一种连续化合成N-乙基乙二胺的工艺方法
文献发布时间:2024-04-18 20:01:23
技术领域
本发明属于化工技术领域,涉及一种以氯乙烷和乙二胺为原料连续化合成N-乙基乙二胺的工艺方法。
背景技术
N-乙基乙二胺(NEED)主要用于生产低毒、高效、广谱的抗菌素药物氧哌嗪青霉素、头孢拉腙、头孢哌酮等医药中间体,也广泛用于农药、表面活性剂的合成。头孢菌素类药物需求量的持续增长,也大大的刺激了其重要中间体N-乙基乙二胺需求量的增长。目前合成N-乙基乙二胺的方法主要有氨乙基硫酸酯法、乙醇胺法、乙酸法、卤代烷氨解法、乙醇法和丙烯腈法。相对而言,卤代烷氨解法具有原料简单易得、反应条件温和、对环境污染较小及适用于工业规模化生产的特点,被广泛研究并作为N-乙基乙二胺的工业化合成路线。
卤代烷氨解法主要是将卤代烷通入2-5倍摩尔量的乙二胺中,反应合成后向反应液中加入NaOH水溶液,然后将有机相从水相中分离出来。然后通过共沸精馏的方式分出有机相中的水和乙二胺的混合相。最后精馏除去正庚烷,并得到质量合格的产品N-乙基乙二胺。近年来,国内各个研究院所及生产单位纷纷对此合成工艺进行改进。2012年,徐庐峰等人在高压反应釜中加入乙二胺、甲醇、甲醇钠,压入氯乙烷后升温至40℃,压力0.2MPa,保温4小时。反应结束过滤除去氯化钠后,取滤液精馏,收集128-130℃馏分,即得目标产物N-乙基乙二胺。
魏文珑等人在25℃下将溴乙烷滴入乙二胺中,乙二胺与溴乙烷的摩尔比为5:1,滴加时间为40min,滴加完毕后保温2h。反应结束后,向反应液中加入NaOH后继续搅拌30min。随后分出有机层,将水层用环己烷萃取,收集有机层和萃取相,常压蒸馏回收环己烷,并精馏收集128~130℃的馏分。产品收率达到64.6%。陈湘君等人将后处理方式改进,首先将溴乙烷滴入乙二胺后,于室温下搅拌3小时后,升温至40度反应3小时,继而再回流2小时。反应结束后加入盐酸,并于70℃减压蒸馏。蒸馏后的馏出液主要是N-乙基乙二胺,通过精馏收集>106℃的组分,即含水的N-乙基乙二胺。然后将使用苯进行共沸脱水,再加入粉状的NaOH脱水,分离有机相后再分馏,收集128-132℃的馏分,即得N-乙基乙二胺。上述两方案的弊端是水与乙二胺分离困难,难于将乙二胺和水通过常压蒸馏的手段分离,需要添加第三组分进行精馏,耗能较高,不利于生产。
卤代烷氨解反应过程需要使用过量的乙二胺避免二取代副产物的生成,但是反应过程中仍有>20%二取代副产生成,进而需要一种副产物少并安全环保的制备方法。
发明内容
本发明的目的在于针对目前氯代烷氨解法合成N-乙基乙二胺的不足,提供一种连续化合成N-乙基乙二胺的工艺方法。
为实现上述目的,本发明采用技术方案为:
一种连续化合成N-乙基乙二胺的工艺方法,采取微通道连续流循环进料方式连续化卤代烷氨解法合成N-乙基乙二胺,合成后通过精馏方式纯化分离,获得高收率、高纯度的N-乙基乙二胺;同时分离出乙二胺回收套用。
