一种四核铜配合物及其制备方法和应用
文献发布时间:2023-06-19 13:45:04
技术领域
本发明涉及合成化合物技术领域,尤其涉及一种四核铜配合物及其制备方法和应用。
背景技术
一氧化氮是氮的化合物,是一种无色、无味、难溶于水的气体。因为一氧化氮带有自由基,这使它的化学性质非常活泼。一氧化氮是一种极不稳定的生物自由基,半衰期只有3~5s,分子小结构简单。在人体中一氧化氮在心、脑血管调节、神经、免疫调节等方面有着十分重要的生物学作用;但是当人体内一氧化氮过量时,会导致人出现晕厥,体内的一氧化氮过高时,会造成血管明显的舒张,导致回心血量减少出现血压减低,甚至血流不畅,严重者会出现呼吸衰竭等症状。在空气中一氧化氮会很快转变为二氧化氮产生刺激作用,产生的氮氧化物会造成呼吸道刺激,损害呼吸道;人体处于过量的氮氧化物环境中,会出现胸闷、呼吸窘迫、紫绀等症状,一氧化氮浓度高可致高铁血红蛋白血症。过量的一氧化氮还会对水体、土壤和大气造成污染。因此,如何调节一氧化氮的含量成为亟需解决的问题。
发明内容
本发明的目的在于克服现有技术中的缺陷,提供一种四核铜配合物及其制备方法和应用。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种四核铜配合物,具有如下所示结构:
所述R为甲基、甲氧基、F、Cl或Br。
本发明还提供了所述四核铜配合物的制备方法,包含下列步骤:
(1)将取代苯酚、乙酸铅和无水乙醇混合,得到混合溶液;
(2)将混合溶液和N,N-二(3-氨丙基)-2-吡啶甲胺的无水甲醇溶液混合后进行配位缩合反应,得到铅配合物;
(3)在碱性条件下,将铅配合物和硫酸铜的无水甲醇溶液混合后进行置换反应,即得所述四核铜配合物;
所述取代苯酚为2,6-二甲酰基-4-甲基苯酚、2,6-二甲酰基-4-甲氧基苯酚、2,6-二甲酰基-4-氟苯酚、2,6-二甲酰基-4-氯苯酚或2,6-二甲酰基-4-溴苯酚。
作为优选,所述步骤(1)中取代苯酚和乙酸铅的摩尔比为0.4~0.6:0.4~0.6;
所述取代苯酚和无水乙醇的用量比为0.4~0.6mmol:10~20mL。
作为优选,所述步骤(1)中混合的方式为搅拌,所述搅拌的转速为200~400rpm,所述搅拌的时间为1.8~2.2h。
作为优选,所述步骤(2)中N,N-二(3-氨丙基)-2-吡啶甲胺和步骤(1)中取代苯酚的摩尔比为0.4~0.6:0.4~0.6;
所述N,N-二(3-氨丙基)-2-吡啶甲胺和N,N-二(3-氨丙基)-2-吡啶甲胺的无水甲醇溶液的摩尔体积比为0.4~0.6mmol:8~12mL。
作为优选,所述步骤(2)中混合为将N,N-二(3-氨丙基)-2-吡啶甲胺的无水甲醇溶液滴加进混合溶液中,所述滴加的速率为2~4滴/秒;
所述步骤(2)中配位缩合反应的时间为2.8~3.2h。
作为优选,所述步骤(3)中硫酸铜和步骤(1)中取代苯酚的摩尔比为1~1.5:0.4~0.6;
所述硫酸铜和硫酸铜的无水甲醇溶液的摩尔体积比为1~1.5mmol:12~18mL;
所述碱性条件的pH值为8~9,所述碱性条件的调节试剂为三乙胺。
