杂环化合物和包含该杂环化合物的有机电致发光器件
文献发布时间:2023-06-19 18:35:48
技术领域
本发明涉及特定的杂环化合物,包含所述特定的杂环化合物的用于有机电致发光器件的材料,优选发光体材料,包含所述特定的杂环化合物的有机电致发光器件,包含所述有机电致发光器件的电子设备,包含至少一种主体和至少一种掺杂剂的发光层,其中所述掺杂剂包含至少一种所述特定的杂环化合物,以及所述杂环化合物在有机电致发光器件中的用途。
当将电压施加到有机电致发光器件(以下可以称为有机EL器件)时,空穴从阳极注入到发光层,电子从阴极注入到发光层。在发光层中,注入的空穴和电子重新结合并形成激子。
有机EL器件包括在阳极和阴极之间的发光层。此外,可以存在这样的情况,其中其具有包括有机层,如空穴注入层、空穴传输层、电子注入层、电子传输层等的叠层结构。
US 2019/0067577A1涉及用于有机电子器件的含硼杂环化合物,例如具有根据下式I的结构的有机发光器件
其中
环A、B、C和D各自独立地为5或6元芳基或杂芳基环;
R
Y为NR、O、PR、S或Se;以及
Z为N或P。
式I的化合物的一个实例是下列化合物
然而,多环化合物的特定结构和取代模式对有机电子器件中的多环化合物的性能具有显著影响。
尽管有上述发展,但仍然需要包含新材料,特别是掺杂剂(=发光体)材料的有机电致发光器件,以提供改进的电致发光器件性能。
因此,相对于上述相关技术,本发明的目的是提供适合于提供有机电致发光器件的材料,其确保有机电致发光器件的良好性能,尤其是良好的EQE和/或长寿命。更特别地,应该可以提供掺杂剂(=发光体)材料,特别是具有窄光谱(较小FWHM)的发蓝光掺杂剂材料,即当用作有机电致发光器件中的掺杂剂时具有良好的色纯度。
根据本发明的一个方面,所述目的通过由式(I)表示的杂环化合物来解决:
其中
环A
或
环C
R
或
R
Y表示直接键、O、S、NR
在Y是直接键的情况下,环B
R
和/或
两个残基R
式(I)的化合物原则上可用于EL器件的任何层中。优选地,式(I)的化合物是有机EL器件中,尤其是发光层中的掺杂剂(=发光体),更优选荧光掺杂剂。特别地,式(I)的化合物在有机EL器件中,尤其是在发光层中用作荧光掺杂剂。
在本申请中,术语有机EL器件(有机电致发光器件)与术语有机发光二极管(OLED)可互换使用。
已经发现,式(I)的特定化合物显示窄的发光特性,优选窄的荧光,更优选窄的蓝色荧光。这种窄的发光特性适于防止由于外耦合而引起的能量损失。根据本发明的式(I)的化合物优选具有低于30 nm,更优选低于25 nm的半峰全宽(FWHM)。
还发现,包含本发明化合物的有机EL器件的特征通常在于高外量子效率(EQE)和长寿命,特别是当式(I)的特定化合物用作掺杂剂(发光材料),尤其是有机电致发光器件中的荧光掺杂剂时。
由上文或下文提到的“取代或未取代”和“可以被取代”表示的任选一个或多个取代基的实例包括具有6-60个,优选6-30个,更优选6-18个环碳原子的芳基,其进而是未取代或取代的,具有5-60个,优选5-30个,更优选5-18个环原子的杂芳基,其进而是未取代或取代的,具有1-20个,优选1-8个碳原子的烷基,具有3-20个,优选3-6个碳原子的环烷基,基团OR
或
两个相邻的取代基一起形成环结构,该环结构进而是未取代或取代的;
R
和/或
两个残基R
或
R
R
R
术语氢、卤素、未取代或取代的具有1-20个碳原子的烷基、未取代或取代的具有1-20个碳原子的卤代烷基、未取代或取代的具有3-20个环碳原子的环烷基、具有6-60个、优选6-30个、更优选6-18个环碳原子的取代或未取代的芳基;具有5-60个,优选5-30个,更优选5-18个环原子的取代或未取代的杂芳基,具有1-20个碳原子,优选1-8个碳原子的羧基烷基,具有1-20个碳原子,优选1-8个碳原子的酰胺基烷基,在芳基残基中具有6-18个环碳原子的羧基芳基,在芳基残基中具有6-18个环碳原子的酰胺基芳基,N(R
是本领域已知的,并且如果所述基团没有在下面提及的具体实施方案中进一步指明,则通常具有以下含义:
在本发明中,氢包括中子数不同的异构体,即,氕、氘和氚。
具有6至60个、优选6至30个、更优选6至18个环碳原子、最优选具有6至13个环碳原子的取代或未取代的芳族基团(也称为芳基)可以是非稠合芳基或稠合芳基。其具体实例包括苯基、萘基、菲基、联苯基、三联苯基、荧蒽基、三亚苯基、菲基、芴基、茚基、蒽基、屈基、螺芴基、苯并[c]菲基,其中优选苯基、萘基、联苯基、三联苯基、菲基、三亚苯基、芴基、茚基和荧蒽基,更优选苯基、1-萘基、2-萘基、联苯-2-基、联苯-3-基、联苯-4-基、菲-9-基、菲-3-基、菲-2-基、三亚苯-2-基、芴-2-基,特别是9,9-二-C
或9,9-二-C
在环A
具有5至60个,优选5至30个,更优选5至18个环原子,最优选具有5至13个环原子的取代或未取代的杂芳族基团(也称为杂芳基)可以是非稠合杂芳基或稠合杂芳基。其具体实例包括吡咯环、异吲哚环、苯并呋喃环、异苯并呋喃环、苯并噻吩、二苯并噻吩环、异喹啉环、喹喔啉环、喹唑啉、菲啶环、菲咯啉环、吡啶环、吡嗪环、嘧啶环、哒嗪环、吲哚环、喹啉环、吖啶环、咔唑环、呋喃环、噻吩环、苯并噁唑环、苯并噻唑环、苯并咪唑环、二苯并呋喃环、三嗪环、噁唑环、噁二唑环、噻唑环、噻二唑环、三唑环、咪唑环、吲哚啉环、咪唑并吡啶环、4-咪唑并[1,2-a ]苯并咪唑基、5-苯并咪唑并[1,2-a ]苯并咪唑基和苯并咪唑并[2,1-b ] [1,3]苯并噻唑基的残基,其中优选苯并呋喃环、吲哚环、苯并噻吩环、二苯并呋喃环、咔唑环和二苯并噻吩环的残基,并且更优选苯并呋喃环、1-苯基吲哚环、苯并噻吩环、二苯并呋喃-1-基、二苯并呋喃-3-基、二苯并呋喃-2-基、二苯并呋喃-4-基、9-苯基咔唑-3-基、9-苯基咔唑-2-基、9-苯基咔唑-4-基、二苯并噻吩-2-基和二苯并噻吩-4-基、二苯并噻吩-1-基和二苯并噻吩-3-基的残基。
在环A
未取代或取代的具有1-20个碳原子的烷基的实例包括甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、新戊基、1-甲基戊基,优选甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基。优选具有1至8个碳原子,更优选1至4个碳原子的烷基。分别具有1-8个碳原子和1-4个碳原子的烷基的合适实例如上所述。
未取代或取代的具有1-20个碳原子的卤代烷基的实例包括作为烷基公开的那些,其中其氢原子部分或全部被卤素原子取代。优选的卤代烷基是具有1-20个碳原子的氟代烷基,包括上述烷基,其中其氢原子部分或全部被氟原子取代,例如CF
未取代或取代的具有3-20个环碳原子的环烷基的实例包括环丙基、环丁基、环戊基、环己基、环辛基和金刚烷基,优选环戊基和环己基。优选具有3-6个碳原子的环烷基。具有3-6个碳原子的环烷基的合适实例如上所述。
卤素原子的实例包括氟、氯、溴和碘,优选氟。
基团OR
基团SR
基团N(R
基团B(R
基团SiR
具有1至20个碳原子,优选1至8个碳原子的羧基烷基的实例包括具有选自上述烷基的烷基部分的那些。
具有1至20个碳原子的氟烷基的实例包括上述烷基,其中其氢原子部分或全部被氟原子取代。
具有1-20个碳原子,优选1-8个碳原子的酰胺基烷基(烷基取代的酰胺基)的实例包括具有选自上述烷基的烷基部分的那些。
具有6-18个碳原子,优选6-13个碳原子的酰胺基芳基(芳基取代的酰胺基)的实例包括具有选自上述芳基的芳基部分的那些。
任选的取代基优选各自独立地表示未取代或取代的具有6至18个环碳原子的芳基;未取代或取代的具有5至18个环原子的杂芳基;未取代或取代的具有1至20个碳原子的烷基;未取代或取代的具有3-20个环碳原子的环烷基;CN;N(R
或
两个相邻的取代基一起形成环结构,该环结构进而是未取代的或取代的;
R
或
R
R
更优选地,任选的取代基各自独立地表示未取代或取代的具有6至18个环碳原子的芳基;未取代或取代的具有5至18个环原子的杂芳基;未取代或取代的具有1至20个碳原子的烷基;未取代或取代的具有3至20个环碳原子的环烷基;CN;或N(R
或
两个相邻的取代基一起形成环结构,该环结构进而是未取代的或取代的;
R
或
R
最优选地,任选的取代基各自独立地表示未取代或取代的具有1至4个碳原子的烷基;未取代或取代的具有3至6个环碳原子的环烷基;未取代或取代的具有6至13个环碳原子的芳基;未取代或取代的具有5至13个环原子的杂芳基;CN;或N(R
或
两个相邻的取代基一起形成环结构,该环结构进而是未取代的或取代的;
R
上述任选的取代基可以进一步被一个或多个上述任选的取代基取代。
任选的取代基的数目取决于被所述一个或多个取代基取代的基团。可能的取代基的最大数目由存在的氢原子的数目限定。优选每个被取代的基团有1、2、3、5、6、7、8或9个任选取代基,更优选1、2、3、5、6或7个任选取代基,最优选1、2、3、4或5个任选取代基,进一步最优选1、2、3、4或5个任选取代基,更进一步最优选1、2、3或4个任选取代基,更进一步最优选每个被取代的基团有1或2个任选取代基。在进一步优选的实施方案中,上述基团中的一些或全部是未取代的。
在进一步优选的实施方案中,式(I)的化合物中的取代基的总数为0、1、2、3、4、5、6、7或8,优选0、1、2、3、4、5或6,即剩余的残基为氢。
在表达“具有a至b个碳原子的取代或未取代的X基团”中的“a至b的碳数”是未取代的X基团的碳数并且不包括任选的取代基的一个或多个碳原子。
由“未取代的或取代的”提及的术语“未取代的”是指氢原子未被上述基团之一取代。
在上述和下述任何式的定义中,指数0是指氢原子存在于由所述指数定义的位置。
式(I)的化合物
在由式(I)表示的杂环化合物中
残基具有以下含义:
环A
或
环C
R
或
R
Y表示直接键、O、S、NR
在Y是直接键的情况下,环B
R
和/或
两个残基R
优选地,环A
其中环C
星形是环C
以及虚线是直接键。
更优选的环A
非稠合芳基或稠合芳基。其具体实例基于苯基、萘基、菲、联苯基、三联苯基、荧蒽、三亚苯基、芴、茚、蒽、屈、螺芴、苯并[c]菲,其中优选苯基、萘基、联苯基、三联苯基、菲、三亚苯基、芴、茚和荧蒽,最优选苯基和萘基;
或
非稠合杂芳基或稠合杂芳基。其具体实例基于吡咯、异吲哚、苯并呋喃、异苯并呋喃、苯并噻吩、二苯并噻吩、异喹啉、喹喔啉、喹唑啉、菲啶、菲咯啉、吡啶、吡嗪、嘧啶、哒嗪、吲哚、喹啉、吖啶、咔唑、呋喃、噻吩、苯并噁唑、苯并噻唑、苯并咪唑、二苯并呋喃、三嗪、噁唑、噁二唑、噻唑、噻二唑、三唑、咪唑、吲哚啉、咪唑并吡啶、4-咪唑并[1,2-a ]苯并咪唑、5-苯并咪唑并[1,2-a ]苯并咪唑和苯并咪唑并[2,1-b ] [1,3]苯并噻唑,其中优选吲哚,尤其是1-苯基吲哚、苯并噻吩、二苯并呋喃、咔唑、二苯并噻吩、苯并呋喃和苯并噻吩。
更优选地,环A
其中虚线是键合位点并且残基R
其中虚线是键合位点并且残基R
其中虚线是键合位点并且残基R
其中环C
其中虚线是键合位点并且残基R
通过两个相邻取代基形成的环结构的实例如下所述(下面的环结构可以被一个或多个上述取代基取代):
其中X为O、CR
X’’和Y’’各自独立地表示O、CR
R
R
E
以及
虚线是键合位点。
R
其中
R
或
两个相邻残基R
X’表示直接键、O、S、NR
环A
R
Y表示直接键、O、S、NR
在Y是直接键的情况下,环B
Y是直接键并且环B
其中Z是O、S、NR
优选地,Y是直接键。
根据本发明的优选杂环化合物由式(II)表示
其中残基和指数如上所述。
在更优选的实施方案中,根据本发明的杂环化合物由式(III)表示
其中残基和指数如上所述。
在一个实施方案中,根据本发明的杂环化合物中的环A
R
其中
R
和/或
两个相邻残基R
和/或
R
并且虚线是键合位点。
最优选地,根据本发明的杂环化合物由式(V)表示
其中
R
或
两个相邻残基R
和/或
两个相邻残基R
和/或
R
R
和/或
两个残基R
或
R
R
通过两个相邻残基R
其中X是O、CR
R
R
R
其中
X’表示直接键、O、S、NR
所有其它残基在上文或下文中定义。
优选地,R
或
两个相邻残基R
和/或
R
R
或
R
R
更优选地,R
或
两个相邻残基R
和/或
R
R
或
R
最优选地,R
或
两个相邻残基R
和/或
R
R
在进一步优选的实施方案中,残基R
在一个优选的实施方案中,根据本发明的杂环化合物由下式之一表示
其中残基如上所述定义,
其中
-在式(VA)和式(VB)中 -
两个相邻残基R
-在式(VC)中 -
两个相邻残基R
更优选地,根据本发明的杂环化合物由下式之一表示
其中残基如上所述定义,
其中
-在式(VAa)和式(VBa)中 -
两个相邻残基R
-在式(VCa)中 -
两个相邻残基R
在一个优选的实施方案中,根据本发明的杂环化合物由式(VA)表示,其中两个相邻残基R
在一个优选的实施方案中,根据本发明的杂环化合物由式(VA)表示,其中R
且R
在进一步优选的实施方式中,根据本发明的杂环化合物由式(VA)表示,其中,R
在一个优选的实施方案中,根据本发明的杂环化合物由式(VA)表示,其中R
且R
在一个优选的实施方案中,根据本发明的杂环化合物由式(VC)表示,其中R
在一个优选的实施方案中,根据本发明的杂环化合物由式(VC)或(VB)表示,其中残基R
下面给出式(I)的化合物的实例:
式(I)的化合物的制备
由式(I)表示的化合物可以根据本申请实例中进行的反应,和通过使用类似于本领域已知的反应和原料的适合预期产物的替代反应或原料合成。
式(I)的化合物例如通过以下步骤制备:
(i)将BHal
其中
Hal表示卤素,优选F、Cl、Br或I,更优选Cl或Br,最优选Br;
R表示C
所有其它残基和指数如上定义。
在本申请的实例中提到合适的反应条件。