所述微通道连续流循环采用的微通道连续流反应装置包括通过管路依次相连的微反应器、延时管道反应器、储罐和背压阀;延时管道反应器与储罐之间管路上设置分流三通阀,通过分流三通阀将循环物料和流出物料构成循环途径。
所述反应器由分散通道和连续相反应通道呈十字相连;连续相反应通道一端为循环液进口(103)与储罐通过管路相连,另一端作为反应器出口(104)通过管路与延时管道反应器相连;分散通道两端分别为原料氯乙烷和乙二胺的进料通道(105,106),两进料通道分别连接分散相进口管路(101,102);所述氯乙烷分散相进口管路通过管路依次连接高压缓冲罐和氯乙烷储罐。
所述微通道反应器的进出口通道尺寸1.5mm,分散孔内径0.2mm,分散管束的个数视流速而定。延时管道内径2mm,壁厚1mm。
所述氯乙烷储罐后设有氯乙烷高压缓冲罐,缓冲罐中的氯乙烷液体使用进料泵和乙二胺同时泵入微反应器中。氯乙烷储罐与缓冲罐之间设有气瓶减压表和质量流量控制器调节压力和流量。
具体为:
通过进料泵将氯乙烷从氯乙烷高压缓冲罐中和过量的无水乙二胺分别同时通过分别通过两分散相进口管路(101,102)泵入微通道反应器中进行烷基化反应,反应生成液从反应器出口(104)流出通过管路依次进入延时管道反应器,再通过分流三通阀,控制反应生成液循环比,使其循环至反应器与原料进行反应,循环的反应生成液进入储罐内,再通过微反应器的循环液进口(103)返回至微反应器,与原料混合,直至氯乙烷全部消耗,得到含N-乙基乙二胺、乙二胺的混合溶液;
(2)将上述获得N-乙基乙二胺、乙二胺的混合溶液利用减压蒸馏和精馏的方式进行对原料乙二胺、产物N-乙基乙二胺和二取代及多取代副产物进行分离纯化;进而获得高收率、高纯度的N-乙基乙二胺;
(3)将减压蒸馏过程中的乙二胺盐酸盐和N-乙基乙二胺盐酸盐中加入碱进行处理,获得质量合格的乙二胺,与上述减压蒸馏和精馏纯化后获得乙二胺合并直接套用。
所述步骤(1)中,所述氯乙烷储罐后设有氯乙烷高压缓冲罐,缓冲罐中的氯乙烷液体使用进料泵和乙二胺同时泵入微反应器中;氯乙烷储罐与缓冲罐之间设有气瓶减压表和质量流量控制器调节压力和流量;反应过程氯乙烷输出压力为0.05-0.1MPa;乙二胺和氯乙烷的卤代烷氨解反应温度30-40℃;所述乙二胺和氯乙烷摩尔比为3:1-2.5:1;所述返回的反应液循环过程的循环比为8-10:1。
所述步骤(2)中减压蒸馏和精馏为首先减压蒸馏分离出乙二胺、N-乙基乙二胺及副产物,而后再通过向体系内加入共沸剂进行精馏,使得乙二胺分离,进而精馏实现N-乙基乙二胺和副产物的纯化分离。
由上述可知,分离纯化过程的减压蒸馏步骤,馏出液为乙二胺、N-乙基乙二胺、二取代副产物以及多取代副产物,釜底液为90-95%乙二胺盐酸盐和5-10%N-乙基乙二胺盐酸盐。
分离纯化过程的精馏步骤,分离乙二胺、N-乙基乙二胺、二取代副产物以及多取代副产物。添加共沸剂,采用共沸精馏的方式先分离乙二胺,然后分出共沸剂,共沸剂可循环使用。最后使用普通精馏的方式分离产物和副产物。