作为优选,所述步骤(3)中混合为将硫酸铜的无水甲醇溶液滴加进铅配合物中,所述滴加的速率为2~4滴/秒。
作为优选,所述步骤(3)中置换反应的时间为3.5~4.5h。
本发明还提供了所述四核铜配合物在制备一氧化氮调节剂产品中的应用。
本发明提供了一种四核铜配合物,本发明以取代苯酚和N,N-二(3-氨丙基)-2-吡啶甲胺为原料,以大配位数的铅离子为模板,进行配位缩合反应得到了铅配合物;将铅配合物和硫酸铜混合,将配合物中的铅离子置换,得到了四核铜配合物,置换出来的铅离子形成硫酸铅沉淀,便于后续的过滤分离。本发明提供的制备方法简单,核心步骤为配位缩合反应和置换反应,能高效的制备得到目标配合物。经过实验验证,本发明制备的四核铜配合物和一氧化氮具有明显的可逆反应进行,并且对DNA无切割伤害效果,可用于制备生物体内和环境中一氧化氮调节剂产品。
附图说明
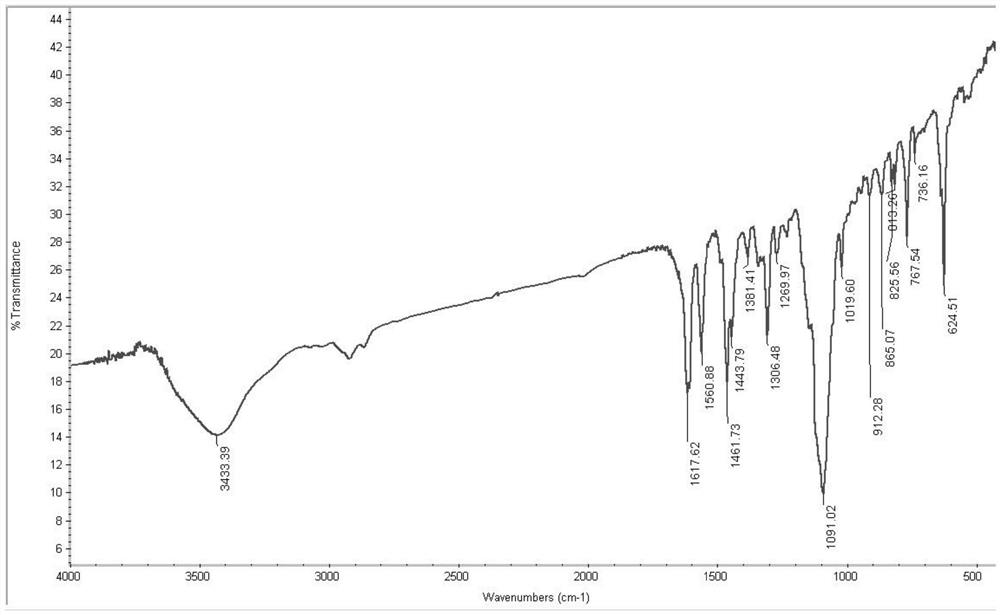
图1为实施例1制备的四核铜配合物的红外光谱图;
图2为实施例1制备的四核铜配合物的分子结构图;
图3为实施例1制备的四核铜配合物的金属配位环境多面体图;
图4为实施例1制备的四核铜配合物的平面定义图;
图5为实施例1制备的四核铜配合物通入NO的颜色对比图;
图6为实施例1制备的四核铜配合物与CT-DNA的循环伏安图;
图7为实施例1制备的四核铜配合物对pBR322 DNA的切割图。
具体实施方式
本发明提供了一种四核铜配合物,具有如下所示结构:
所述R为甲基、甲氧基、F、Cl或Br。
本发明提供了所述四核铜配合物的制备方法,包含下列步骤:
(1)将取代苯酚、乙酸铅和无水乙醇混合,得到混合溶液;
(2)将混合溶液和N,N-二(3-氨丙基)-2-吡啶甲胺的无水甲醇溶液混合后进行配位缩合反应,得到铅配合物;
(3)在碱性条件下,将铅配合物和硫酸铜的无水甲醇溶液混合后进行置换反应,即得所述四核铜配合物;
所述取代苯酚为2,6-二甲酰基-4-甲基苯酚、2,6-二甲酰基-4-甲氧基苯酚、2,6-二甲酰基-4-氟苯酚、2,6-二甲酰基-4-氯苯酚或2,6-二甲酰基-4-溴苯酚。
在本发明中,所述步骤(1)中乙酸铅优选为四水合乙酸铅。
在本发明中,所述步骤(1)中取代苯酚和乙酸铅的摩尔比优选为0.4~0.6:0.4~0.6,进一步优选为0.45~0.55:0.45~0.55,更优选为0.48~0.52:0.48~0.52。
在本发明中,所述取代苯酚和无水乙醇的用量比优选为0.4~0.6mmol:10~20mL,进一步优选为0.45~0.55mmol:12~18mL,更优选为0.48~0.52mmol:14~16mL。
在本发明中,所述步骤(1)中混合的方式优选为搅拌,所述搅拌的转速优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm;所述搅拌的时间优选为1.8~2.2h,进一步优选为1.9~2.1h,更优选为1.95~2.05h。
在本发明中,所述步骤(2)中N,N-二(3-氨丙基)-2-吡啶甲胺和步骤(1)中取代苯酚的摩尔比优选为0.4~0.6:0.4~0.6,进一步优选为0.45~0.55:0.45~0.55,更优选为0.48~0.52:0.48~0.52。