中间体(II)例如从式(III)的化合物开始制备
并且
(i)使化合物(III)的Hal
(ii)使化合物(III)的Hal
其中
Hal
Hal
R表示C
所有其它残基和指数如上定义。
通常,首先进行步骤(i),然后进行步骤(ii)。
其可以如下改性:
其中X’是直接键(即R
其中所有残基和指数如上定义。
式(V)的优选化合物例如通过以下步骤制备:
(i)将BHal
其中
Hal表示卤素,优选F、Cl、Br或I,更优选Cl或Br,最优选Br;
R表示C
所有其它残基和指数如上定义。
中间体(VI)例如从式(VII)的化合物开始制备
并且
(i)使化合物(VII)的Hal
(ii)使化合物(VII)的Hal
其中
Hal
Hal
R表示C
所有其它残基和指数如上定义。
通常,首先进行步骤(i),然后进行步骤(ii)。
其可以如下改性:
其中所有残基和指数如上定义。
在进一步的实施方案中,式(I)的化合物例如如下制备:
ia)将BHal
其中
Hal表示卤素,优选F、Cl、Br或I,更优选Cl或Br,最优选Br;
以及
所有其它残基和指数如上定义。
在本申请的实例中提及合适的反应条件。
中间体(IIa)例如从式(IIIa)的化合物开始制备
并且
(i)使化合物(IIIa)的Hal
(ii)使化合物(IIIa)的Hal
其中
Hal
Hal
所有其它残基和指数如上定义。
通常,首先进行步骤(i),然后进行步骤(ii)。
其可以如下改性:
其中X’是直接键(即R
其中所有残基和指数如上定义。
式(Va)的优选化合物例如通过以下步骤制备:
Ia)将BHal
其中
Hal表示卤素,优选F、Cl、Br或I,更优选Cl或Br,最优选Br;
R
所有其它残基和指数如上定义。
中间体(VIa)例如从式(VIIa)的化合物开始制备
并且
(i)使化合物(VIIa)的Hal
(ii)使化合物(VIIa)的Hal
其中
Hal
Hal
R
所有其它残基和指数如上定义。
通常,首先进行步骤(i),然后进行步骤(ii)。
其可以如下改性:
其中所有残基和指数如上定义。
下面描述合适的制备方法的实例。
有机电致发光器件
根据本发明的一个方面,提供了包含至少一种式(I)的化合物的用于有机电致发光器件的材料。
根据本发明的另一个方面,提供了包含至少一种式(I)的化合物的有机电致发光器件。
根据本发明的另一个方面,提供了以下有机电致发光器件:一种有机电致发光器件,其包括阴极、阳极和一个或多个有机薄膜层,所述有机薄膜层包括设置在阴极和阳极之间的发光层,其中所述有机薄膜层的至少一层包含至少一种式(I)的化合物。
根据本发明的另一个方面,提供了有机电致发光器件,其中发光层包含至少一种式(I)的化合物。
根据本发明的另一个方面,提供了有机电致发光器件,其中发光层包含作为掺杂剂材料的至少一种式(I)的化合物和作为主体材料的蒽化合物。
根据本发明的另一个方面,提供了设置有根据本发明的有机电致发光器件的电子设备。
根据本发明的另一个方面,提供了包含至少一种式(I)的化合物的发光体材料。
根据本发明的另一个方面,提供了包含至少一种主体和至少一种掺杂剂的发光层,其中所述掺杂剂包含至少一种式(I)的化合物。
根据本发明的另一个方面,提供了根据本发明的式(I)的化合物在有机电致发光器件中的用途。
在一个实施方案中,有机EL器件包括在阳极和发光层之间的空穴传输层。
在一个实施方案中,有机EL器件包括在阴极和发光层之间的电子传输层。
在本说明书中,关于“发光层和阳极之间的一个或多个有机薄膜层”,如果在发光层和阳极之间仅存在一个有机层,则其是指该层,如果存在多个有机层,则其是指该层的至少一层。例如,如果在发光层和阳极之间存在两个或更多个有机层,则更靠近发光层的有机层被称为“空穴传输层”,更靠近阳极的有机层被称为“空穴注入层”。“空穴传输层”和“空穴注入层”中的每一个可以是单层或者可以由两个或更多个层形成。这些层中的一个可以是单层,而其它层可由两个或更多个层形成。
类似地,关于“发光层和阴极之间的一个或多个有机薄膜层”,如果在发光层和阴极之间仅存在一个有机层,则其是指该层,如果存在多个有机层,则其是指该层的至少一层。例如,如果在发光层和阴极之间存在两个或更多个有机层,则更靠近发光层的有机层被称为“电子传输层”,更靠近阴极的有机层被称为“电子注入层”。“电子传输层”和“电子注入层”中的每一个可以是单层或者可以由两个或更多个层形成。这些层中的一个可以是单层,而其它层可由两个或更多个层形成。
上述“包括发光层的一个或多个有机薄膜层”,优选发光层,包含由式(I)表示的化合物。由式(I)表示的化合物优选用作发光体材料,更优选用作荧光发光体材料,最优选用作蓝色荧光发光体材料。通过在有机EL器件中,优选在发光层中存在式(I)的化合物,提供了特征在于高外量子效率(EQE)和长寿命的有机EL器件。
根据本发明的另一个方面,提供了有机电致发光器件的发光层,其包含至少一种式(I)的化合物。
优选地,发光层包含至少一种发光材料(掺杂剂材料)和至少一种主体材料,其中发光材料是至少一种式(I)的化合物。
在一个实施方案中,主体不选自CBP (4,4' -双- (N-咔唑基) -联苯),mCP,mCBPSif87 (二苯并[ b,d ]噻吩-2-基三苯基硅烷),CzSi,Sif88 (二苯并[ b,d ]噻吩-2-基)二苯基硅烷)、DPEPO (双[2- (二苯基膦基)苯基]醚氧化物)、9- [3- (二苯并呋喃-2-基)苯基] -9H-咔唑,9- [3- (二苯并呋喃-2-基)苯基] -9H-咔唑,9- [3- (二苯并噻吩-2-基)苯基] -9H-咔唑,9- [3,5-双(2-二苯并呋喃基)苯基] -9H-咔唑,9- [3,5-双(2-二苯并噻吩基)苯基] -9H-咔唑,T2T (2,4,6-三(联苯-3-基) -1,3,5-三嗪),T3T (2,4,6-三(三苯基-3-基) -1,3,5-三嗪)和/或TST (2,4,6-三(9,9' -螺二芴-2-基) -1,3,5-三嗪)。
优选的主体材料是取代或未取代的多环芳烃(PAH)化合物、取代或未取代的多杂芳族化合物、取代或未取代的蒽化合物、或取代或未取代的芘化合物。
更优选地,根据本发明的有机电致发光器件在发光层中包含作为掺杂剂材料的至少一种式(I)的化合物和至少一种主体材料,该主体材料选自取代或未取代的多环芳烃(PAH)化合物、取代或未取代的多杂芳族化合物、取代或未取代的蒽化合物和取代或未取代的芘化合物。优选地,所述至少一种主体为至少一种取代或未取代的蒽化合物。
在另一个优选的实施方案中,根据本发明的有机电致发光器件在发光层中包含作为掺杂剂材料的至少一种式(I)的化合物和至少一种主体材料,该主体材料选自取代或未取代的多环芳烃(PAH)化合物、取代或未取代的蒽化合物和取代或未取代的芘化合物。优选地,所述至少一种主体为至少一种取代或未取代的蒽化合物。
根据本发明的另一个方面,提供了有机电致发光器件的发光层,其包含作为掺杂剂材料的至少一种式(I)的化合物和作为主体材料的蒽化合物。
合适的蒽化合物由下式(10)表示:
其中
一对或多对的两个或更多个相邻的R
不形成取代或未取代的、饱和或不饱和环的R
R
条件是不形成取代或未取代、饱和或不饱和环的R
其中,在式(31)中,
L
Ar
化合物(10)中的各取代基、“取代或未取代”的取代基和卤素原子的具体实例与上述相同。
将解释“一对或多对的两个或更多个相邻的R
“一对两个或更多个相邻R
饱和或不饱和环的“取代或未取代”中的“取代”中的取代基与式(10)中提及的“取代或未取代”中的那些相同。
“饱和或不饱和环”是指,当R
“任意元素”优选为C元素、N元素、O元素或S元素。在任意元素(例如C元素或N元素)中,不形成环的原子键可以由氢原子等封端。
“一个或多个任意元素”优选为2个或更多个且15个或更少,更优选3个或更多个且12个或更少,并且进一步优选3个或更多个且5个或更少的任意元素。
例如,R
在一个实施方案中,R
优选地,R
更优选地,R
最优选地,R
进一步最优选地,R
在一个实施方案中,化合物(10)是由下式(10-1)表示的化合物:
其中在式(10-1)中,R
在一个实施方案中,化合物(10)是由下式(10-2)表示的化合物:
其中在式(10-2)中,R
在一个实施方案中,化合物(10)是由下式(10-3)表示的化合物:
其中,在式(10-3)中,
R
L
Ar
在一个实施方案中,化合物(10)是由下式(10-4)表示的化合物:
其中,在式(10-4)中,
L
R
X
R
R
未与L
不与L
在一个实施方案中,化合物(10)是由下式(10-4A)表示的化合物:
其中在式(10-4A)中,
L
R
X
R
一对或多对的相邻的两个或更多个R
不形成取代或未取代的饱和或不饱和环的R
其中在式(10-4A-1)中,
两个原子键*的每一个与相邻的R
R
不与L
在一个实施方案中,化合物(10)是由下式(10-6)表示的化合物:
其中在式(10-6)中,
L
R
R
X
在一个实施方案中,由式(10-6)表示的化合物是由下式(10-6H)表示的化合物:
其中在式(10-6H)中,
L
R
X
在一个实施方案中,由式(10-6)和(10-6H)表示的化合物是由下式(10-6Ha)表示的化合物:
其中在式(10-6Ha)中,
L
X
在一个实施方案中,由式(10-6)、(10-6H)和(10-6Ha)表示的化合物是由下式(10-6Ha-1)或(10-6Ha-2)表示的化合物:
其中在式(10-6Ha-1)和(10-6Ha-2)中,
L
X
在一个实施方案中,化合物(10)是由下式(10-7)表示的化合物:
其中,在式(10-7)中,
L
R
X
R
在一个实施方案中,化合物(10)是由下式(10-7H)表示的化合物:
其中在式(10-7H)中,
L
X
R
在一个实施方案中,化合物(10)是由下式(10-8)表示的化合物:
其中,在式(10-8)中,
L
R
X
R
在一个实施方案中,由式(10-8)表示的化合物是由下式(10-8H)表示的化合物:
在式(10-8H)中,L
R
X
在一个实施方案中,对于由式(10-7)、(10-8)或(10-8H)表示的化合物,R
其中在式(10-8-1)和(10-8-2)中,
两个原子键*独立地与R
R
X
在一个实施方案中,化合物(10)是由下式(10-9)表示的化合物:
其中,在式(10-9)中,
L
R
R
X
在一个实施方案中,化合物(10)选自由下式(10-10-1)至(10-10-4)表示的化合物。
在式(10-10-1H)至(10-10-4H)中,L
对于由式(10)表示的化合物,可以给出下列化合物作为具体实例。
在发光层包含作为掺杂剂的式(I)表示的化合物和至少一种主体的情况下,其中优选的主体如上所述,并且主体更优选为至少一种式(10)表示的化合物,相对于发光层的总质量,至少一种式(I)表示的化合物的含量优选为0.5质量%至70质量%,更优选为0.5质量%至30质量%,进一步优选为1质量%至30质量%,更进一步优选为1质量%至20质量%,特别优选为1质量%至10质量%,进一步特别优选为1质量%至5质量%。
相对于发光层的总质量,至少一种主体(其中优选的主体如上所述,优选为至少一种式(10)表示的化合物)的含量优选为30质量%至99.9质量%,更优选为70至99.5质量%,进一步优选为70至99质量%,更进一步优选为80至99质量%,特别优选为90至99质量%,进一步特别优选为95至99质量%。
将对根据本发明的一个方面的有机EL器件的层结构进行解释。
根据本发明的一个方面的有机EL器件包括阴极、阳极和一个或多个有机薄膜层,该有机薄膜层包括设置在阴极和阳极之间的发光层。有机层包括至少一个由有机化合物组成的层。或者,通过层叠由有机化合物组成的多个层来形成有机层。除了有机化合物之外,有机层还可以包含无机化合物。
至少一个有机层是发光层。有机层可以构成例如单个发光层,或者可以包括可以在有机EL器件的层结构中采用的其它层。对可在有机EL器件的层结构中采用的层没有特别限制,但其实例包括空穴传输区(包括至少一个空穴传输层,优选另外包括空穴注入层、电子阻挡层、激子阻挡层等中的至少一种)、发光层、间隔层和设置在阴极和发光层之间的电子传输区(包括至少一个电子传输层,优选另外包括电子注入层、空穴阻挡层等中的至少一种)。
根据本发明的一个方面的有机EL器件可以是例如荧光或磷光单色发光器件或荧光/磷光混合白色发光器件。优选地,有机EL器件是荧光单色发光器件,更优选蓝色荧光单色发光器件或荧光/磷光混合白色发光器件。蓝色荧光是指在400至500 nm (最大峰值)的荧光,优选在430 nm至490 nm (最大峰值)的荧光。
此外,其可以是具有单个发光单元的简单类型的器件或具有多个发光单元的串联类型的器件。
本说明书中的“发光单元”是包括有机层的最小单元,其中至少一个有机层是发光层,并且通过注入的空穴和电子的复合来发光。
另外,本说明书中描述的“发光层”是具有发光功能的有机层。发光层例如是磷光发光层、荧光发光层等,优选荧光发光层,更优选蓝色荧光发光层,并且可以是单层或多层的堆叠。
发光单元可以是具有多个磷光发光层或荧光发光层的堆叠型单元。在这种情况下,例如,可以在各发光层之间设置用于防止在磷光发光层中产生的激子扩散到荧光发光层中的间隔层。
作为简单类型的有机EL器件,可以给出诸如阳极/发光单元/阴极的器件结构。
以下示出了发光单元的代表性层结构的实例。括号中的层是任意提供的。
(a)(空穴注入层/)空穴传输层/荧光发光层(/电子传输层/电子注入层)
(b)(空穴注入层/)空穴传输层/磷光发光层(/电子传输层/电子注入层)
(c)(空穴注入层/)空穴传输层/第一荧光发光层/第二荧光发光层(/电子传输层/电子注入层)
(d)(空穴注入层/)空穴传输层/第一磷光层/第二磷光层(/电子传输层/电子注入层)
(e)(空穴注入层/)空穴传输层/磷光发光层/间隔层/荧光发光层(/电子传输层/电子注入层)
