9(1),e87472.
Bochukova EG,H.N.-S.-S.(2010),“与严重的早发性肥胖症相关的大且罕见的染色体缺失”(Large,rare chromosomal deletions associated with severe early-onsetobesity),Nature,463(7281),666-70.
Brand H,P.V.(2014),“早发性神经精神障碍中的隐蔽和复杂的染色体畸变”(Cryptic and complex chromosomal aberrations in early-onset neuropsychiatricdisorders),Am J Hum Genet.,95(4),454-61.
Butcher NJ,K.T.(2013),“早发性帕金森氏病与22q11.2缺失综合征之间的关联:帕金森氏病的新的遗传形式的鉴定及其临床意义”(Association between early-onsetParkinson disease and 22q11.2 deletion syndrome:identification of a novelgenetic form of Parkinson disease and its clinical implications),JAMANeurol.,70(11),1359-66.
Chaisson MJ,H.J.(2015),“使用单分子测序解析人类基因组的复杂性”(Resolving the complexity of the human genome using single-moleculesequencing),Nature,517(7536),608-11.
Chen B,Gilbert LA,Cimini BA等(2013),“通过优化的CRISPR/Cas系统在活的人类细胞中对基因组位点进行动态成像”(Dynamic Imaging of Genomic Loci in LivingHuman Cells by an Optimized CRISPR/Cas System),Cell.2013;155(7):1479-1491
Deininger PL,Batzer MA.(1999),“Alu重复序列和人类疾病”(Alu repeats andhuman disease),Mol Genet Metab.1999Jul;67(3):183-93.综述。
Deng,W,Shi X,Tjian R,Lionnet T,Singer.(2015),“固定的细胞中基因组位点的CASFISH:CRISPR/Cas9介导的原位标记”(CASFISH:CRISPR/Cas9-mediated in situlabeling of genomic loci in fixed cells),Proc.Natl.Acad.Sci.,112(38),11870-11875
Dominguez AA,Lim WA,Qi LS.(2016),“超越编辑:改造CRISPR-Cas9用于精确基因组调控和质询”(Beyond editing:repurposing CRISPR-Cas9 for precision genomeregulation and interrogation),Nat Rev Mol Cell Biol.2016Jan;17(1):5-15
Dumas,LJ.等(2012),“与人类脑尺寸病理学和进化有牵连的DUF1220结构域拷贝数”(DUF1220-Domain Copy Number Implicated in Human Brain-Size Pathology andEvolution),American Journal of Human Genetics 91.3:444–454
Gasiunas G,Barrangou R,Horvath P,Siksnys V.(2012),“Cas9-crRNA核糖核蛋白复合体介导特异性DNA切割,以实现细菌中的适应性免疫”(Cas9-crRNAribonucleoprotein complex mediates specific DNA cleavage for adaptiveimmunity in bacteria),Proc.Natl.Acad.Sci.109,E2579–E2586
Jinek M,Chylinski K,Fonfara I,Hauer M,Doudna JA,Charpentier E.(2012),“适应性细菌免疫中的可编程的双RNA指导的DNA核酸内切酶”(A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity),Science 337,816–821
Lo A和Qia L.(2017),“CRISPR–Cas系统对基因表达的遗传和表观遗传控制”(Genetic and epigenetic control of gene expression by CRISPR–Cas systems),Version 1.F1000Res.2017;6:F1000 Faculty Rev-747.doi:10.12688/f1000research.11113.1
McCaffrey J,Young E,Lassahn K,Sibert J,Pastor S,Riethman H和Xiao M(2017),“高通量单分子端粒表征”(High-throughput single-molecule telomerecharacterization),Genome Res.2017.27:1904-1915
Qi LS,Larson MH,Gilbert LA,Doudna JA,Weissman JS,Arkin AP,Lim WA.(2013),“改造CRISPR作为用于基因表达的序列特异性控制的RNA指导的平台”(Repurposing CRISPR as an RNA-guided platform for sequence-specific controlof gene expression),Cell.2013Feb28;152(5):1173-83.
Sternberg SH,Doudna JA(2015),“使用CRISPR-Cas9扩展生物学家的工具箱”(Expanding the Biologist’s Toolkit with CRISPR-Cas9),Mol Cell 58(4):568–574.