在本发明中,所述N,N-二(3-氨丙基)-2-吡啶甲胺和N,N-二(3-氨丙基)-2-吡啶甲胺的无水甲醇溶液的摩尔体积比优选为0.4~0.6mmol:8~12mL,进一步优选为0.45~0.55mmol:9~11mL,更优选为0.48~0.52mmol:9.5~10.5mL。
在本发明中,所述步骤(2)中混合优选为将N,N-二(3-氨丙基)-2-吡啶甲胺的无水甲醇溶液滴加进混合溶液中,所述滴加的速率优选为2~4滴/秒,更优选为3滴/秒。
在本发明中,所述步骤(2)中配位缩合反应的时间优选为2.8~3.2h,进一步优选为2.9~3.1h,更优选为2.95~3.05h;所述配位缩合反应优选在搅拌条件下进行,所述搅拌的速率优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm。
在本发明中,以R为甲基的情况为例,所述步骤(2)的配位缩合反应如下所示:
在本发明中,所述步骤(3)中硫酸铜优选为五水硫酸铜。
在本发明中,所述步骤(3)中硫酸铜和步骤(1)中取代苯酚的摩尔比优选为1~1.5:0.4~0.6,进一步优选为1.1~1.4:0.45~0.55,更优选为1.2~1.3:0.48~0.52。
在本发明中,所述硫酸铜和硫酸铜的无水甲醇溶液的摩尔体积比优选为1~1.5mmol:12~18mL,进一步优选为1.1~1.4mmol:13~17mL,更优选为1.2~1.3mmol:14~16mL。
在本发明中,所述碱性条件的pH值优选为8~9,进一步优选为8.2~8.8,更优选为8.4~8.6;所述碱性条件的调节试剂优选为三乙胺。
在本发明中,所述步骤(3)中混合优选为将硫酸铜的无水甲醇溶液滴加进铅配合物中,所述滴加的速率优选为2~4滴/秒,更优选为3滴/秒。
在本发明中,滴加完毕硫酸铜的无水甲醇溶液后,滴加三乙胺调节pH值。
在本发明中,所述步骤(3)中置换反应的时间优选为3.5~4.5h,进一步优选为3.6~4.4h,更优选为3.8~4.2h;所述置换反应优选在搅拌条件下进行,所述搅拌的速率优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm。
在本发明中,所述置换反应为硫酸铜中的铜离子置换铅配合物中的铅离子,铅离子和硫酸根离子生成硫酸铅沉淀,置换反应搅拌结束后进行砂芯过滤,将过滤得到的硫酸铅进行洗涤,所述洗涤所用试剂优选为无水甲醇和乙腈的混合溶液,所述混合溶液和步骤(1)中取代苯酚的体积摩尔比优选为20~30mL:0.4~0.6mmol,进一步优选为22~28mL:0.45~0.55mmol,更优选为24~26mL:0.48~0.52mmol;所述无水甲醇和乙腈的体积比优选为0.8~1.2:0.8~1.2,更优选为0.9~1.1:0.9~1.1;洗涤结束后合并滤液和洗液,得到混合体系。
在本发明中,将混合体系和高氯酸钠的无水甲醇溶液混合后得到沉淀。