(f)(空穴注入层/)空穴传输层/第一磷光发光层/第二磷光发光层/间隔层/荧光发光层(/电子传输层/电子注入层)
(g)(空穴注入层/)空穴传输层/第一磷光层/间隔层/第二磷光发光层/间隔层/荧光发光层(/电子传输层/电子注入层)
(h)(空穴注入层/)空穴传输层/磷光发光层/间隔层/第一荧光发光层/第二荧光发光层(/电子传输层/电子注入层)
(i)(空穴注入层/)空穴传输层/电子阻挡层/荧光发光层(/电子传输层/电子注入层)
(j)(空穴注入层/)空穴传输层/电子阻挡层/磷光发光层(/电子传输层/电子注入层)
(k)(空穴注入层/)空穴传输层/激子阻挡层/荧光发光层(/电子传输层/电子注入层)
(l)(空穴注入层/)空穴传输层/激子阻挡层/磷光发光层(/电子传输层/电子注入层)
(m) (空穴注入层/)第一空穴传输层/第二空穴传输层/荧光发光层(/电子传输层/电子注入层)
(n) (空穴注入层/)第一空穴传输层/第二空穴传输层/荧光发光层(/第一电子传输层/第二电子传输层/电子注入层)
(o) (空穴注入层/)第一空穴传输层/第二空穴传输层/磷光发光层(/电子传输层/电子注入层)
(p) (空穴注入层/)第一空穴传输层/第二空穴传输层/磷光发光层(/第一电子传输层/第二电子传输层/电子注入层)
(q) (空穴注入层/)空穴传输层/荧光发光层/空穴阻挡层(/电子传输层/电子注入层)
(r) (空穴注入层/)空穴传输层/磷光发光层/空穴阻挡层(/电子传输层/电子注入层)
(s) (空穴注入层/)空穴传输层/荧光发光层/激子阻挡层(/电子传输层/电子注入层)
(t) (空穴注入层/)空穴传输层/磷光发光层/激子阻挡层(/电子传输层/电子注入层)。
根据本发明的一个方面的有机EL器件的层结构不限于上述实例。
例如,当有机EL器件具有空穴注入层和空穴传输层时,优选在空穴传输层和阳极之间提供空穴注入层。此外,当有机EL器件具有电子注入层和电子传输层时,优选在电子传输层和阴极之间提供电子注入层。此外,空穴注入层、空穴传输层、电子传输层和电子注入层中的每一个可以由单层形成或由多层形成。
多个磷光发光层、多个磷光发光层和荧光发光层可以是发射多种不同颜色的发光层。例如,发光单元(f)可以包括空穴传输层/第一磷光层(发射红光) /第二磷光发光层(发射绿光) /间隔层/荧光发光层(发射蓝光) /电子传输层。
电子阻挡层可以设置在每个发光层和空穴传输层或间隔层之间。此外,空穴阻挡层可以设置在每个发光层和电子传输层之间。通过设置电子阻挡层或空穴阻挡层,可以将电子或空穴限制在发光层中,从而提高载流子在发光层中的复合概率,并提高发光效率。
作为串联型有机EL器件的代表性器件结构,例如,可以给出诸如阳极/第一发光单元/中间层/第二发光单元/阴极的器件结构。
第一发光单元和第二发光单元例如独立地选自上述发光单元。
中间层通常也被称为中间电极、中间导电层、电荷产生层、吸电子层、连接层、连接件层或中间绝缘层。中间层是向第一发光单元提供电子并向第二发光单元提供空穴的层,并且可以由已知材料形成。
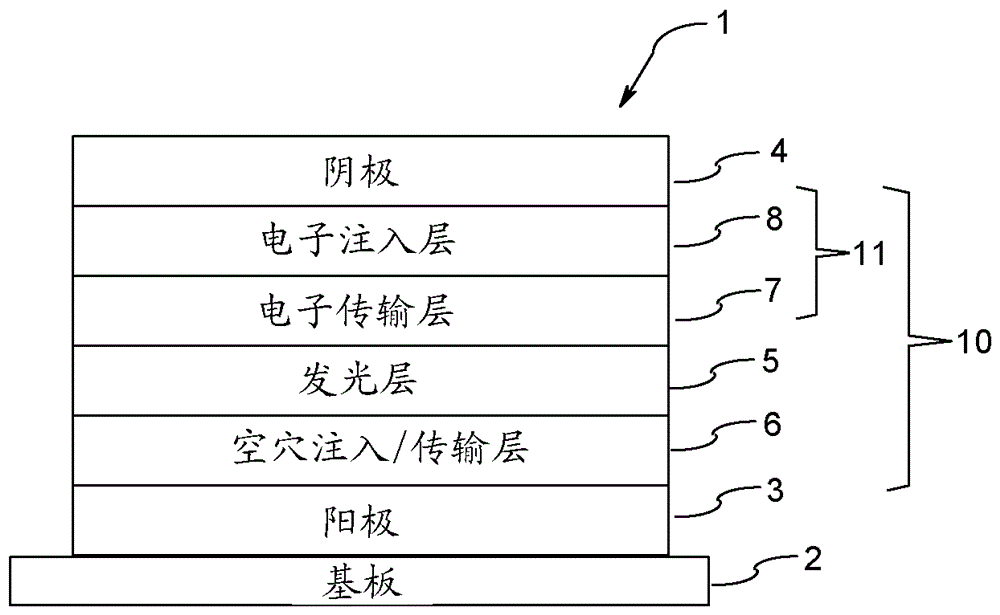
图1显示本发明的有机EL器件的一个实例的示意结构。有机EL器件1包括基板2、阳极3、阴极4和设置在阳极3和阴极4之间的发光单元10。发光单元10包括发光层5,其优选包含主体材料和掺杂剂。在发光层5和阳极3之间可以提供空穴注入和传输层6等,在发光层5和阴极4之间可以提供电子注入层8和电子传输层7等(电子注入和传输单元11),可以在发光层5的阳极3侧提供电子阻挡层,可以在发光层5的阴极4侧提供空穴阻挡层,由于这种结构,可以将电子或空穴限制在发光层5中,由此可以提高在发光层5中产生激子的可能性。
以下,将对构成本说明书中描述的有机EL器件的各层的功能、材料等进行解释。
(基板)
该基板用作该有机EL器件的支撑物。基板优选在波长400至700 nm的可见光区域中具有50%或更大的透光率,优选光滑的基板。基板的材料的实例包括钠钙玻璃、铝硅酸盐玻璃、石英玻璃、塑料等。作为基板,可以使用柔性基板。柔性基板是指可以弯曲(柔性)的基板,并且其实例包括塑料基板等。用于形成塑料基板的材料的具体实例包括聚碳酸酯、聚烯丙酯、聚醚砜、聚丙烯、聚酯、聚氟乙烯、聚氯乙烯、聚酰亚胺、聚萘二甲酸乙二醇酯等。此外,可以使用无机气相沉积膜。
(阳极)
作为阳极,例如,优选使用具有高逸出功(具体地,4.0 eV或更高)的金属、合金、导电化合物、其混合物等。阳极材料的具体实例包括氧化铟-氧化锡(ITO:氧化铟锡)、含硅或氧化硅的氧化铟-氧化锡、氧化铟-氧化锌、含氧化钨或氧化锌的氧化铟、石墨烯等。另外,也可以使用金、银、铂、镍、钨、铬、钼、铁、钴、铜、钯、钛和这些金属的氮化物(例如氧化钛)。
阳极通常通过溅射方法将这些材料沉积在基板上而形成。例如,氧化铟-氧化锌可以通过溅射法使用相对于氧化铟添加了1至10质量%的氧化锌的靶来形成。另外,含有氧化钨或氧化锌的氧化铟可以通过溅射法使用相对于氧化铟添加了0.5至5质量%的氧化钨或0.1至1质量%的氧化锌的靶形成。
作为形成阳极的其它方法,可以给出真空沉积法、涂布法、喷墨法、旋涂法等。当使用银膏等时,可以使用涂布法、喷墨法等。
与阳极接触形成的空穴注入层通过使用允许容易的空穴注入而与阳极的逸出功无关的材料形成。因此,在阳极中,可以使用普通电极材料,例如金属、合金、导电化合物及其混合物。具体地,具有小逸出功的材料,例如碱金属,如锂和铯;碱土金属,例如钙和锶;含有这些金属的合金(例如,镁-银和铝-锂);稀土金属,例如铕和镱;和含有稀土金属的合金。
(空穴传输层)/(空穴注入层)
空穴传输层是在发光层和阳极之间形成的有机层,并具有将空穴从阳极传输到发光层的功能。如果空穴传输层由多层构成,有时可将更接近阳极的有机层定义为空穴注入层。空穴注入层具有从阳极向有机层单元有效地注入空穴的功能。所述空穴注入层通常用于稳定从阳极到通常由有机材料组成的空穴传输层的空穴注入。与阳极具有良好接触的有机材料或具有p型掺杂的有机材料优选用于空穴注入层。
p-掺杂通常由一种或多种p-掺杂剂材料和一种或多种基质材料组成。基质材料优选具有较浅的HOMO能级,p-掺杂剂优选具有较深的LUMO能级,以提高层的载流子密度。p-掺杂剂的具体实例是下面提到的受主材料。合适的基质材料是下面提到的空穴传输材料,优选芳族或杂环胺化合物。
具有高平面性的受主材料或稠合芳烃材料或稠合杂环优选用作空穴注入层的p-掺杂剂材料。
受主材料的具体实例是具有一个或多个吸电子基团的醌化合物,例如F
相对于基质材料,p型掺杂剂的比率优选小于20%摩尔比,更优选小于10%,例如1%、3%或5%。
空穴传输层通常用于有效地注入和传输空穴,并且优选使用芳族或杂环胺化合物。
用于空穴传输层的化合物的具体实例由通式(H)表示,
其中
Ar
优选地,Ar
第二空穴传输层优选插在第一空穴传输层和发光层之间,以通过阻挡过量的电子或激子来提高器件性能。
第二空穴传输层的具体实例与第一空穴传输层相同。优选第二空穴传输层具有较高的三重态能量以阻挡三重态激子,尤其是对于磷光器件,例如双咔唑化合物、联苯胺化合物、三苯胺(triphenylenyl amine)化合物、芴胺化合物、咔唑取代的芳胺化合物、二苯并呋喃取代的芳胺化合物和二苯并噻吩取代的芳胺化合物。
(发光层)
发光层是包含具有高发光性质的物质(发光体材料或掺杂剂材料)的层。作为掺杂剂材料,可以使用各种材料。例如,可以使用发荧光化合物(荧光掺杂剂)、发磷光化合物(磷光掺杂剂)等。发荧光化合物是能够从单重激发态发光的化合物,含有发荧光化合物的发光层被称为荧光发光层。此外,发磷光化合物是能够从三重激发态发光的化合物,并且包含发磷光化合物的发光层被称为磷光发光层。
优选地,本申请的有机EL器件中的发光层包含式(I)的化合物作为掺杂剂材料。
发光层优选包含至少一种掺杂剂材料和至少一种允许其有效发光的主体材料。在一些文献中,掺杂剂材料被称为客体材料、发光体或发光材料。在一些文献中,主体材料被称为基质材料。
单个发光层可以包含多种掺杂剂材料和多种主体材料。此外,可以存在多个发光层。
在本说明书中,将与荧光掺杂剂组合的主体材料称为“荧光主体”,将与磷光掺杂剂组合的主体材料称为“磷光主体”。注意,荧光主体和磷光主体不仅仅通过分子结构分类。磷光主体是用于形成含有磷光掺杂剂的磷光发光层的材料,但不表示其不能用作用于形成荧光发光层的材料。同样情况可应用于荧光主体。
在一个实施方案中,优选发光层包含根据本发明的由式(I)表示的化合物(以下,这些化合物可以称为“化合物(I)”)。更优选地,其作为掺杂剂材料被包含。另外,优选化合物(I)作为荧光掺杂剂包含在发光层中。另外,化合物(I)优选作为蓝色荧光掺杂剂包含在发光层中。
在一个实施方案中,对发光层中作为掺杂剂材料的化合物(I)的含量没有特别限制。就充分发光和浓度淬灭而言,相对于发光层的质量,含量优选为0.5至70质量%,更优选为0.8至30质量%,进一步优选为1至30质量%,更进一步优选为1至20质量%,特别优选为1至10质量%,进一步特别优选为1至5质量%,更进一步特别优选为2至4质量%。
(荧光掺杂剂)
作为化合物(I)以外的荧光掺杂剂,例如可以给出稠合多环芳族化合物、苯乙烯胺化合物、稠环胺化合物、含硼化合物、吡咯化合物、吲哚化合物、咔唑化合物。其中,优选稠环胺化合物、含硼化合物、咔唑化合物。
作为稠环胺化合物,可以给出二氨基芘化合物、二氨基屈化合物、二氨基蒽化合物、二氨基芴化合物、稠合有一个或多个苯并呋喃并骨架的二氨基芴化合物等。
作为含硼化合物,可以给出吡咯亚甲基化合物、三苯基硼烷化合物等。
作为蓝色荧光掺杂剂,例如可以给出芘化合物、苯乙烯胺化合物、屈化合物、荧蒽化合物、芴化合物、二胺化合物、三芳基胺化合物等。具体地,可以给出N,N ' -双[4- (9H-咔唑-9-基)苯基] -N,N ' -二苯基芪-4,4' -二胺(缩写:YGA2S)、4- (9H-咔唑-9-基) -4'- (10-苯基-9-蒽基)三苯基胺(缩写:YGAPA)、4- (10-苯基-9-蒽基) -4' - (9-苯基-9H-咔唑-3-基)三苯基胺(缩写:PCBAPA)等。
作为绿色荧光掺杂剂,例如可以给出芳族胺化合物等。具体地,例如可以给出N-(9,10-二苯基-2-蒽基) -N,9-二苯基-9H-咔唑-3-胺(缩写:2PCAPA)、N- [9,10-双(1,1 '–联苯-2-基) -2-蒽基] -N,9-二苯基-9H-咔唑-3-胺(缩写:2PCABPhA)、N- (9,10-二苯基-2-蒽基) -N,N ',N ' -三苯基-1,4-苯二胺(缩写:2DPAPA)、N- [9,10-双(1,1 ' –联苯-2-基) -2-蒽基] -N,N ',N ' -三苯基-1,4-苯二胺(缩写:2DPABPhA)、N- [9,10-双(1,1 ' -联苯-2-基) ] -N- [4- (9H-咔唑-9-基)苯基] -N-苯基蒽-2-胺(缩写:2YGABPhA)、N,N,9-三苯基蒽-9-胺(缩写:DPhAPhA)等。
作为红色荧光掺杂剂,可以给出并四苯化合物、二胺化合物等。具体地,可以给出N,N,N ',N ' -四(4-甲基苯基)并四苯-5,11-二胺(缩写:p-mPhTD)、7,14-二苯基-N,N,N',N ' -四(4-甲基苯基)苊并[1,2-a ]荧蒽-3,10-二胺(缩写:p-mPhAFD)等。
(磷光掺杂剂)
作为磷光掺杂剂,可以给出发磷光重金属配合物和发磷光稀土金属配合物。
作为重金属配合物,可以给出铱配合物、锇配合物、铂配合物等。重金属配合物是例如选自铱、锇和铂的金属的邻位金属化配合物。
稀土金属配合物的实例包括铽配合物、铕配合物等。具体地,可以给出三(乙酰丙酮) (单菲咯啉)铽(III) (缩写:Tb(acac)
作为蓝色磷光掺杂剂,例如可以给出铱配合物、锇配合物、铂配合物等。具体地,可以给出四(1-吡唑基)硼酸双[2- (4',6' -二氟苯基)吡啶-N,C2' ]铱(III) (缩写:FIr6)、吡啶甲酸双[2- (4',6' -二氟苯基)吡啶-N,C2' ]铱(III) (缩写:Ir(CF
作为绿色磷光掺杂剂,例如可以给出铱配合物等。具体地,可以给出三(2-苯基吡啶-N,C2 ')铱(III) (缩写:Ir(ppy)
作为红色磷光掺杂剂,可以给出铱配合物、铂配合物、铽配合物、铕配合物等。具体地,可以给出乙酰丙酮双[2- (2 ' -苯并[4,5-α]噻吩基)吡啶-N,C3 ' ]铱(III) (缩写:Ir(btp)
如上所述,发光层优选包含至少一种化合物(I)作为掺杂剂。
(主体材料)
作为主体材料,可以给出例如金属配合物,例如铝配合物、铍配合物和锌配合物;杂环化合物,例如吲哚化合物、吡啶化合物、嘧啶化合物、三嗪化合物、喹啉化合物、异喹啉化合物、喹唑啉化合物、二苯并呋喃化合物、二苯并噻吩化合物、噁二唑化合物、苯并咪唑化合物、菲咯啉化合物;稠合多环芳烃(PAH)化合物,例如萘化合物、三亚苯基化合物、咔唑化合物、蒽化合物、菲化合物、芘化合物、屈化合物、并四苯化合物、荧蒽化合物;和芳族胺化合物,例如三芳基胺化合物和稠合多环芳族胺化合物。可以组合使用多种类型的主体材料。
作为荧光主体,具有比荧光掺杂剂更高的单重态能级的化合物是优选的。例如,可以给出杂环化合物、稠合芳族化合物等。作为稠合芳族化合物,优选蒽化合物、芘化合物、屈化合物、并四苯化合物等。蒽化合物优选用作蓝色荧光主体。
在化合物(I)用作至少一种掺杂剂材料的情况下,优选的主体材料是取代或未取代的多环芳烃(PAH)化合物、取代或未取代的多杂芳族化合物、取代或未取代的蒽化合物、或取代或未取代的芘化合物,优选取代或未取代的蒽化合物或取代或未取代的芘化合物,更优选取代或未取代的蒽化合物,最优选由上述式(10)表示的蒽化合物。