实施例
上文讨论的实施方式的某些方面在下面的实施例中进一步详细公开,所述实施例不打算以任何方式限制本公开的范围。
使用CRISPR结合体来靶向1号染色体上存在的Duff1220重复序列。所述Duff1220重复序列以可变的拷贝数存在,组织在所述重复序列单元的几个串联的阵列中,在灵长动物中已将所述拷贝数与脑尺寸相关联。长的串联重复序列可能对测量具有挑战性,因为它们比读出序列更长并且对于通过FISH或其他细胞遗传学方法进行可视化来说过短。跨越所述重复序列阵列的超长分子的光学作图可以允许对所述重复序列单元进行计数并将所述阵列放置在背景中;然而,所述重复序列通常不被常规的标记酶标记。
在Mccaffery等人的基础上合成了靶向探针,并使用荧光标记的tracrRNA产生CRISPR复合体。将所述复合体与线性化后的预期含有13个Duff1220拷贝的BAC DNA温育。对CRISPR结合复合体浓度进行滴定以确定在这种测定法中的理想浓度,其为约15nM。在这个浓度下,信噪比(SNR)允许将所述样品直接装载到SAPHYR芯片上,而不必清除过量的荧光团,这被认为简化了所述方法。对于杂交来说,每150ng DNA 120nM(每ng DNA约0.8nM)被确定是理想的浓度(数据未示出)。从所述图像测量在每个结合位点处标记物检测的效率以及信号被称为在预期的Duff1220位点之外的频率。靶结合效率为86%,而约16%的标记物被发现是脱靶的。这种性能与使用Nt.BspQI的标准切口标记相近。
在B19的BAC上的dCas9/Duf1220-Atto647标记的结果示出在图2A-B中。预期的标记图案和标记物之间的距离在上方示出,对应于荧光显微术图像。如图2A中所示,DUF1220三联体的单个拷贝为4.7kb。所述dCas9标记方法能够使用被设计成与所述DUF1220重复序列单元互补的gRNA来标记这个区域。正如所预测的,所述串联重复序列相隔大约4.7kb。图2B示出了所述DUF1220 gRNA标记物和簇集分子的柱状图。表1示出了图2B的对齐率的计算。
表1.对齐率
因此,根据本文中的某些实施方式,用CRISPR-dCas标记长的DNA产生了准确标记,其具有与切口标记相近的准确度。
为了测试CRISPR结合体对人类基因组DNA的特异性和效率,选择了另一个靶序列,其与整个基因组中约四分之一的Alu重复序列结合。这个靶的显著优势在于它在基因组中非常频繁地出现,并且也值得研究,因为它是可移动的并涉及结构变异和疾病。Alu重复序列是人类基因组中最丰富的可转座元件,并且它们可以拷贝自身并插入到新的位置中,有时改变基因表达并甚至引起疾病(Deininger 1999)。不同于某些其他潜在的~20-bp的CRISPR靶,这个Alu重复序列在基因组中非常频繁地出现,允许在整个基因组中相对高的标记物密度。CRISPR结合体-Alu的使用与单独的切口标记相比可以为标准的基因组作图实验增添另外70%的标记物,或者与单独使用DLE-1的DLS相比增添另外约50%的标记物,可能为基因组变异分析提供更高的信息密度。此外,由于CRISPR结合体可以采取可选的颜色,因此所述第二种颜色信号可以提供另外的图案唯一性。
为了估算结合和检测的特异性和效率,首先将Nt.BspQI位点用使用绿色染料的标准的基于切口平移的标记进行标记,然后将我们的靶向Alu的CRISPR结合复合体(红色染料atto647)结合在人类假单倍体葡萄胎细胞系CHM1上。收集数据并在Nt.BspQI图案的基础上作图到人类参比。图3A示出了这些对齐的分子,所述Nt.BspQI信号采用深紫色,所述Alu信号用紫红色示出。所述紫红色信号在所有分子中以一致的图案出现,以致于作图到特定区域的所有分子中大约90%的分子在作图的切口标记物附近含有相同的信号。当对已被所述复合体靶向的Alu重复序列的位置进行注释时,它们形成协调的图案。尽管在某些情况下分子的共有区显示出与参比位点不一致的标记物,但这与个体之间Alu图案的高度可变性相一致。图3A示出了通过dCas9/Alu-Atto647在CHM1上进行的基因组作图。
图3B描绘了通过dCas9/Alu在CHM1上进行的Alu重复序列作图。将分子用NLRS和+CRISPR结合体两者标记,将标记的分子通过它们的切口标记物图案与hg19参比的使用Nt.BspQI的计算机模拟消化对齐。预测的Alu靶位点在上方示出。Nt.BspQI信号用紫色显示在所述分子上,Alu信号用橙色显示。作图到特定区域的所有分子中大约90%的分子在作图的切口标记物附近含有相同的橙色信号。所述Alu重复序列信号形成与预测的Alu位点协调的图案。