在本发明中,所述高氯酸钠优选为一水高氯酸钠,所述高氯酸钠和步骤(1)中取代苯酚的质量摩尔比优选为0.3~0.5g:0.4~0.6mmol,进一步优选为0.35~0.45g:0.45~0.55mmol,更优选为0.38~0.42g:0.48~0.52mmol;所述高氯酸钠和高氯酸钠的无水甲醇溶液的质量体积比优选为0.3~0.5g:8~12mL,进一步优选为0.35~0.45g:9~11mL,更优选为0.38~0.42g:9.5~10.5mL。
在本发明中,将高氯酸钠的无水甲醇溶液滴加进行混合体系中,所述滴加的速率优选为2~4滴/秒,更优选为3滴/秒。
在本发明中,所述高氯酸钠的无水甲醇溶液和混合体系的混合在搅拌条件下进行,所述搅拌的速率优选为200~400rpm,进一步优选为250~350rpm,更优选为280~320rpm;所述高氯酸钠的无水甲醇溶液滴加完毕后继续搅拌,所述继续搅拌的时间优选为3.5~4.5h,进一步优选为3.6~4.4h,更优选为3.8~4.2h。搅拌结束后,将沉淀进行砂芯抽滤获得沉淀。
在本发明中,将沉淀顺次进行水洗涤和乙醇洗涤,所述水洗涤中单次水和步骤(1)中取代苯酚的体积摩尔比优选为8~12mL:0.4~0.6mmol,进一步优选为9~11mL:0.45~0.55mmol,更优选为9.5~10.5mL:0.48~0.52mmol;所述水洗涤的次数优选大于等于2次,进一步优选大于等于3次,更优选大于等于4次。
在本发明中,所述乙醇洗涤中单次乙醇和步骤(1)中取代苯酚的体积摩尔比优选为8~12mL:0.4~0.6mmol,进一步优选为9~11mL:0.45~0.55mmol,更优选为9.5~10.5mL:0.48~0.52mmol;所述乙醇洗涤的次数优选大于等于2次,进一步优选大于等于3次,更优选大于等于4次。
在本发明中,将洗涤后的沉淀溶于乙醇乙腈溶液中静置,所述乙醇乙腈溶液和步骤(1)中取代苯酚的质量摩尔比优选为1.6~2.0g:0.4~0.6mmol,进一步优选为1.7~1.9g:0.45~0.55mmol,更优选为1.75~1.85g:0.48~0.52mmol;所述乙醇乙腈溶液中乙腈和乙醇的体积比优选为0.8~1.2:0.8~1.2,更优选为0.9~1.1:0.9~1.1;所述静置的时间优选大于等于28天,进一步优选大于等于30天,更优选大于等于32天。
在本发明中,静置结束后得到深绿色块状单晶即为四核铜配合物。
在本发明中,以R为甲基的情况为例,所述步骤(3)中置换反应如下所示:
本发明还提供了所述四核铜配合物在制备一氧化氮调节剂产品中的应用。
在本发明中,所述一氧化氮调节剂产品用于生物医疗、食品检测、环境监测中一氧化氮的调节、检测和监测;使被测对象的氮氧化物处于可控地步和合理水平。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
在本发明中,pBR 322 DNA购自TOYOBO,小牛胸腺DNA(CT-DNA)购自Sigma。
实施例1