作为磷光主体,优选具有比磷光掺杂剂更高的三重态能级的化合物。例如,可以给出金属配合物、杂环化合物、稠合芳族化合物等。其中,可以给出吲哚化合物、咔唑化合物、吡啶化合物、嘧啶化合物、三嗪化合物、喹啉(quinolone)化合物、异喹啉化合物、喹唑啉化合物、二苯并呋喃化合物、二苯并噻吩化合物、萘化合物、三亚苯基化合物、菲化合物、荧蒽化合物等。
(电子传输层)/(电子注入层)
电子传输层是在发光层和阴极之间形成的有机层,具有将电子从阴极传输到发光层的功能。当电子传输层由多层形成时,更靠近阴极的有机层或无机层通常被定义为电子注入层(例如参见图1中的层8,其中电子注入层8和电子传输层7形成电子注入和传输单元11)。电子注入层具有将电子从阴极有效地注入到有机层单元的功能。优选的电子注入材料是碱金属、碱金属化合物、碱金属配合物、碱土金属配合物和稀土金属配合物。
根据一个实施方案,优选电子传输层还包括一个或多个层,如第二电子传输层、提高器件效率和寿命的电子注入层、空穴阻挡层、激子阻挡层或三重态阻挡层。
根据一个实施方案,优选在阴极和发光单元之间的界面区域中包含给电子掺杂剂。由于这种结构,有机EL器件可以具有增加的亮度或长寿命。这里,给电子掺杂剂是指具有逸出功为3.8 eV或更小的金属的掺杂剂。作为其具体实例,可以提及选自碱金属、碱金属配合物、碱金属化合物、碱土金属、碱土金属配合物、碱土金属化合物、稀土金属、稀土金属配合物和稀土金属化合物等中的至少一种。
作为碱金属,可以给出Li (逸出功:2.9 eV)、Na (逸出功:2.36 eV)、K (逸出功:2.28 eV)、Rb (逸出功:2.16 eV)、Cs (逸出功:1.95 eV)等。特别优选逸出功为2.9 eV或更小的材料。其中,优选K、Rb和Cs。进一步优选Rb或Cs。最优选Cs。作为碱土金属,可以给出Ca(逸出功:2.9 eV),Sr (逸出功:2.0 eV至2.5 eV),Ba (逸出功:2.52 eV)等。特别优选逸出功为2.9 eV或更小的材料。作为稀土金属,可以给出Sc、Y、Ce、Tb、Yb等。特别优选逸出功为2.9 eV或更小的材料。
碱金属化合物的实例包括碱金属氧化物如Li
对碱金属配合物、碱土金属配合物和稀土金属配合物没有特别限制,只要它们含有碱金属离子、碱土金属离子和稀土金属离子中的至少一种作为金属离子。同时,配体的优选实例包括但不限于,羟基喹啉、苯并羟基喹啉、羟基吖啶、羟基菲啶、羟基苯基噁唑、羟基苯基噻唑、羟基二芳基噁二唑、羟基二芳基噻二唑、羟基苯基吡啶、羟基苯基苯并咪唑、羟基苯并三唑、羟基氟硼烷(fluborane)、联吡啶、菲咯啉、酞菁、卟啉、环戊二烯、β-二酮和偶氮甲川。
关于给电子掺杂剂的添加形式,优选的是,在界面区域中以层或岛的形状形成给电子掺杂剂。优选的形成方法是这样的方法,其中通过电阻加热沉积法在沉积给电子掺杂剂的同时沉积用于形成界面区域的有机化合物(发光材料或电子注入材料),从而将给电子掺杂剂分散在有机化合物中。
在将给电子掺杂剂形成层的形状的情况下,将用作界面中的有机层的发光材料或电子注入材料形成为层的形状。然后,通过电阻加热沉积法单独沉积还原性掺杂剂,以形成优选具有0.1 nm至15 nm厚度的层。在将给电子掺杂剂形成岛的形状的情况下,将用作界面中的有机层的发光材料或电子注入材料形成为岛的形状。然后,通过电阻加热沉积法单独沉积给电子掺杂剂,以形成优选厚度为0.05 nm至1 nm的岛。作为电子传输层中使用的除式(I)的化合物以外的电子传输材料,可以优选使用分子中具有一个或多个杂原子的芳族杂环化合物。特别地,优选含氮的杂环化合物。
根据一个实施方案,优选电子传输层包含含氮杂环金属螯合物。
根据另一个实施方案,优选电子传输层包含取代或未取代的含氮杂环化合物。用于电子传输层的优选杂环化合物的具体实例是6元吖嗪化合物;例如吡啶化合物、嘧啶化合物、三嗪化合物、吡嗪化合物,优选嘧啶化合物或三嗪化合物;6-元稠合吖嗪化合物,如喹啉化合物、异喹啉化合物、喹喔啉化合物、喹唑啉化合物、菲咯啉化合物、苯并喹啉化合物、苯并异喹啉化合物、二苯并喹喔啉化合物,优选喹啉化合物、异喹啉化合物、菲咯啉化合物;5-元杂环化合物,例如咪唑化合物、噁唑化合物、噁二唑化合物、三唑化合物、噻唑化合物、噻二唑化合物;稠合咪唑化合物,例如苯并咪唑化合物、咪唑并吡啶化合物、萘并咪唑化合物、苯并咪唑并菲啶化合物、苯并咪唑并苯并咪唑化合物,优选苯并咪唑化合物、咪唑并吡啶化合物或苯并咪唑并菲啶化合物。
根据另一个实施方案,优选电子传输层包含表示为Ar
Ar
根据另一个实施方案,优选电子传输层包含芳烃化合物。用于电子传输层的优选芳烃化合物的具体实例是低聚亚苯基化合物、萘化合物、芴化合物、荧蒽基、蒽化合物、菲化合物、芘化合物、三亚苯基化合物、苯并蒽化合物、屈化合物、苯并菲化合物、并四苯化合物和苯并屈化合物,优选蒽化合物、芘化合物和荧蒽化合物。
(阴极)
对于阴极,优选使用金属、合金、导电化合物及其混合物,它们各自具有小的逸出功(具体地,逸出功为3.8 eV或更小)。用于阴极的材料的具体实例包括碱金属如锂和铯;碱土金属,例如镁、钙和锶;铝、含有这些金属的合金(例如,镁-银、铝-锂);稀土金属,例如铕和镱;以及含有稀土金属的合金。
阴极通常通过真空气相沉积或溅射方法形成。此外,在使用银膏等的情况下,可以采用涂布法、喷墨法等。
此外,可以使用各种导电材料,例如银、ITO、石墨烯、含硅或氧化硅的氧化铟-氧化锡(独立地选择逸出功)以形成阴极。使用溅射法、喷墨法、旋涂法等将这些导电材料制成膜。
(绝缘层)
在有机EL器件中,由于电场被施加到薄膜,因此容易产生基于泄漏或短路的像素缺陷。为了防止这种情况,优选在一对电极之间插入绝缘薄层。用于绝缘层中的材料的实例包括氧化铝、氟化锂、氧化锂、氟化铯、氧化铯、氧化镁、氟化镁、氧化钙、氟化钙、氮化铝、氧化钛、氧化硅、氧化锗、氮化硅、氮化硼、氧化钼、氧化钌和氧化钒。其混合物可以用于绝缘层,并且包含这些材料的多层的层压体也可以用于绝缘层。
(间隔层)
间隔层是当荧光发光层和磷光发光层堆叠时在荧光发光层和磷光发光层之间提供的层,以防止在磷光发光层中产生的激子扩散到荧光发光层或以调节载流子平衡。此外,可以在多个磷光发光层之间提供间隔层。
由于在发光层之间提供间隔层,因此用于间隔层的材料优选是具有电子传输能力和空穴传输能力的材料。为了防止三重态能量在相邻磷光发光层中扩散,优选间隔层具有2.6 eV或更大的三重态能量。作为用于间隔层的材料,可以给出与用于上述空穴传输层的那些相同的材料。
(电子阻挡层、空穴阻挡层、激子阻挡层)
电子阻挡层、空穴阻挡层、激子(三重态)阻挡层等可以与发光层相邻设置。
电子阻挡层具有防止电子从发光层泄漏到空穴传输层的功能。空穴阻挡层具有防止空穴从发光层泄漏到电子传输层的功能。为了提高空穴阻挡能力,优选使用具有深HOMO能级的材料。激子阻挡层具有防止发光层中产生的激子扩散到相邻层并将激子限制在发光层内的功能。为了提高三重态阻挡能力,优选使用具有高三重态能级的材料。
(形成层的方法)
除非另有说明,对形成本发明的有机EL器件的各层的方法没有特别的限制。可以使用已知的成膜方法,例如干式成膜法、湿式成膜法等。干式成膜法的具体实例包括真空沉积法、溅射法、等离子体法、离子电镀法等。湿式成膜法的具体实例包括各种涂布法,例如旋涂法、浸渍法、流涂法、喷墨法等。
(膜厚度)
除非另有说明,对本发明的有机EL器件的各层的膜厚度没有特别限制。如果膜厚度太小,则可能出现缺陷如针孔,从而难以获得足够的亮度。如果膜厚度太大,则需要施加高的驱动电压,导致效率降低。在这一方面,膜厚度优选为0.1 nm至10μm,更优选为5 nm至0.2μm。
(电子装置(电子设备))
本发明还涉及包括根据本申请的有机电致发光器件的电子设备(电子装置)。电子装置的实例包括显示部件,例如有机EL面板模块;电视机、移动电话、智能电话和个人计算机等的显示装置;以及照明装置和车辆照明装置的发光装置。
实施例
下面,根据下列合成实施例、实施例和对比例更详细地解释本发明,这些实施例不应被解释为限制本发明的范围。
除非另有说明,以下实施例中提到的百分比和比率是重量%和重量比。
I 合成实施例
在保护性气体气氛中进行所有实验。
化合物1
中间体1-1
在惰性气氛下,将23.2ml正丁基锂(2.7M,在己烷中)加入6.34g (62.7mmol) N,N-二异丙基胺中,同时保持温度低于25℃。在室温下搅拌20分钟后,将反应混合物用10ml无水四氢呋喃稀释,以得到新鲜制备的LDA (二异丙基氨基锂)溶液。
在惰性气氛下,将10.00g (52.2mmol) 1-溴-3-氯苯和6.81g (62.7mmol)氯三甲基硅烷溶解在30ml无水四氢呋喃中。将澄清的无色溶液冷却至-78℃,并向其中缓慢加入新鲜制备的LDA溶液。将温度在-78℃保持10分钟,然后将其升高到-30℃并保持1.5小时。然后将该亮橙色溶液缓慢温热至室温,并搅拌17小时以得到黄色乳状溶液。将反应混合物倒入水中,用乙酸乙酯萃取。然后将有机萃取物用MgSO
中间体1-2
将5.00g (18.97mmol)中间体1-1、2.97g (19.91mmol) 4-叔丁基苯胺和7.29g(76.00mmol)叔丁醇钠加入100ml甲苯中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将347mg (2mol%)三(二亚苄基丙酮)二钯(0)和439mg (8mol%)四氟硼酸三-叔丁基鏻加入到反应混合物中。在两个额外的冷冻-抽吸-解冻循环之后,将反应混合物加热至60℃持续25小时。将反应物冷却至室温,用甲苯稀释。将有机萃取物用水洗涤并用MgSO
中间体1-3
将3.00g (9.04mmol)中间体1-2、2.12g (9.94mmol) 1-溴-4-叔丁基苯和3.47g(36.1mmol)叔丁醇钠加入50ml甲苯中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将166mg (2mol%)三(二亚苄基丙酮)二钯(0)和209mg (8mol%)四氟硼酸三-叔丁基鏻添加到反应混合物中。在两个额外的冷冻-抽吸-解冻循环之后,将反应混合物加热至60℃持续19小时。将反应物冷却至室温,用甲苯稀释。将有机萃取物用水洗涤并用MgSO
中间体1-4
将10.0g (40.6mmol) 1-溴-9H-咔唑、13.4g (52.8mmol)双(频哪醇合)二硼和16.0g (168.2mmol)乙酸钾悬浮在100ml无水N,N-二甲基甲酰胺中。通过用高真空将反应容器排气并用氩气回填,使悬浮液脱气。重复该过程7次,并在重复排气-回填2次之前,将2.32g (7mol%)的与二氯甲烷的配合的[1,1' -双(二苯基膦基)二茂铁-钯(II)加入到反应混合物中。然后将反应混合物加热至80℃持续19小时。冷却至室温后,用10ml乙醚和50ml环己烷稀释反应物,并在小硅胶垫上过滤。用300ml的环己烷和乙醚的5:1混合物洗涤该垫。在旋转蒸发仪上除去溶剂,并通过硅胶柱色谱法,使用环己烷作为洗脱剂纯化残余物。合并含有产物的级分,并在旋转蒸发仪上除去溶剂,直到沉淀出白色固体。过滤悬浮液,得到作为白色固体的10.25g (86%产率)中间体1-4。
中间体1-5
将3.65g (7.86mmol)中间体1-3、2.54g (8.65mmol)中间体1-4和6.68g(31.5mmol) K
化合物1
将0.50g (0.84mmol)中间体1-5溶解在10 ml的1,2-二氯苯中,并使用3个冷冻-抽吸-解冻循环脱气。向反应混合物中加入0.34g (3.36mmol)三乙胺,随后缓慢加入1.68ml(1.68mmol)三氯硼烷(1M庚烷溶液)。将反应混合物加热至180℃持续42小时,产生澄清油状溶液。冷却至室温后,用70ml环己烷稀释凝胶状混合物,并通过二氧化硅垫过滤。用200ml环己烷洗涤该垫以除去溶剂,使用100ml甲苯,然后使用100ml二氯甲烷将所需产物洗脱到不同的级分中。在旋转蒸发仪上除去溶剂,并通过硅胶柱色谱法,使用庚烷和甲苯的混合物(0-20%梯度)纯化粗产物,得到作为油的产物,使用几滴乙醚将其结晶。通过过滤收集固体以得到作为亮黄色粉末的0.13g (29%产率)化合物1。
化合物2
中间体2-1
将5.00g (18.97mmol)中间体1-1、5.83g (2.86mmol) 3,6-二叔丁基-9H-咔唑和7.29g (76.00mmol)叔丁醇钠加入150ml二甲苯中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将347mg (2mol%)三(二亚苄基丙酮)二钯(0)和329mg (3mol%) Xantphos (4,5-双(二苯基膦基) -9,9-二甲基呫吨)加入反应混合物中。在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物加热至120℃持续15小时。向反应混合物中加入另外的347mg (2mol%)的三(二亚苄基丙酮)二钯(0)和329mg (3mol%)的Xantphos,并且将反应物进一步加热总共50小时。然后将反应物冷却至室温,用甲苯萃取,将有机萃取物用无水MgSO
中间体2-2
将5.00g (17.89mmol) 3,6-二叔丁基-9H-咔唑溶解在50ml乙酸中,并向白色悬浮液中分批加入3.18g (17.89mmol) N-溴琥珀酰亚胺。4小时后,加入200ml水,再搅拌反应物30分钟。过滤所得沉淀物,并将固体用水、饱和NaHCO
中间体2-3
将3.40g (9.49mmol)中间体2-2、3.13g (12.34mmol)双(频哪醇合)二硼和3.73g(39.20mmol)乙酸钾悬浮于40ml无水N,N-二甲基甲酰胺中。通过用高真空将反应容器排气并用氩气回填,使悬浮液脱气。重复该过程7次,并在重复排气回填2次之前,向反应混合物中加入542mg (7mol%)与二氯甲烷配合的[1,1' -双(二苯基膦基)二茂铁]二氯钯(II)。然后将反应混合物加热至80℃持续21小时。冷却至室温后,用乙醚稀释反应物,用水洗涤,用MgSO