因此,根据本文中的某些实施方式,DNA的CRISPR-dCas标记可以标记重复的元件。
DNA样品、纯化
超高分子量DNA从BAC克隆CH17-353B19和RP11-963J21(invitrogen)和人类细胞系CHM1制备。使用Bionano plug裂解流程和试剂,将细胞固定化在低熔点琼脂糖中,裂解,用蛋白酶K处理,然后清洗,用A garase(Thermo Fisher,Wilmington,MA)溶解并进行液滴透析。
CRISPR-RNA(crRNA)制备
20nt的crRNA靶序列从crispr.mit.edu网站设计。用于DUF1220结构域的基因组序列(AAGUUCCUUUUAUGCAUUGG,SEQ ID NO:1)的DUF crRNA和用于序列(UGUAAUCCCAGCACUUUGGG,SEQ ID NO:2)的Alu crRNA两者均由Synthego(Menlo Park,CA)合成。
荧光向导RNA的制备
通用的ATTO
荧光CRISPR-dCas9 RNP组装体
含有双核酸酶突变(D10A和H840A)的酿脓链球菌(S.pyogenes)Cas9蛋白(dCas9)购自PNA Bio Inc(#CD01,Thousand Oaks,CA),并用提供的稀释剂稀释到1mg/mL或6μM的储用物浓度。将荧光gRNA(50pmol)与600ng dCas9(3.7pmol)、1×NEB缓冲液3和1×BSA(NEB)混合,并在37℃下温育60min。该荧光CRISPR RNP在4℃下稳定长达4周,或者在-80℃下长期稳定。
BAC DNA或人类基因组DNA的dCas9荧光结合
将150ng线性化的DNA与50ng ATTO647-RNP混合,并在37℃下温育60min。将所述DNA骨架用YOYO-1染色,并用蓝色显示。将所述染色的样品装载到纳米通道内并成像。
使用dCas9荧光结合和序列基序标记的双色基因组作图
将人类基因组DNA样品用Nt.BspQI(NEB)在37℃产生切口2小时。然后将所述切口的DNA用Taq DNA聚合酶(NEB)、ATTO532dUTP dAGC和1×Thermopol缓冲液(NEB)在72℃标记60min。将所述切口用NAD+、dNTPs和Taq DNA连接酶在37℃修复30min。然后将所述样品用Qiagen蛋白酶K处理30min,并用PMSF失活30min。将所述样品在0.1μm NC膜(Millipore)上在TE中透析2小时。在透析后,将60-100ng DNA样品与1×NEB缓冲液3中的50ng ATTO647-RNP和1×BSA(NEB)在37℃温育60min。将DNA骨架用YOYO-1染色,并用蓝色显示。所述DNA骨架染色步骤被延迟到CRISPR结合之后。将所述染色的样品装载到纳米通道内并成像。
DNA的RNP结合和DNA骨架染色
将150ng DNA(来自于BAC和人类,分别地)与0.4μL(0.15pmol)RNP合并,并在37℃温育60min。使用来自于Bionano NLRS试剂盒的DNA染色剂将DNA骨架染色。将所述样品在室温储存过夜,并转移到4℃用于长期储存。
数据获取和可视化
DUF1220作图:将16μL样品装载到IRYS芯片流动池中。启动IRYS运行,并在电泳和成像的20个循环后,使用IrysView将分子与参比序列对齐。
Alu重复序列作图:将19μL样品装载到SAPHYR芯片流动池中。通过Bionano Access软件启动SAPHYR运行。在电泳和成像的30个循环后,通过Bionano Solve软件进行从头组装。使用Bionano Access将共有序列图谱和分子与参比序列对齐并进行可视化。
序列表
<110> 生物纳米基因公司(BioNano Genomics, Inc.)
<120> DNA的标记
<130> BNGEN.049WO
<150> 62/689,650
<151> 2018-06-25
<150> 62/696,696
<151> 2018-07-11
<160> 2
<170> PatentIn version 3.5
<210> 1
<211> 20
<212> RNA
<213> 人工序列
<220>
<223> 合成的寡核苷酸
<400> 1
aaguuccuuu uaugcauugg 20
<210> 2
<211> 20
<212> RNA
<213> 人工序列
<220>
<223> 合成的寡核苷酸
<400> 2
uguaauccca gcacuuuggg 20
- 血浆游离DNA双分子标记、标记和检测血浆cfDNA的方法及其用途
- 基于cfDNA的测序及数据分析的癌症相关生物标记及其在cfDNA样品分类中的应用