将0.5mmol的2,6-二甲酰基-4-甲基苯酚、0.5mmol的四水合乙酸铅和15mL的无水乙醇混合在300rpm转速下搅拌2h获得混合溶液;将含0.5mmolN,N-二(3-氨丙基)-2-吡啶甲胺的10mL无水甲醇溶液以3滴/秒的速度滴加进混合溶液中,滴加完毕后溶液由亮黄色浑浊变为黄色透明溶液,在300rpm的转速下搅拌进行配位缩合反应,3h后获得铅配合物;然后将含1.3mmol五水硫酸铜的15mL无水甲醇溶液以3滴/秒的速度滴加进行铅配合物中,滴加完毕后溶液变成浅绿色浑浊,滴加三乙胺调节pH值为8.5,在300rpm的转速下搅拌进行置换反应,搅拌4h后使用砂芯过滤;将获得的硫酸铅沉淀使用25mL的无水甲醇和乙腈的混合溶液洗涤,无水甲醇和乙腈的体积比为1:1,合并滤液和洗液得到混合体系;控制转速为300rpm,将混合体系和高氯酸钠的无水甲醇溶液混合,混合的方式为滴加,将含0.4g一水高氯酸钠的10mL无水甲醇溶液以3滴/秒的速度滴加进混合体系中,滴加完毕后继续搅拌4h得到沉淀;将沉淀用10mL的水洗涤3次,然后用10mL的乙醇洗涤3次获得洗涤后的沉淀,将洗涤后的沉淀溶于1.8g乙醇乙腈溶液中,乙腈和乙醇的体积比为1:1,静置挥发28天后得到0.166g墨绿色片状单晶,产率为48%,即为四核铜配合物。
将本实施例制备得到的四核铜配合物进行红外光谱分析,结果如图1所示。元素分析(%):实测值:C为43.45,H为4.36,N为10.01;计算值(Cu
四核铜配合物的晶体结构数据和结构精修数据如表1所示,部分键长键角的数据如表2所示。
表1
表2
四核铜配合物的分子结构图如图2所示,从图中可以看出,该配合物为四核铜配合物,为中心对称,有两种配位环境不同的铜原子Cu1和Cu2。Cu1为五配位,为近似四方锥结构,酚氧原子O1、氢氧根氧原子O2、醋酸根氧原子O3和亚胺氮原子N2构成四方锥的底面,Cu1与他们的键长为
四核铜配合物的金属配位环境多面体图如图3所示,从图3中可以看出,Cu2的配位环境也是近似四方锥结构,但是锥顶方向与Cu1的四方锥锥顶相反,酚氧原子O1、氢氧根氧原子O2、亚胺氮原子N1和叔胺氮原子N3构成四方锥的底面,Cu2与他们的键长为
四核铜配合物的平面定义图如图4所示,从图中可以看出四个铜原子在一个平面上,构成一个平行四边形。平行四边形的短边和长边的Cu–Cu长度分别为
将四核铜配合物配制成浓度为5×10
将配合物和DNA结合后进行电化学性质测试,结果如图6所示,图中曲线a是没有加入DNA的配合物的循环伏安曲线,曲线b是加入DNA后的配合物的循环伏安曲线;没加入DNA时,阴极电位(E
将不同浓度的配合物和pBR322 DNA混合静置3h,结果如图7所示,图中1为空白DNA,2为混合50μM浓度配合物后的DNA,3为混合100μM浓度配合物后的DNA,4为混合200μM浓度配合物后的DNA,5为混合400μM浓度配合物后的DNA,6为混合800μM浓度配合物后的DNA。从图中可以看出,随着配合物浓度的增加,DNA环状结构Form I并不能被配合物切割成其他形式,这表明在实验条件下,配合物对DNA不会造成破坏。
实施例2