所述减压蒸馏的温度为80-100℃,减压蒸馏真空压力为0.095--0.1Mpa;
所述精馏塔板数30-40块,温度128-130℃。
所述精馏塔顶端出料口设有一个两相分液器,首先在分液器中填充满共沸剂,随着精馏过程的发生,重组分乙二胺自下层放料口接出,共沸剂自上流回塔内。
所述共沸剂为正庚烷、环己烷,共沸剂加入量约为精馏液体积的1/3;乙二胺和正庚烷乙二胺和正庚烷、环己烷馏出的塔顶温度分别为86-98℃、74-80℃。
将减压蒸馏过程中的乙二胺盐酸盐和N-乙基乙二胺盐酸盐中加入与氯乙烷等摩尔量的碱进行处理,获得合格的可套用的乙二胺,5-10%的N-乙基乙二胺随着乙二胺一起回用;其中,碱为含30%甲醇钠的甲醇溶液(即,30%甲醇钠+70%甲醇)或氢氧化钠水溶液。
所述甲醇钠回收乙二胺的方法为首先使用与氯化氢等摩尔量的30%的甲醇钠的甲醇溶液中和氯化氢,中和过程产生的氯化钠废渣通过过滤的方式与反应液进行分离,分离后的乙二胺-甲醇混合液首先采用常压蒸馏的方式去除大部分甲醇,使用精馏的方式除尽甲醇,最终得到含有少量N-乙基乙二胺的乙二胺,完成套用实验。
其中,常压蒸馏和精馏过程回收的甲醇,可用于反复清洗一次过滤的氯化钠废渣,将废渣中的乙二胺洗净,直至洗液为中性。所述清洗次数为2-3次。
所述氢氧化钠回收方式为使用氢氧化钠作为中和剂回收乙二胺,首先添加与氯化氢等摩尔量50%的氢氧化钠水溶液,完成中和反应。采用初步过滤并分相的方式,获得油相和水相,水相为氯化钠水溶液,油相为含水、氯化钠的乙二胺混合液。向获得的油相中逐渐加入氢氧化钠固体至饱和,吸附其中的水分并析出溶解的氯化钠,继续进行过滤分相处理,获得的水相进行套用中和过程,获得的油相采用减压蒸馏的方式收集馏出组分,组分含有水和乙二胺,最后采用甲苯作为共沸精馏的共沸剂除水,获得质量合格的含有少量N-乙基乙二胺的乙二胺。
其中,氢氧化钠回收过程中二次过滤所得氢氧化钠溶液可作为中和剂完成中和反应,完成套用过程。
其中,甲苯和水的共沸温度为84-104℃,采用分相的方式可将甲苯和水进行分离;甲苯和乙二胺的共沸温度为104-116℃,采出的混合液作为下次精馏的共沸剂。
所述两种回收方式收回的乙二胺中,含水量不得高于2%,甲醇含量不得高于5%,否则影响乙二胺套用过程。
本发明的有益效果:
本发明烷基化合成过程、产品与原料的分离纯化过程、乙二胺回收套用过程;其中,烷基化合成过程采用连续化微通道循环进料合成N-乙基乙二胺,一方面微反应器能够增加反应的传质、避免反混,能够避免二取代副产的生成;另一方面反应液循环进料也能够提高反应的选择性;具体为:
1、本发明烷基化过程采用微通道连续化循环进料的方法增加传质、避免反混,避免副产物的生成,提升反应收率,减小后处理的难度、降低能耗、减少三废量、节约成本。
2、本发明N-乙基乙二胺合成工艺具有明确、全面且系统的特点,包含了烷基化反应过程、产品与原料的分离纯化过程和乙二胺回收套用过程,是一条适用于工业化的绿色环保工艺。