中间体2-4
将2.00g (4.33mmol)中间体2-1、2.46g (6.06mmol)中间体2-3和3.67g(17.3mmol) K
化合物2
将2.44g (3.46mmol)中间体2-4溶解在70 ml的1,2-二氯苯中,并用氮气吹扫反应容器。在室温下加入2.42ml (13.84mmol) N,N-二异丙基乙胺,然后滴加5.20ml(5.20mmol)三溴硼烷(1M庚烷溶液)。将所得澄清浅橙色溶液加热至145℃持续20小时,然后冷却至室温。缓慢加入15ml甲醇使反应淬灭,将所得溶液倒入200ml甲醇中。将黄色沉淀物搅拌5分钟,然后过滤,用甲醇洗涤并干燥,得到作为黄色固体的1.11g (50%产率)化合物2。
化合物3
中间体3-1
将6.00g (22.76 mmol)中间体1-1、10.81g (25.03 mmol) 3,6-双(4- (叔丁基)苯基) -9H-咔唑(其根据New Journal of Chemistry,2019,16629中所述的方法合成)和4.37g (45.5 mmol)叔丁醇钠加入175 mL二甲苯中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将417mg (2mol%)三(二亚苄基丙酮)二钯(0)和527 mg (4mol%) Xantphos (4,5-双(二苯基膦基) -9,9-二甲基呫吨)加入反应混合物中。在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物加热至120℃持续15小时。向反应混合物中加入另外347mg (2mol%)的三(二亚苄基丙酮)二钯(0)和329mg (3mol%)的Xantphos,并且将反应物进一步加热总共41小时。然后将反应物冷却至室温,用甲苯萃取,将有机萃取物用无水硫酸镁干燥,并用二氧化硅小垫过滤。用甲苯洗涤该垫,并在旋转蒸发仪上除去滤液的溶剂。将粗产物通过硅胶柱色谱法,使用庚烷/ THF 95/5纯化,得到作为浅黄色泡沫的1.6g (11%产率)中间体3-1。
ESI-MS: 614.3 [M+H]
中间体3-2
将2.46g (4.00 mmol)中间体3-1、2.11g (5.21 mmol)中间体2-3和3.40g (16.0mmol)磷酸钾悬浮在40mL甲苯、10 mL二氧杂环己烷和10 mL水的混合物中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将18 mg (2mol%)乙酸钯(II)和197 mg (12mol%) SPhos加入反应混合物中。在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物加热至90℃持续15小时。然后将反应物冷却至室温,用甲苯萃取,将有机萃取物用无水硫酸镁干燥,干沉积在二氧化硅上。将粗产物通过硅胶柱色谱法,使用庚烷和甲苯的混合物(0-35%梯度)纯化,得到作为浅黄色泡沫的1.4g (41%产率)中间体3-2。
ESI-MS: 857.5 [M+H]+。
化合物3
将3.70g (4.32 mmol)中间体3-2溶于80 mL的1,2-二氯苯中,并用氮气吹扫反应容器。在室温下加入3.02 mL (17.26 mmol) N,N-二异丙基乙胺,然后滴加8.50 mL (8.50mmol)三溴硼烷(1M庚烷溶液)。将得到的澄清浅橙色溶液加热至160℃持续16小时,然后冷却至室温。缓慢加入5mL乙酸钠10%水溶液淬灭反应。用甲苯(2×20 mL)萃取水相。将合并的有机相用二氧化硅塞过滤,将二氧化硅塞用甲苯(40 mL)冲洗。将滤液倒入500ml甲醇中。将黄色沉淀物搅拌5分钟,然后过滤,用甲醇洗涤并干燥,得到作为黄色固体的2.24g (66%产率)化合物3。
ESI-MS: 793.5 [M+H]
化合物4
中间体4-1
将41.0g (0.13 mol) 1-溴-3-氯-5-碘苯、23.0g (0.13 mol) 4-叔丁基苯基硼酸、4.48g (3.88 mmol)四(三苯基膦)钯(0)和300g 的10%碳酸钠水溶液悬浮于120 mL甲苯和120 mL乙醇中。将悬浮液抽真空三次,用氩气回填,并在73℃下加热22小时。将浅黄色悬浮液冷却至室温,用200 mL水淬灭。将有机相用水(2×200 mL)洗涤,并用硫酸钠干燥。将产物进一步通过MPLC,使用CombiFlash Companion (硅胶,庚烷)纯化,得到作为白色固体的39.6g (93%产率)中间体4-1。
中间体4-2
将16 mL (0.11 mol)二异丙胺溶于100 mL四氢呋喃中,并用45 mL正丁基锂(2.5M,在己烷中)在-30℃下滴加处理,将该溶液在-70℃的最高温度下缓慢加入到30.0g(93 mmol)中间体4-1和14.1 mL (0.11 mol)氯三甲基硅烷在200 mL四氢呋喃中的预冷却溶液中。在完全添加之后,在-75℃下进一步搅拌浅黄色溶液45分钟。加入100 mL的5%氯化铵水溶液,搅拌反应混合物直至达到室温。用200 mL庚烷稀释该溶液,用200 mL水和100 mL饱和氯化钠水溶液洗涤有机相。将有机相用硫酸钠干燥,并真空浓缩。将产物进一步通过MPLC,使用CombiFlash Companion (硅胶,庚烷)纯化,得到作为无色油的36.7g (98%产率)的中间体4-2。
中间体4-3
将2.98g (7.52 mmol)中间体4-2、2.31g (8.27 mmol) 3,6-二叔丁基-9H-咔唑、0.28g (0.3 mmol)三(二亚苄基丙酮)二钯(0)、0.35g (0.6 mmol) 4,5-双(二苯基膦基) -9,9-二甲基呫吨(Xantphos)和2.9g (30 mmol)叔丁醇钠悬浮于30 mL邻二甲苯中。将橙色悬浮液抽真空三次,并用氩气回填,在117℃下搅拌22小时。将深棕色反应混合物冷却至室温,用100 mL甲苯稀释,然后用100 mL水萃取。将有机相用100 ml水和100 ml饱和氯化钠水溶液洗涤,然后用硫酸钠干燥并真空浓缩。将产物进一步通过MPLC,使用CombiFlashCompanion(硅胶,庚烷)纯化。用100 mL甲醇处理所得油,并在40℃搅拌直至形成悬浮液,得到作为白色固体的1.37g (30%产率)中间体4-3。
ESI-MS (阳性,m/z):C
中间体 4-4
将2.00g (3.4 mmol)中间体4-3、1.64g (4.0 mmol)中间体2-3、16 mg (0.07mmol)乙酸钯(II)、171 mg (0.42 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和2.86g (13.4 mmol)磷酸三钾溶于25 mL甲苯、15 mL1,4-二氧杂环己烷和7 mL水的混合物中。将溶液抽真空三次并用氩气回填,并在77℃下加热20小时。将反应混合物冷却至室温,倒入100 mL水中,搅拌10分钟。将通过的有机物经3 cm硅胶层过滤,随后用200 mL庚烷冲洗硅胶层。将收集的洗脱液在真空下浓缩。将产物溶解在乙醇中,加入水,形成悬浮液。将悬浮液搅拌30分钟,然后过滤,并用水洗涤固体。将固体溶解在二氯甲烷中,然后用硫酸钠干燥,并在真空下浓缩,得到作为白色固体的1.8g (64%)的中间体4-4。
ESI-MS (阴性,m/z):C
化合物4
将1.80g (2.15 mmol)中间体4-4溶解在40 ml的1,2-二氯苯中。滴加1.5 mL (8.6mmol) N,N-二异丙基乙胺和3.2 mL三溴硼烷(1.0M,在庚烷中)。将浅黄色溶液在145℃下加热24小时,冷却至室温并缓慢倒入300 mL甲醇中。将悬浮液搅拌10分钟,过滤,并用甲醇和乙醇洗涤固体。将固体真空干燥,得到作为黄色固体的1.15g (69%产率)化合物4。
ESI-MS (阳性,m/z):C
化合物5
中间体5-1
将12.0g (47.7 mmol) 8-氯-7H-苯并[ c ]咔唑、18.2g (71.5 mmol)双(频哪醇合)二硼、1.81g (3.8 mmol) 2-二环己基膦基-2 ',4 ',6 ' -三异丙基联苯(XPhos)和9.4g (95 mmol)乙酸钾悬浮于200mL二氧杂环己烷中。加入873 mg (0.95 mmol)三(二亚苄基丙酮)二钯(0),并将悬浮液在101℃下加热40分钟。将悬浮液冷却并用40 mL的二氧杂环己烷稀释。将悬浮液通过3 cm硅胶层过滤,随后用100 mL二氧杂环己烷洗涤硅胶层。将收集的洗脱液真空浓缩,将固体从50 mL庚烷中重结晶。将固体用30 ml冷庚烷洗涤,并进一步在真空下干燥,得到作为白色固体的12.3g (75%产率)中间体5-1。
ESI-MS (阳性,m/z):C
中间体5-2
将3.00g (6.49 mmol)中间体2-1、2.45g (7.14 mmol)中间体5-1、29 mg (0.13mmol)乙酸钯(II)、320 mg (0.78 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhoS)和5.51g (26.0 mmol)磷酸三钾溶于55 mL邻二甲苯、30 mL1,4-二氧杂环己烷和15mL水的混合物中。将反应混合物抽真空三次并用氩气回填,并在84℃下加热3小时。冷却反应混合物,加入40 mL甲苯和40 mL水。将有机相用水(3×40 mL)洗涤,然后用硫酸钠干燥,并真空浓缩。将产物进一步通过MPLC,使用CombiFlash Companion(硅胶,甲苯)纯化。将得到的白色泡沫与50 mL一起加热,并将得到的混浊溶液冷却到室温。加入10mL水,加热混合物直至形成悬浮液。将悬浮液搅拌20分钟,冷却至室温并过滤。将固体溶解在二氯苯中,用硫酸镁干燥,并将溶液真空浓缩,得到作为白色固体的3.40g (82%产率)中间体5-2。
ESI-MS (阴性,m/z):C
化合物5
将3.40g (5.29 mmol)中间体5-2溶解在70 mL1,2-二氯苯中。滴加3.7 mL (21.2mmol) N,N-二异丙基乙胺和10.6 mL三溴硼烷(1.0M,在庚烷中)。将黄色溶液在150℃下加热18小时。将橙色溶液冷却,缓慢加入4 mL的10%乙酸钠水溶液。将混合物滴加到600 mL甲醇中。过滤黄色悬浮液,用乙醇和庚烷洗涤固体。在150 mL二氯甲烷和100 mL异丙醇的混合物中加热固体,然后缓慢冷却至室温。过滤悬浮液,用异丙醇洗涤固体,得到作为黄色固体的2.12g (69%产率)化合物5。
ESI-MS (阴性,m/z):C
化合物6
中间体6-1
将17.3g (70.0mmol) 4-溴二苯并[ b,d ]呋喃、12.48g (77.0 mmol) 2,6-二氯苯胺、10.09g (105 mmol)叔丁醇钠悬浮于150 mL邻二甲苯中。用Ar对悬浮液进行脱气,并将2.62g (6mol%) BINAP和471 mg (3mol%)三(二亚苄基丙酮)二钯(0)加入到反应混合物中。将反应混合物加热至155℃持续3小时。将反应物冷却至室温,用甲苯/水稀释,并经硅藻土过滤。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为白色固体的19.26g (84%产率)中间体6-1。
ESI-MS: 328.3 [M+H]
中间体6-2
将17.72g (54.0 mmol)中间体6-1、14.93g (108 mmol)碳酸钾悬浮于N,N-二甲基乙酰胺中。用Ar使该悬浮液脱气,并向反应混合物中加入485 mg (4mol%)乙酸钯和1.59g(8mol%)四氟硼酸三环己基鏻。将反应混合物加热至130℃持续3.5小时。将反应物冷却至室温,用甲苯/水稀释,并经硅藻土过滤。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为白色固体的10.39g (66%产率)中间体6-2。
ESI-MS: 290.0 [M-H]
中间体6-3
将9.63g (33.0 mmol)中间体6-2、10.06g (39.6 mmol)双(频哪醇合)二硼和8.10g (83.0 mmol)乙酸钾悬浮于125 mL的1,4-二氧杂环己烷中。用Ar对悬浮液进行脱气,并向反应混合物中加入453 mg (1.5mol%)三(二亚苄基丙酮)二钯(0)和406 mg (3mol%)2-二环己基膦基-2 ',6 ' -二甲氧基联苯。将反应混合物加热至105℃持续4小时。将反应物冷却至室温,用甲苯/水稀释,并经硅藻土过滤。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物在70 mL庚烷中回流15分钟,冷却至室温,然后过滤橙色悬浮液并真空干燥。得到作为米黄色固体的11.40g (90%产率)中间体6-3。
ESI-MS: 382.3 [M-H]
中间体6-4
将3.86g (6.5 mmol)中间体4-3、2.74g (7.15 mmol)中间体6-3、4.24g (13.0mmol)碳酸铯悬浮于甲苯/乙醇/水(60/20/10mL)的混合物中。用Ar对悬浮液脱气,并将44mg (3mol%)乙酸钯和186 mg (6mol%) 2-二环己基膦基-2 ',4 ',6 ' -三异丙基联苯加入到反应混合物中。将反应混合物加热至60℃持续2.5小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为白色泡沫的4.46g (84%产率)的中间体6-4。
ESI-MS: 813.6 [M-H]
化合物6