将0.4mmol的2,6-二甲酰基-4-溴苯酚、0.6mmol的四水合乙酸铅和14mL的无水乙醇混合在200rpm转速下搅拌2.2h获得混合溶液;将含0.4mmolN,N-二(3-氨丙基)-2-吡啶甲胺的8mL无水甲醇溶液以2滴/秒的速度滴加进混合溶液中,滴加完毕后溶液由亮黄色浑浊变为黄色透明溶液,在200rpm的转速下搅拌进行配位缩合反应,3.2h后获得铅配合物;然后将含1.4mmol五水硫酸铜的12mL无水甲醇溶液以2滴/秒的速度滴加进行铅配合物中,滴加完毕后溶液变成浅绿色浑浊,滴加三乙胺调节pH值为8.5,在200rpm的转速下搅拌进行置换反应,搅拌3.5h后使用砂芯过滤;将获得的硫酸铅沉淀使用28mL的无水甲醇和乙腈的混合溶液洗涤,无水甲醇和乙腈的体积比为1:1,合并滤液和洗液得到混合体系;控制转速为200rpm,将混合体系和高氯酸钠的无水甲醇溶液混合,混合的方式为滴加,将含0.3g一水高氯酸钠的8mL无水甲醇溶液以2滴/秒的速度滴加进混合体系中,滴加完毕后继续搅拌3.5h得到沉淀;将沉淀用8mL的水洗涤2次,然后用8mL的乙醇洗涤2次获得洗涤后的沉淀,将洗涤后的沉淀溶于1.6g乙醇乙腈溶液中,乙腈和乙醇的体积比为1:1,静置挥发30天后得到0.087g墨绿色片状单晶,产率为41%,即为四核铜配合物。本实施例制备的四核铜配合物进行了与实施例1相同的试验,表现出了优异的与NO可逆结合的性能。
实施例3
将0.6mmol的2,6-二甲酰基-4-甲氧基苯酚、0.4mmol的四水合乙酸铅和16mL的无水乙醇混合在400rpm转速下搅拌1.9h获得混合溶液;将含0.6mmolN,N-二(3-氨丙基)-2-吡啶甲胺的12mL无水甲醇溶液以3滴/秒的速度滴加进混合溶液中,滴加完毕后溶液由亮黄色浑浊变为黄色透明溶液,在400rpm的转速下搅拌进行配位缩合反应,2.8h后获得铅配合物;然后将含1.5mmol五水硫酸铜的18mL无水甲醇溶液以3滴/秒的速度滴加进行铅配合物中,滴加完毕后溶液变成浅绿色浑浊,滴加三乙胺调节pH值为8.2,在400rpm的转速下搅拌进行置换反应,搅拌3.5h后使用砂芯过滤;将获得的硫酸铅沉淀使用22mL的无水甲醇和乙腈的混合溶液洗涤,无水甲醇和乙腈的体积比为1:1,合并滤液和洗液得到混合体系;控制转速为400rpm,将混合体系和高氯酸钠的无水甲醇溶液混合,混合的方式为滴加,将含0.5g一水高氯酸钠的12mL无水甲醇溶液以3滴/秒的速度滴加进混合体系中,滴加完毕后继续搅拌4.5h得到沉淀;将沉淀用11mL的水洗涤4次,然后用11mL的乙醇洗涤4次获得洗涤后的沉淀,将洗涤后的沉淀溶于1.9g乙醇乙腈溶液中,乙腈和乙醇的体积比为1:1,静置挥发29天后得到0.183g墨绿色片状单晶,产率为43%,即为四核铜配合物。本实施例制备的四核铜配合物进行了与实施例1相同的试验,表现出了优异的与NO可逆结合的性能。
由以上实施例可知,本发明提供了一种四核铜配合物,经过配位缩合反应和置换反应后即得。本发明提供的配合物与NO呈现可逆性结合,与DNA作用的研究表明,该配合物能有效与DNA结合,同时没有DNA毒性,能长时间发挥平衡NO浓度的效力,作为NO水平调节剂改善目标中氮氧化物的含量。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。