3、本发明在烷基化过程和减压蒸馏过程中,不添加溶剂和酸类水溶液物质,降低了精制过程分离的难度;同时,减压蒸馏与共沸精馏方式的结合,一方面降低了分离原料和产品的能耗,另一方面,也为乙二胺回收过程提供了便利条件。
4、本发明回收套用过程中采用甲醇钠和氢氧化钠回收乙二胺,并确定了回收乙二胺套用过程原料液的质量要求,最终完成了乙二胺套用实验,结果与新鲜乙二胺类似。同时,乙二胺的系统回收极大地降低了原料成本。
附图说明
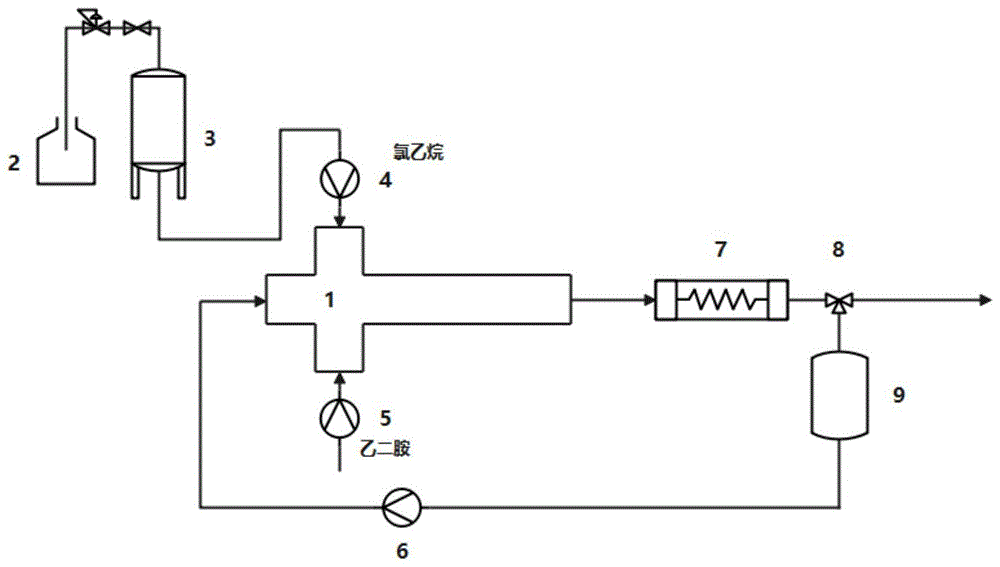
图1为本发明烷基化反应工艺流程简图,
图2为图1中的微反应器结构示意图,
图3为图2中微反应器分散通道的横截面,
其中,1为微反应器,两个分散相进口管路(101,102)、连续相进口管路(103)、反应器出口管路(104)和两个分散通道(105,106),2为氯乙烷储罐,3为高压缓冲罐,4为第一恒流泵,5为第二恒流泵,6为第二恒流泵,7为延时反应器,8为分流三通阀,9为储罐。
具体实施方式
本发明合成N-乙基乙二胺的全流程工艺,相比于报道工艺,本发明工艺流程简单,能耗低,在高转化率的同时,保证产品质量;全流程过程系统、安全,为产业化过程提供了坚实的参考依据。
本发明包括微通道连续化烷基化合成过程合成N-乙基乙二胺、产品与原料的分离纯化、乙二胺的回收套用过程;其中,微通道连续化合成N-乙基乙二胺能够增强传质、提升反应收率,减小后处理的难度、降低能耗、减少三废量、节约成本。由于乙二胺过量很大,且乙二胺和N-乙基乙二胺沸点相近、相对挥发度接近于1,精馏需要很高的塔板数,能耗过高。采用先减压蒸馏分离乙二胺盐酸盐,后共沸精馏分离乙二胺的方法,最大程度降低成本、节约能耗。最后将乙二胺盐酸盐中和成乙二胺,完成套用,实现N-乙基乙二胺的合成、后处理以及套用的全流程高效、环保的技术方案。
实施例1
如图1-3所示,所述微通道连续流循环采用的微通道连续流反应装置包括通过管路依次相连的微反应器、延时管道反应器、储罐和背压阀;延时管道反应器与储罐之间管路上设置分流三通阀,通过分流三通阀将循环物料和流出物料构成循环途径。
所述反应器由分散通道和连续相反应通道呈十字相连;连续相反应通道一端为循环液进口(103)与储罐通过管路相连,另一端作为反应器出口(104)通过管路与延时管道反应器相连;分散通道两端分别为原料氯乙烷和乙二胺的进料通道(105,106),两进料通道分别连接分散相进口管路(101,102);所述氯乙烷分散相进口管路通过管路依次连接高压缓冲罐和氯乙烷储罐。
上述各进口管路上设置进料泵。
实施例2
(1)烷基化反应过程:利用实施例装置,氯乙烷储罐通过管路与高压缓冲罐相连,通过第一平流泵将氯乙烷液体,通过第二平流泵将乙二胺分别通过两分散相进口管路(101,102)泵入微通道反应器中。