将3.26g (4.00 mmol)中间体6-4溶于50 mL的1,2-二氯苯中,并用Ar脱气。将2.79mL (16.0 mmol) N-乙基-N-异丙基丙-2-胺加入到反应混合物中,随后缓慢加入6.00 mL(6.00 mmol)三溴硼烷(1M庚烷溶液)。将反应混合物加热至160℃持续28.5小时。然后,再加入2.00 mL (2.00 mmol)三溴硼烷(1M庚烷溶液),然后加热至160℃持续16.5小时。冷却至室温后,用水淬灭反应物,并用1,2-二氯苯萃取。将合并的有机层用盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为黄色固体的1.36g (45%产率)化合物6。
ESI-MS: 751.9 [M+H]
化合物7
中间体7-1
将48.0g (0.16 mol) 1,3-二溴-5- (叔丁基)苯溶解在500 ml四氢呋喃中。在45分钟内,在-71℃的最高温度下滴加70.0 ml正丁基锂(2.5M,在己烷中)。在15分钟内,在-55℃的最高温度下分批加入45.9g (0.18 mol)碘,并在-78℃下进一步搅拌所得悬浮液45分钟。加入400 ml的10%亚硫酸钠水溶液,进一步搅拌反应混合物直至达到室温。分离有机相,将水相用环己烷(2×150 ml)萃取。将合并的有机相用水(2×200mL)和饱和氯化钠水溶液洗涤。将有机相用硫酸钠干燥,并真空浓缩,得到作为橙色油的54.7g (83%产率)中间体7-1。
中间体7-2
将3.22g (10.00 mmol) 9,9-二甲基-2,7-二(叔丁基) -9,10-二氢吖啶、3.73g(11.00 mmol)中间体7-1和2.88g (30.00 mmol)乙酸钠悬浮在57 mL二甲苯中。在使用3个冷冻-抽吸-解冻循环使悬浮液脱气之后,将225 mg (1.00 mmol)乙酸钯和554 mg (1.00mmol) 1,1 ' -双(二苯基膦基)二茂铁加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物搅拌至100℃持续1小时。将反应物冷却至室温并浓缩。将残余物通过硅胶柱色谱法,用环己烷作为洗脱剂纯化,得到作为白色固体的3.42g (64%产率)中间体7-2。
ESI-MS: 534.6 [M+H]
中间体7-3
将3.42g (6.42 mmol)中间体7-1、2.86g (7.06 mmol)中间体2-3和5.45g (25.68mmol)磷酸钾溶于54 mL甲苯、27 mL二氧杂环己烷和16 mL水中。在使用3个冷冻-抽吸-解冻循环将溶液脱气之后,将29 mg (0.13 mmol)乙酸钯和316 mg (0.77 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至85℃持续14.5小时。将反应物冷却至室温,用甲苯稀释。将有机萃取物用水洗涤,用硫酸钠干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合溶剂作为洗脱液纯化,得到作为白色固体的4.17g (产率89%)中间体7-3。
ESI-MS: 731.9 [M+H]
化合物7
将4.14g (5.66 mmol)中间体7-3溶于190mL二氯苯中。然后,向溶液中加入11.9mL (11.9 mmol) 1.0M三溴化硼的庚烷溶液,接着加入4.2 mL (23.78 mmol) N,N-二异丙基乙胺,将混合物在185℃搅拌40小时。将反应物冷却至室温,用甲苯稀释。用1.0M乙酸钠水溶液淬灭反应混合物。将水层用甲苯萃取。将有机萃取物用水洗涤,用硫酸镁干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用庚烷作为洗脱剂纯化,得到作为黄色固体的2.49g (54%产率)化合物7。
ESI-MS: 739.9 [M+H]
化合物8
中间体8-1
将5.00g (14.8 mmol)中间体7-1、5.09g (11.8 mmol) 3,6-双(4- (叔丁基)苯基) -9H-咔唑、0.28g (1.5 mmol)碘化铜(I)、0.51g (4.42 mmol)环己烷-1,2-二胺和9.39g (44.2 mmol)磷酸三钾悬浮于75 ml的1,4-二氧杂环己烷中,在91℃下加热五个小时。将悬浮液通过3 cm硅胶层过滤,随后用100ml二氧杂环己烷冲洗硅胶。真空浓缩洗脱液,将产物进一步通过MPLC使用CombiFlash Companion (硅胶,庚烷/0-5%二氯甲烷梯度)纯化,得到6.9g (91%)中间体8-1。
ESI-MS (阳性,m/z):C
中间体8-1
将2.20g (3.42 mmol)中间体8-2、1.53g (3.77 mmol)中间体2-3、15 mg (0.07mmol)乙酸钯(II)、154 mg (0.41 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和2.91g (13.7 mmol)磷酸三钾溶于30 mL甲苯、15 mL的1,4-二氧杂环己烷和10mL水的混合物中。将溶液抽真空三次并用氩气回填,并在82℃下加热三小时。将反应混合物用100mL甲苯稀释,并用100mL水处理。将有机相用水(3×50mL)洗涤,用硫酸钠干燥并真空浓缩。用30 mL二氯甲烷和50mL乙醇稀释所得油。将溶液真空浓缩至50mL体积,过滤所得悬浮液,用50mL乙醇洗涤固体,得到作为白色固体的2.19g (76%产率)中间体8-3。
ESI-MS (阴性,m/z):C
化合物8
将2.00g (2.38 mmol)中间体8-2溶解在40 mL的1,2-二氯苯中。滴加1.7 mL (9.5mmol) N,N-二异丙基乙胺和4.75 mL三溴硼烷(1.0M,在庚烷中)。在172℃下加热该棕色溶液2.5小时。冷却反应混合物,加入100mL甲醇。将悬浮液搅拌15分钟,然后过滤。用50mL甲醇,然后用30 mL水洗涤固体,接着用50mL甲醇和30 mL庚烷洗涤。将固体进一步通过MPLC,使用CombiFlash Companion (硅胶,二氯甲烷)纯化,得到作为黄色固体的1.84g (91%产率)化合物8。
ESI-MS (阳性,m/z):C
化合物9
中间体9-1
用175 mL四氢呋喃稀释175 mL氯化锌溶液(1.9M,在2-甲基四氢呋喃中),冷却至0℃。在10分钟内,在最高温度25℃下加入300 mL环己基氯化镁溶液(1M,在2-甲基四氢呋喃中)。将反应混合物在0℃下进一步搅拌10分钟,然后在最高温度15℃下缓慢加入到54.0g(0.24 mol) 6-溴-2-四氢萘酮、0.34g (1.5 mmol)乙酸钯(II)和1.30g (3.0 mmol) 2-二环己基膦基-2 ',6 ' -双(N,N-二甲基氨基)联苯(CPhos)在540 mL四氢呋喃中的预冷却溶液中。将所得橙色悬浮液在0℃下搅拌一小时,并在31℃下加热另外一小时。加入0.34g(1.5 mmol)乙酸钯(II)和1.30g (3.0 mmol)CPhos,并继续加热另外两个小时。将黑色悬浮液冷却至室温,并在硅藻土助滤剂垫上过滤,随后用500 mL环己烷冲洗助滤剂。将合并的洗脱液与300 mL水混合,真空除去有机溶剂。将残余物与600 mL环己烷和600 mL乙酸乙酯一起搅拌。分离有机相,用300 mL水和200 mL饱和氯化钠水溶液洗涤。将有机相用硫酸钠干燥,用硅胶垫过滤,然后用300 mL的环己烷和乙酸乙酯(2:1)的溶剂混合物冲洗硅胶,再用300 mL乙酸乙酯冲洗。将合并的洗脱液真空浓缩,得到59.6g (87%产率)中间体9-1。
中间体9-2
将30.0g (0.13 mol) 1-溴-4- (叔丁基)苯胺悬浮于300 ml的37%盐酸水溶液中并冷却至0℃。在最大温度2℃下,在15分钟内滴加60.5g (0.13 mol) 15%亚硝酸钠水溶液。在5℃的最高温度下,用40分钟滴加74.8g (0.40 mol)氯化锡在74.8g的37%盐酸水溶液中的溶液。在0℃下搅拌浓稠悬浮液90分钟。过滤悬浮液,用150 mL饱和氯化钠水溶液和200mL庚烷洗涤灰白色残余物。将剩余固体在40℃下真空干燥18小时,得到作为白色粉末的31g(84%产率)中间体9-2,其直接用于下一反应步骤。
中间体9-3
将29.8g (48.7 mmol)中间体9-1和15.0g (48.7 mmol)中间体9-2与150 mL的4N盐酸溶液(在二氧杂环己烷中)和100mL二氧杂环己烷混合。将黄色悬浮液在110℃加热90分钟。将橙色悬浮液冷却至室温并过滤。用二氧杂环己烷洗涤白色固体,将收集的洗脱液用水和250 mL甲苯稀释。分离有机相,用碳酸氢钠溶液洗涤,直至达到碱性pH,然后用饱和氯化钠水溶液洗涤,用硫酸钠干燥。将混合物在硅胶塞上过滤,随后用环己烷冲洗硅胶层。将收集的洗脱液在真空下浓缩。将产物通过MPLC使用CombiFlash Companion (硅胶,庚烷/0-2%乙酸乙酯梯度)纯化,得到作为橙色固体的14.7g (69%产率)中间体9-3。
ESI-MS (阴性,m/z):C
中间体 9-4
将邻二甲苯中的10.3g (23.6 mmol)中间体9-3和6.10g (24.8 mmol)对氯醌(p-choranil)在138℃加热六小时。将反应混合物冷却至室温,并用乙酸乙酯稀释直至形成溶液。将溶液与20g硅胶混合,并在真空下浓缩。将固体进一步通过MPLC,使用
ESI-MS (阳性,m/z):C
中间体9-5
将11.7g (26.9 mmol)中间体9-4、10.3g (40.4 mmol)双(频哪醇合)二硼和5.40g(55.0 mmol)乙酸钾悬浮于110 mL二氧杂环己烷中。加入520 mg (1.09 mmol) 2-二环己基膦基-2 ',4 ',6 ' -三异丙基联苯(XPhos)和250 mg (0.27 mmol)三(二亚苄基丙酮)二钯(0),并将该悬浮液在66℃下加热15小时。将橙色悬浮液冷却并用140 mL水稀释。在室温下搅拌悬浮液并过滤。将固体溶解在乙酸乙酯中,加入50g硅藻土助滤剂。将混合物真空浓缩,并通过MPLC使用CombiFlash Companion(硅胶,庚烷/乙酸乙酯9:1)进行纯化,得到作为浅黄色固体的9.3g (72%产率)中间体9-5。
ESI-MS (阳性,m/z):C
中间体9-6
将40.0g (0.12 mol) 3,6-二溴-9H-咔唑、43.8g (0.25 mol) 3-叔丁基苯基硼酸、2.13g (1.85 mmol)四(三苯基膦)钯(0)和574g 的10%碳酸钠水溶液悬浮于260 mL甲苯和260 mL乙醇中。将悬浮液抽真空三次,用氩气回填,并在74℃下加热两小时。将橙色悬浮液冷却至室温并过滤。用甲苯和水洗涤固体,然后溶于热甲苯中。将热溶液在硅胶垫上过滤,随后用热甲苯冲洗二氧化硅。将合并的洗脱液在真空下浓缩直至形成悬浮液,并冷却至室温。过滤悬浮液,将固体用甲苯洗涤,得到作为白色固体的33.0g (55%产率)中间体9-6。
ESI-MS (阳性,m/z):C
中间体9-7
将4.00g (11.8 mmol)中间体7-1、4.07g (9.44 mmol)中间体9-6、225 mg (1.18mmol)碘化铜(I)、404 mg (3.54 mmol)环己烷-1,2-二胺和7.51g (35.4 mmol)磷酸三钾悬浮于75 mL的1,4-二氧杂环己烷中,在91℃加热六小时。将该悬浮液通过3 cm硅胶层过滤,然后用100mL二氧杂环己烷冲洗硅胶。将洗脱液在真空下浓缩,将产物进一步通过MPLC,使用CombiFlash Companion(硅胶,庚烷/0-20%梯度的二氯甲烷)纯化,得到5.46g (90%)中间体9-7。
ESI-MS (阳性,m/z):C
中间体9-8
将5.00g (7.78 mmol)中间体9-7、4.12g (8.56 mmol)中间体9-5、35 mg (0.16mmol)乙酸钯(II)、383 mg (0.93 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和6.61g (31.1 mmol)磷酸三钾溶解在30 ml甲苯、15 ml1,4-二氧杂环己烷和10ml水的混合物中。将溶液抽真空三次并用氩气回填,并在82℃下加热三小时。将反应混合物冷却至室温,用100ml甲苯稀释,用100ml水处理。将有机相用水(3×50mL)洗涤,用硫酸钠干燥并真空浓缩。用30 ml二氯甲烷和100ml乙醇稀释所得油。将溶液真空浓缩至100ml体积,过滤所得悬浮液,用50ml乙醇洗涤固体,得到作为白色固体的4.7g (66%产率)中间体9-8。
ESI-MS (阳性,m/z):C
化合物9
将4.50g (4.91 mmol)中间体9-8溶解在120 ml的1,2-二氯苯中。滴加3.4 ml(19.6 mmol) N,N-二异丙基乙胺和9.8 ml三溴硼烷(1.0M,在庚烷中)。在172℃下加热该棕色溶液四个小时。将反应混合物冷却,并加入300 ml甲醇。真空浓缩溶液,将产物通过MPLC,使用
ESI-MS (阳性,m/z):C
化合物10
中间体10-1
将13.2g (47.2 mmol) 3,6-二-叔-9H-咔唑和20.0g (59.0 mmol)中间体7-1溶解在230 mL二氧杂环己烷中。向该溶液中加入1.12g (5.90 mmol)碘化铜(I)和2.02g (17.7mmol)环己烷-1,2-二胺和37.6g (177 mmol)磷酸钾。将混合物在95℃搅拌6.5小时。将反应混合物冷却至室温后,过滤固体并用甲苯洗涤。用3-氨基-2-丙醇的水溶液洗涤溶液。用硫酸钠干燥有机层并除去溶剂。将残余物通过硅胶柱色谱法,使用庚烷作为洗脱剂纯化,得到作为米黄色固体的19.8g (86%产率)中间体10-1。