通过控制进料流速调节乙二胺:氯乙烷摩尔比=3:1,氯乙烷与乙二胺进料质量比为0.36:1,进行烷基化反应,反应温度为40℃,反应生成液从反应器出口(104)流出通过管路依次进入延时管道反应器,再通过分流三通阀,按照一定的循环比将反应生成液分为循环物料和流出物料。循环物料进入储罐内,再通过微反应器的循环液进口(103)返回至微反应器,与原料混合,控制返回的反应液的循环比为9:1,直至氯乙烷全部消耗,流出物料气相色谱检测:N-乙基乙二胺的纯度为92.1%,两种二取代副产物的纯度分别为2.5%和5.4%(反应液中乙二胺约占52.03%)。
(2)产品与原料的分离纯化过程:
①减压蒸馏过程:将烷基化过程得到的反应液转移至油浴加热装置中,首先设置油浴温度为70-80℃,同时开启真空装置,真空度为0.095--0.1Mpa。当油浴温度升至58℃,冷凝器出口开始有馏出液馏出,随着温度的升高,出液速度逐渐加快。在80℃的油浴下减压蒸馏1h,馏出液采出速度减慢,将油浴温度升至110℃,将残留的未被缚酸的胺尽可能蒸出,收集馏出液。经过气相分析结果测定,馏出液中乙二胺含量为28.9%,N-乙基乙二胺含量为64.8%,两种二取代副产物的含量分别为1.8%和4.5%。釜底液即被缚酸的乙二胺和少量的N-乙基乙二胺,其中乙二胺的含量为94.1%,N-乙基乙二胺的含量为5.9%。
②共沸精馏过程:将上述获得减压蒸馏后的馏出液与共沸剂正庚烷混合,其中,馏出液与共沸剂体积比为3:1。采用精馏的方式对组分进行分离提纯,共沸精馏分理出乙二胺,常规精馏分离N-乙基乙二胺,具体为首先正庚烷与乙二胺发生共沸,共沸点为86-98℃,根据正庚烷和乙二胺不互溶的现象,下层分出乙二胺,轻组分正庚烷流回塔内。当乙二胺全部除尽后,继续用精馏的方式采出正庚烷,温度约为98℃。随后,在精馏塔塔顶温度128-130℃采出产物N-乙基乙二胺。经过气相检测,馏出乙二胺纯度为99.3%,产物N-乙基乙二胺纯度为99.5%,经过计算单批次产物收率为87.3%。
(3)乙二胺回收套用过程:向减压蒸馏过程得到的釜底液中加入360g含30%的甲醇钠的甲醇溶液进行中和,常温下搅拌后过滤除去固体氯化钠,并使用甲醇清洗滤饼3次。收集滤液和清洗液,首先在85℃水浴加热下常压蒸馏蒸馏除甲醇,然后使用普通精馏的方式除尽甲醇,使用塔板数约为20块的精馏塔,分离塔顶温度65℃甲醇组分。两步去除的甲醇占比约为74%和26%。经过气相色谱检测可知,回收液中乙二胺含量为93.9%,N-乙基乙二胺为5.6%,甲醇含量为0.5%,乙二胺回收率为98%。
实施例3
与实施例2中的烷基化反应过程、产品与原料的分离纯化过程相同,在乙二胺回收套用过程中本实施例采用氢氧化钠作为中和剂去除氯化氢。首先,向减压蒸馏过程得到的釜底液中加入160g 50%的氢氧化钠水溶液,常温下搅拌1h,然后常温过滤除去生成的氯化钠,然后初步分离出水相和油相。继续向油相中加入80g氢氧化钠固体做干燥水处理,搅拌30min后过滤并作分相处理。所得的水相作为中和剂进行套用中和过程,所得油相采用100℃油浴加热、真空度为0.095--0.1Mpa的减压蒸馏的方式收集馏出组分,组分含有水和乙二胺,最后采用甲苯作为共沸精馏的共沸剂进行除水,84-104℃收集甲苯和水,采用分相的方式可将甲苯和水进行分离;104-116℃收集甲苯和乙二胺,采出的混合液作为下次精馏的共沸剂,116℃收集乙二胺。获得N-乙基乙二胺含量6.5%、乙二胺含量93.5%的混合液,乙二胺回收率为91.7%。
实施例4
(1)烷基化反应过程:利用实施例装置,氯乙烷储罐通过管路与高压缓冲罐相连,通过第一平流泵将氯乙烷液体,通过第二平流泵将乙二胺分别通过两分散相进口管路(101,102)泵入微通道反应器中。
通过控制进料流速调节乙二胺:氯乙烷摩尔比=3:1,氯乙烷与乙二胺进料质量比为0.36:1,进行烷基化反应,反应温度为10℃,反应生成液从反应器出口(104)流出通过管路依次进入延时管道反应器,再通过分流三通阀,按照一定的循环比将反应生成液分为循环物料和流出物料。循环物料进入储罐内,进入储罐的循环物料通过微反应器的循环液进口(103)返回至微反应器,与原料混合,控制返回的反应液的循环比为12:1,直至氯乙烷全部消耗,流出物料气相色谱检测:N-乙基乙二胺的纯度为93.0%,两种二取代副产物的纯度分别为2.1%和4.9%(反应液中乙二胺约占54.4%)。
实施例5
(1)烷基化反应过程:利用实施例装置,氯乙烷储罐通过管路与高压缓冲罐相连,通过第一平流泵将氯乙烷液体,通过第二平流泵将实施例2中回收乙二胺(乙二胺含量为93.9%)分别通过两分散相进口管路(101,102)泵入微通道反应器中。
通过控制进料流速调节乙二胺:氯乙烷摩尔比=3:1,氯乙烷与乙二胺进料质量比为0.34:1,进行烷基化反应,反应温度为40℃,反应生成液从反应器出口(104)流出通过管路依次进入延时管道反应器,再通过分流三通阀,按照一定的循环比将反应生成液分为循环物料和流出物料。循环物料进入储罐内,进入储罐的循环物料通过管路通过微反应器的循环液进口(103)返回至微反应器,与原料混合,控制返回的反应液的循环比为9:1,直至氯乙烷全部消耗,流出物料气相色谱检测:N-乙基乙二胺的纯度为91.7%,两种二取代副产物的纯度分别为2.7%和5.6%(反应液中乙二胺约占51.6%)。
(2)产品与原料的分离纯化过程:
①减压蒸馏过程:同实施例2过程,将得到的烷基化反应液倒入减压蒸馏装置中,将未被缚酸胺蒸出。经过气相分析结果测定,馏出液中乙二胺含量为28%,N-乙基乙二胺含量为65.2%,两种二取代副产物的含量分别为2.1%和4.7%。釜底液即被缚酸的乙二胺和少量的N-乙基乙二胺,其中乙二胺的含量为93.4%,N-乙基乙二胺的含量为6.6%。
②共沸精馏过程:将上述获得的减压蒸馏后的馏出液与共沸剂正庚烷混合,混合体积比为3:1。采用精馏的方式对组分进行分离提纯,共沸精馏分理出乙二胺,常规精馏分离N-乙基乙二胺,具体为首先正庚烷与乙二胺发生共沸,共沸点为86-98℃,根据正庚烷和乙二胺不互溶的现象,下层分出乙二胺,轻组分正庚烷流回塔内。当乙二胺全部除尽后,继续用精馏的方式采出正庚烷,温度约为98℃。随后,在精馏塔塔顶温度128-130℃采出产物N-乙基乙二胺。经过气相检测,馏出乙二胺纯度为99.3%,产物N-乙基乙二胺纯度为99.5%,经过计算单批次产物收率为86.8%。