ESI-MS: 491 [M+H]
中间体10-2
将2.60g (5.30 mmol)中间体10-1、1.38g (5.45 mmol)双(频哪醇合)二硼和1.04g (10.60 mmol)乙酸钠悬浮在27 mL甲苯中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将120 mg (0.13 mmol)三(二亚苄基丙酮)二钯(0)和152 mg (0.51 mmol) 2-二环己基膦基-2 ',4 ',6 ' -三异丙基联苯加入到混合物中。在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物加热至110℃持续6小时。将反应物冷却至室温,用甲苯和水稀释。将水层用甲苯萃取,将有机层用盐水洗涤,用硫酸镁干燥,过滤,浓缩溶液。将粗产物用二氯甲烷和乙腈重结晶,得到作为白色固体的2.69g (80%产率)中间体10-2。
ESI-MS: 538.8 [M+H]
中间体10-3
将10.15g (23.52 mmol) 3,6-双(4- (叔丁基)苯基) -9H-咔唑悬浮于THF中,并分批加入4.19g (23.52 mmol) N-溴琥珀酰亚胺。将混合物在室温下搅拌50分钟后,滤出反应混合物。浓缩滤液。将粗产物通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合溶剂作为洗脱剂纯化。将产物在二氯甲烷和庚烷的混合溶剂中沉淀,得到作为白色固体的10.21g (85%产率)中间体10-3。
ESI-MS: 508 [M-H]
中间体10-4
将1.37g (2.68 mmol)中间体10-3、2.55g (4.03 mmol)中间体10-2和2.28g(10.73 mmol)磷酸钾溶于18 mL甲苯、9 mL二氧杂环己烷和6 mL水中。在使用3个冷冻-抽吸-解冻循环将溶液脱气之后,将12 mg (0.05 mmol)乙酸钯和132 mg (0.32 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至85℃持续16.5小时。将反应物冷却至室温,用甲苯稀释。将有机萃取物用水洗涤,用硫酸钠干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用庚烷和甲苯的混合溶剂作为洗脱液纯化,得到作为白色固体的2.11g (产率93%)中间体7-2。
ESI-MS: 839.8 [M+H]
化合物10
将2.11g (2.51 mmol)中间体10-4溶解在36 mL二氯苯中。然后,向溶液中加入5.14 mL (5.12 mmol) 1.0M三溴化硼的庚烷溶液,接着加入1.8 mL (10.28 mmol) N,N-二异丙基乙胺,并将混合物在180℃搅拌15小时。将反应物冷却至室温,并用甲醇稀释。通过过滤收集沉淀物,用乙醇和水洗涤。将粗产物溶解在二氯甲烷中,用异丙醇沉淀,然后过滤,得到作为黄色固体的1.77g (83%产率)化合物10。
ESI-MS: 849.7 [M+H]
化合物11
中间体11-1
将23.5g (112mmol) 4-溴-2-氯-1-氟苯、20.2g (112mmol) 4- (叔丁基)苯基硼酸悬浮在130 mL甲苯、130 mL乙醇和250 mL的10%碳酸钠水溶液的混合物中。通过鼓泡N
中间体11-2
将260g (1.26 mol) 2,4-二叔丁基苯酚和330g (1.89 mol) 1-溴-2-氟苯加入到5.70L N-甲基吡咯烷酮中,并加入821g (2.52 mol)碳酸铯。将混合物在170℃搅拌90小时。将反应物冷却至室温并向其中加入水。收集有机层。浓缩后,将残余物通过硅胶柱色谱法,使用庚烷作为洗脱剂纯化,得到作为米黄色固体的409g (90%产率)中间体11-2。
产物不经进一步纯化即使用。
中间体11-3
将399g (1.10 mol)中间体11-2溶于1.40L N-甲基吡咯烷酮中,然后加入821g(2.52 mol)碳酸铯。在氩气气氛下,加入17.38g (66.3 mmol)三苯基膦和7.44g (33.1mmol)乙酸钯(II)。将混合物在120℃下搅拌3小时。将反应物冷却至室温并加入水。收集有机层,并用盐水洗涤。浓缩后,将残余物通过硅胶柱色谱法,使用甲苯作为洗脱剂纯化。将主要级分部分浓缩,通过用庚烷置换溶剂沉淀,然后过滤,得到作为白色固体的227g (73%产率)中间体11-3。
ESI-MS: 280 [M+H]
中间体11-4
将118g (421 mmol)中间体11-3溶解在1.20L THF中,并将该溶液在5℃下冷却。在氩气气氛下,在5℃下滴加400ml (620mmol) 1.55M正丁基锂的己烷溶液。在将反应混合物冷却到-60℃后,加入118g (631 mmol) 1,2-二溴乙烷,并将该混合物搅拌17小时。向反应混合物中加入500ml水,将水相用甲苯萃取。收集有机相,用盐水洗涤。浓缩后,将残余物通过硅胶柱色谱法,用甲苯作为洗脱剂纯化。浓缩主要级分。将产物溶于热庚烷中,用冰水浴重结晶,得到作为白色固体的77g (51%产率)中间体11-4。
ESI-MS: 360 [M+H]
中间体11-5
在惰性气氛下,将9.57 mL正丁基锂(1.6M,在己烷中)滴加到5.00g (13.9 mmol)中间体11-4在50 mL四氢呋喃中的溶液中,同时使用丙酮-干冰浴将温度保持低于-60℃。添加完成后,将反应物在-78℃下搅拌15分钟,然后缓慢加入2.20 mL (19.7 mmol)硼酸三甲酯,同时保持温度低于-60℃。添加完成后,将反应物在-78℃下搅拌15分钟,然后缓慢温热至室温,并搅拌17小时,得到乳状溶液。向反应物中加入50 mL10% HCl溶液,搅拌黄色双相混合物1小时。用乙酸乙酯萃取所得混合物,并用水和盐水洗涤有机萃取物,经硫酸镁干燥,并经硅胶短垫过滤。在旋转蒸发仪上除去溶剂,得到作为白色固体的4.25g (60%产率)中间体11-5。
中间体11-6
将7.00g (29.6 mmol) 1-溴-4-氯-2-硝基苯、10.1g (31.1 mmol)中间体11-5悬浮于70 mL甲苯、70 mL乙醇和70 mL10%碳酸钠水溶液的混合物中。通过鼓泡N
中间体11-7
将10.4g (23.9 mmol)中间体11-6和15.8g (59.6 mmol)三苯基膦溶于100 mL1,2-二氯苯中,并加热至回流3小时。然后在减压下蒸馏1,2-二氯苯和三苯基膦,冷却红色油并在搅拌下加入庚烷。将所得的橙色悬浮液在室温下搅拌,然后在0℃下搅拌30分钟,然后过滤。在旋转蒸发仪上从滤液中除去溶剂,并通过硅胶柱色谱法,使用庚烷和甲苯的混合物纯化粗产物,得到灰白色固体。将固体溶解在回流的乙醇中,并通过在室温下加入水而沉淀。过滤所得悬浮液,合并后续产物,得到作为白色固体的8.0g (83%产率)中间体11-7。
ESI-MS: 402.4 [M-H]
中间体11-8
将10.4g (39.6mmol)中间体11-1、8.00g (19.8mmol)中间体11-7和8.41g(39.6mmol)磷酸钾悬浮在80ml N,N-二甲基甲酰胺中并加热至110℃持续5小时。然后将悬浮液冷却至100℃,并缓慢加入水。将所得灰白色悬浮液冷却至室温并过滤。将粗固体在热乙醇/水的9:1混合物中研磨3次,得到作为白色固体的12.5g (96%产率)中间体11-8。
ESI-MS: 646.6 [M+H]
中间体11-9
将4.00g (6.20 mmol)中间体11-8和5.2g (24.8 mmol)磷酸钾溶于120 mL二氧杂环己烷和30 mL水的混合物中,通过鼓泡N
ESI-MS: 889.9 [M+H]
化合物11
在惰性气氛下,将3.50 mL叔丁基锂(1.9M,在己烷中)滴加到1.95g (2.19 mmol)中间体11-9在200 mL叔丁基苯中的溶液中,同时使用丙酮-干冰浴将温度保持低于-50℃。添加完成后,将反应物加热到45℃持续1小时,冷却到-78℃,然后缓慢加入0.35 mL (3.70mmol)三溴化硼,同时保持温度低于-60℃,将反应物温热到室温,加入1.10 mL (6.58mmol) N,N-二异丙基乙胺,并将混合物加热到150℃持续17小时。然后将反应物冷却至室温,用水淬灭并过滤。用甲苯萃取双相滤液,并且用10%碳酸钠水溶液洗涤有机相两次,随后用盐水洗涤。将有机萃取物用硫酸镁干燥,用硅胶垫过滤。在旋转蒸发仪上将亮橙色溶液浓缩至约100 mL,并向其中加入300 mL乙醇。将沉淀物冷却至室温并搅拌17小时,然后过滤。将所得固体通过硅胶色谱法,使用庚烷和二氯甲烷的混合物纯化。将所得树脂溶于200 mL二氯甲烷和200 mL乙醇中,并在旋转蒸发仪上在60℃浓缩直至形成悬浮液。然后趁热过滤,用一些冷乙醇洗涤固体,得到作为亮黄色固体的215 mg (11.4%产率)化合物11。
ESI-MS: 864.0 [M+H]
化合物12
中间体12-1
在80℃和氮气下,向在270 mL乙酸中的30.0g (134 mmol) 4-(溴苯基)肼盐酸盐中滴加20.7g (134 mmol) 4- (叔丁基)环己-1-酮。然后将反应混合物在100℃搅拌5小时。
真空除去溶剂,将反应混合物溶解在甲苯中。用水,然后用碳酸氢钠溶液洗涤有机相。用硫酸镁干燥有机相,并在真空中除去溶剂。产物不经纯化用于下一反应步骤。产量41.0 g。
中间体12-2
在氮气下,在10分钟内向250 mL甲苯中的41.0g (134 mmol) 6-溴-3- (叔丁基)-2,3,4,9-四氢-1H-咔唑中加入60.8g (268 mmol) 2,3-二氯-5,6-二氰基醌。反应是放热的。然后将反应混合物在25℃搅拌1小时。过滤固体并用甲苯洗涤。将有机相用10%氢氧化钠水溶液洗涤。将有机相用水、盐水洗涤,用硫酸镁干燥。真空除去溶剂。使用庚烷/乙酸乙酯95/5的硅胶柱色谱法得到产物。产量21.6g (52%)
中间体12-3
向在300 mL二氧杂环己烷和50 mL水中的17.9g (59 mmol) 6-溴-3- (叔丁基) -2,3,4,9-四氢-1H-咔唑中加入27.1g (107 mmol) 4,4,4',4',5,5,5',5' -八甲基-2,2' -二(1,3,2-二氧杂硼杂环戊烷)和17.4g (178 mmol)乙酸钾。将反应混合物用氩气脱气。加入542 mg (0.592 mmol)三(二亚苄基丙酮)二钯(0)和564 mg (1.18 mmol) 2-二环己基膦基-2',4',6' -三异丙基联苯(XPhos)。将反应混合物用氩气脱气。将反应混合物在110℃下在氩气下搅拌8小时。过滤固体并除去水相。在真空中除去溶剂。使用庚烷/乙酸乙酯90/10的硅胶柱色谱法得到产物。产量11.7g (55%)
中间体12-4
向120 mL二甲苯、70 mL二氧杂环己烷和50 mL水中的11.7g (33.4 mmol) 3- (叔丁基) -6- (4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基) -9H-咔唑中加入9.81g(36.8 mmol) 2-氯-4,6-二苯基嘧啶和11.6g (84.0 mmol)碳酸钾。将反应混合物用氩气脱气。加入1.16g (1.00 mmol)四(三苯基膦)钯(0)。将反应混合物用氩气脱气。在110℃和氩气下搅拌反应混合物8小时。除去水相。真空除去溶剂。使用庚烷/乙酸乙酯95/5和庚烷/乙酸乙酯90/10的硅胶柱色谱法得到产物。产量6.75g (44%)
ESI-MS: 454 [M+1]
中间体12-5
向在60 mL乙酸中的6.75g (14.9 mmol) 3- (叔丁基) -6- (4,6-二苯基嘧啶-2-基) -9H-咔唑中加入2.65g (14.9 mmol) N-溴琥珀酰亚胺,并在20℃和氮气下搅拌反应混合物。2.5小时后,将产物过滤,用乙酸,然后用甲醇洗涤。使用庚烷/乙酸乙酯97/3的硅胶柱色谱法得到产物。产量4.00g (39%)。将产物从甲苯中结晶。
中间体12-6
向在30 ml甲苯、15 ml二氧杂环己烷和10 ml水中的1.52g (2.85 mmol) 1-溴-3-(叔丁基) -6- (4,6-二苯基嘧啶-2-基) -9H-咔唑中加入1.61g (3.00 mmol) 3,6-二叔丁基-9- (3- (叔丁基) -5- (4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)苯基) -9H-咔唑和1.82g (8.56 mmol)磷酸三钾。将反应混合物用氩气脱气。加入94 mg (0.23 mmol)二环己基(2 ',6 ' -二甲氧基[1,1 ' -联苯] -2-基)膦SPhos和26 mg (0.114 mmol)乙酸钯(II)。将反应混合物用氩气脱气。在70℃和氩气下搅拌反应混合物1小时,过滤固体并用庚烷洗涤。将有机相用硫酸镁干燥,真空除去溶剂。使用庚烷/乙酸乙酯95/5的硅胶柱色谱法得到产物。产量2.19g (88%)。
化合物12
在氩气下,向26 mL邻二氯苯中的1.98g (2.29 mmol) 3- (叔丁基) -1- (3- (叔丁基) -5- (3,6-二叔丁基-9H-咔唑-9-基)苯基) -6- (4,6-二苯基嘧啶-2-基) -9H-咔唑中加入1.19g (9.18 mmol) N-乙基-N-异丙基丙-2-胺。在氩气下,在5分钟内向反应混合物中加入4.59 mL (4.50 mmol) 1M的三溴硼烷的庚烷溶液。在氩气下在185℃搅拌反应混合物2.5小时。将反应混合物冷却至25℃并加入甲醇。将产物过滤并用甲醇洗涤。使用二氯甲烷100%的硅胶柱色谱法得到产物。产量1.71g (77%)。