实施例6
(1)烷基化反应过程:利用实施例装置,氯乙烷储罐通过管路与高压缓冲罐相连,通过第一平流泵将氯乙烷液体,通过第二平流泵将实施例3中回收乙二胺(乙二胺含量为93.5%)分别通过两分散相进口管路(101,102)泵入微通道反应器中。
通过控制进料流速调节乙二胺:氯乙烷摩尔比=3:1,氯乙烷与乙二胺进料质量比为0.34:1,进行烷基化反应,反应温度为40℃,反应生成液从反应器出口(104)流出通过管路依次进入延时管道反应器,再通过分流三通阀,按照一定的循环比将反应生成液分为循环物料和流出物料。循环物料进入储罐内,进入储罐的循环物料通过管路通过微反应器的循环液进口(103)返回至微反应器,与原料混合,控制返回的反应液的循环比为9:1,直至氯乙烷全部消耗,流出物料气相色谱检测:N-乙基乙二胺的纯度为91.1%,两种二取代副产物的纯度分别为2.9%和6.0%(反应液中乙二胺约占52.9%)。
(2)产品与原料的分离纯化过程:
①减压蒸馏过程:同实施例2过程,将得到的烷基化反应液倒入减压蒸馏装置中,将未被缚酸胺蒸出,收集馏出液。经过气相分析结果测定,馏出液中乙二胺含量为28.5%,N-乙基乙二胺含量为65.0%,两种二取代副产物的含量分别为2.2%和4.3%。釜底液即被缚酸的乙二胺和少量的N-乙基乙二胺,其中乙二胺的含量为94.1%,N-乙基乙二胺的含量为5.9%。
②共沸精馏过程:将减压蒸馏后的馏出液与共沸剂正庚烷混合,混合体积比为3:1。采用精馏的方式对组分进行分离提纯,共沸精馏分理出乙二胺,常规精馏分离N-乙基乙二胺,具体为首先正庚烷与乙二胺发生共沸,共沸点为86-98℃,根据正庚烷和乙二胺不互溶的现象,下层分出乙二胺,轻组分正庚烷流回塔内。当乙二胺全部除尽后,继续用精馏的方式采出正庚烷,温度约为98℃。随后,在精馏塔塔顶温度128-130℃采出产物N-乙基乙二胺。经过气相检测,馏出乙二胺纯度为99.4%,产物N-乙基乙二胺纯度为99.5%,经过计算单批次产物收率为87.0%。
对比例
首先在反应釜中加入360g乙二胺,使用水浴加热装置将原料液调至40℃。使用压力调节阀和质量流量控制器调节自氯乙烷储罐中流出的氯乙烷。并通过进料泵将氯乙烷液体通入反应釜内,控制一定的进料速度,保证进料时间为1h,调节乙二胺:氯乙烷的反应摩尔比为3:1,氯乙烷共计通入130g。然后在40℃下保温搅拌30min,使未转化的氯乙烷反应完全。经过气相色谱检测氯乙烷转化率为100%,生成物中N-乙基乙二胺的纯度为83.6%,两种二取代副产物的纯度分别为5.5%和10.9%。经过后处理单批次产物收率为74.5%。
- 一种基于声波探测地下硐室围岩破裂范围的钻测装置
- 一种基于声波探测地下硐室围岩破裂范围的钻测装置