ESI-MS: 871.8 [M+1]
化合物13
中间体13-1
将16.62g (47.70 mmol) (2-溴-4-碘苯基)肼盐酸盐和7.36g (47.70 mmol) 4-(叔丁基)环己酮加入到95 mL乙酸中,将混合物在100℃下搅拌2小时,将反应混合物冷却至室温后,通过过滤收集固体,并用乙酸乙酯洗涤。浓缩滤液,然后通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合溶剂作为洗脱剂纯化残余物,得到作为白色固体的11.2g (54%产率)中间体13-1。
ESI-MS: 433 [M+H]
中间体13-2
将7.03g (16.27 mmol)中间体13-1和7.39g (32.50 mmol) 2,3-二氯-5,6-二氰基醌加入60 mL甲苯中,将混合物在100℃搅拌2.5小时。将反应混合物冷却至室温后,通过过滤除去固体,并用甲苯洗涤。浓缩滤液,然后通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合溶剂作为洗脱剂纯化残余物,得到作为米黄色粉末的4.73g (68%产率)中间体13-2。
ESI-MS: 427 [M+H]
中间体13-3
将4.28g (10.00 mmol)中间体13-2、1.78g (10.00 mmol) 4-叔丁基苯硼酸和2.76g (19.99 mmol)碳酸钾溶解在50 mL甲苯、10 mL乙醇和10 mL水中。在使用3个冷冻-抽吸-解冻循环使溶液脱气之后,将578mg(0.50mmol)四(三苯基膦)钯添加至混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至70℃持续20小时。将反应物冷却至室温并用甲苯稀释。将有机萃取物用水洗涤,用硫酸钠干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用庚烷和甲苯的混合溶剂作为洗脱液纯化,得到作为白色固体的3.56g (产率82%)中间体13-3。
ESI-MS: 433 [M-H]
中间体13-4
将3.40g (5.30 mmol)中间体8-1、1.38g (5.45 mmol)双(频哪醇合)二硼和1.04g(10.60 mmol)乙酸钠悬浮在27 mL甲苯中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将120 mg (0.13 mmol)三(二亚苄基丙酮)二钯(0)和152 mg (0.51 mmol) 2-二环己基膦基-2 ',4 ',6 ' -三异丙基联苯加入到混合物中。在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物加热至110℃持续6小时。将反应物冷却至室温,用甲苯和水稀释。将水层用甲苯萃取,将有机层用盐水洗涤,用硫酸镁干燥,过滤,浓缩溶液。将粗产物用二氯甲烷和乙腈重结晶,得到作为白色固体的2.74g (75%产率)中间体13-4。
ESI-MS: 690 [M+H]。
中间体13-5
将1.39g (3.22 mmol)中间体13-3、3.06g (4.84 mmol)中间体10-2和2.73g(12.8 mmol)磷酸钾溶于21 mL甲苯、11 mL二氧杂环己烷和7 mL水中。在使用3个冷冻-抽吸-解冻循环将溶液脱气之后,将15 mg (0.06 mmol)乙酸钯和158 mg (0.38 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至85℃持续16.5小时。将反应物冷却至室温,用甲苯稀释。将有机萃取物用水洗涤,用硫酸钠干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用庚烷和甲苯的混合溶剂作为洗脱液纯化,得到作为白色固体的2.15g (79%产率)中间体13-5。
ESI-MS: 918 [M+H]
化合物13
将1.83g (2.00 mmol)中间体13-5溶解在28 mL二氯苯中。然后,向溶液中加入4.10 mL (4.10 mmol) 1.0M三溴化硼的庚烷溶液,接着加入1.4 mL (8.19 mmol) N,N-二异丙基乙胺,将混合物在180℃搅拌15小时。将反应物冷却至室温,并用甲醇稀释。通过过滤收集沉淀物,用乙醇和水洗涤。将粗产物溶解在二氯甲烷中,用异丙醇沉淀,然后过滤,得到作为黄色固体的1.39g (75%产率)化合物13。
ESI-MS: 925 [M+H]
化合物14
中间体14-1
将40.0g (178 mmol) 6-溴-3,4-二氢萘-2(1H) -酮、38.0g (213 mmol)(3- (叔丁基)苯基)硼酸和38.6g (364 mmol)碳酸钠溶于523 mL甲苯、261 mL乙醇和105 mL水中。在使用3个冷冻-抽吸-解冻循环使溶液脱气之后,将3.08g (2.67 mmol)四(三苯基膦)钯加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至80℃持续1.5小时。将反应物冷却至室温,加入溶解在50 mL水中的5g氰化钠后,将反应混合物搅拌30分钟。将有机萃取物用水洗涤,经硫酸钠干燥,过滤,将溶液浓缩,得到作为白色固体的21.0g (42%产率)中间体14-1。其不经进一步纯化而用于下一反应。
中间体14-2
将27.9g (71.8 mmol) (2-溴-4-碘苯基)肼盐酸盐和20.0g (71.8 mmol)中间体14-1加入198 mL的4N HCl的二氧杂环己烷溶液中,将混合物在110℃搅拌3小时。将反应混合物冷却到室温后,将反应混合物倒入500 mL水中。加入600 mL二氯甲烷后,用二氯甲烷萃取水层,用硫酸钠干燥收集的有机层。过滤后,浓缩溶液,得到作为白色固体的18.8g (47%产率)中间体14-2。
ESI-MS: 556.2 [M-H]
中间体14-3
将18.0g (32.4 mmol)中间体14-2和8.75g (35.6 mmol) 2,3-二氯-5,6-二氰基醌加入180 mL邻二甲苯中,并将该混合物在130℃下搅拌2.5小时。将反应混合物冷却至室温后,将反应混合物悬浮在200mL庚烷中。过滤悬浮液,用庚烷洗涤,浓缩滤液。将粗产物在回流下溶解于甲苯中。在将溶液在室温下冷却后,通过过滤收集形成的固体,用庚烷洗涤,得到作为浅灰色固体的11.25g (63%产率)的中间体14-3。
ESI-MS: 552.2 [M-H]
中间体14-4
将11.0g (19.85 mmol)中间体14-3、3.53g (19.85 mmol)(4- (叔丁基)苯基)硼酸和4.63g (43.7 mmol)碳酸钠溶解在120 mL甲苯、120 mL乙醇和40 mL水中。在使用3个冷冻-抽吸-解冻循环将溶液脱气后,将688 mg (0.60 mmol)四(三苯基膦)钯加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至80℃持续4小时。将反应物冷却至室温,加入溶解在50 mL水中的1g氰化钠后,将反应混合物搅拌30分钟。将有机萃取物用水洗涤,用硫酸钠干燥,过滤,将溶液浓缩。将粗产物通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合溶剂作为洗脱剂纯化,得到作为米黄色固体的7.33g (65%产率)中间体14-4。
ESI-MS: 560.5 [M-H]
中间体14-5
将1.50g (2.68 mmol)中间体14-4、2.15g (4.01 mmol)中间体10-2和2.28g(10.70 mmol)磷酸钾溶于22 mL甲苯、11 mL二氧杂环己烷和7 mL水中。在使用3个冷冻-抽吸-解冻循环将溶液脱气之后,将21 mg (0.09 mmol)乙酸钯和231 mg (0.56 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至85℃持续21小时。将反应物冷却至室温,用甲苯稀释。将有机萃取物用水洗涤,用硫酸钠干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用庚烷和甲苯的混合溶剂作为洗脱液纯化,得到作为白色固体的1.72g (产率72%)中间体14-5。
ESI-MS: 891 [M-H]
化合物14
将1.78g (2.00 mmol)中间体14-5溶解在28 mL二氯苯中。然后,向溶液中加入4.10 mL (4.10 mmol) 1.0M三溴化硼的庚烷溶液,接着加入1.4 mL (8.19 mmol) N,N-二异丙基乙胺,将混合物在180℃搅拌15小时。将反应物冷却至室温,并用甲醇稀释。通过过滤收集沉淀物,用乙醇和水洗涤。将粗产物溶解在二氯甲烷中,用异丙醇沉淀,然后过滤,得到作为黄色固体的1.41g (83%产率)化合物14。
ESI-MS: 900 [M+H]
化合物15
中间体15-1
向26.4g (78.0 mmol)中间体7-1在250 mL1,4-二氧杂环己烷中的溶液中加入15.35g (65.0 mmol) 3,6-二氯-9H-咔唑、41.4g (195.0 mmol)磷酸钾、1.55g (8.13mmol)碘化铜、2.73 mL (22.75 mmol)环己-1,2-二胺。用Ar将悬浮液脱气,然后加热至85℃持续1.5小时。冷却至室温后,将悬浮液用硅藻土过滤,用温甲苯(4×100 mL)洗涤。蒸发滤液,并通过硅胶柱色谱法,使用庚烷作为洗脱剂纯化所得残余物。所得白色固体进一步从环己烷(2×150 mL)中重结晶,得到作为白色固体的14.66g (80%产率)中间体15-1。
中间体15-3
将12.30g (27.5 mmol)中间体15-1、11.15g (27.5 mmol)中间体2-3、2.20g(55.0 mmol)氢氧化钠悬浮于四氢呋喃/水(120/60 mL)的混合物中。用Ar将悬浮液脱气,并将477 mg (1.5mol%)四(三苯基膦)钯(0)加入到反应混合物中。将反应混合物回流1小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为白色泡沫的17.76g (100%产率)的中间体15-2。
ESI-MS: 643.4[M-H]
中间体15-3
将17.43g (27.0 mmol)中间体15-2溶解在225 ml1,2-二氯苯中,并用Ar脱气。将18.49 mL (108 mmol) N-乙基-N-异丙基丙-2-胺加入到反应混合物中,随后缓慢加入54.0mL (54.0 mmol)三溴硼烷(1M庚烷溶液)。将反应混合物加热至180℃持续5小时。冷却至室温后,过滤反应中形成的沉淀物,并用1,2-二氯苯、甲醇和庚烷冲洗,得到作为黄色固体的13.76g (78%产率)中间体15-3。通过LC-MS [M + H] 653.3证实产物的分子量。
ESI-MS: 653.3[M+H]
化合物15
将2.35g (3.6 mmol)中间体15-3、2.80g (14.4 mmol) (4- (三甲基甲硅烷基)苯基)硼酸、4.69g (14.4 mmol)碳酸铯悬浮在甲苯/乙醇/水(36/12/6mL)混合物中。用Ar将该悬浮液脱气,并将40 mg (5mol%)乙酸钯和148 mg (10mol%) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到该反应混合物中。将反应混合物加热至80℃持续1.5小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为黄色固体的2.98g (94%产率)化合物15。
ESI-MS: 881.5[M+H]
化合物16
将2.61g (4.0 mmol)中间体15-3、2.62g (16.0mmol) (2-异丙基苯基)硼酸、5.21g (16.0mmol)碳酸铯悬浮在甲苯/乙醇/水(36/12/6mL)的混合物中。用Ar对悬浮液进行脱气,并将45 mg (5mol%)乙酸钯和164mg (10mol%) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到反应混合物中。将反应混合物加热至80℃持续5小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱,使用庚烷/甲苯作为洗脱剂纯化,得到作为黄色固体的3.15g (96%产率)化合物16。
ESI-MS: 821.5 [M+H]
化合物17
中间体17-1
将40.0g (0.15 mol) 6-溴-1,1,4,4-四甲基-1,2,3,4-四氢化萘悬浮于150 ml乙酸酐中。在室温下经三小时滴加78 mL (1.12 mol)硝酸。搅拌黄色悬浮液30分钟,然后用1.5L水处理,并再搅拌一小时。过滤悬浮液,用500 mL水洗涤固体。将固体悬浮在400 mL10%碳酸钠水溶液中。过滤悬浮液,用500 mL水洗涤固体。将固体进一步悬浮于150 mL乙醇中,然后过滤,并用50 mL乙醇洗涤固体,得到作为白色固体的43.2g (93%产率)中间体17-1。
中间体17-2
在-78℃下,在15分钟内用25.9 mL正丁基锂(2.5M,在己烷中)滴加处理200 mL四氢呋喃中的20.0g (61.7 mmol) 2-溴-N,N-二苯基苯胺。在-78℃加入32.5 mL氯化锌溶液(1.9M,在2-甲基四氢呋喃中),在45分钟内将黄色溶液温热至室温。加入18.3g (58.6mmol)中间体17-2、565 mg (0.62 mmol)三(二亚苄基丙酮)二钯(0)和358 mg (1.23 mmol)四氟硼酸三-叔丁基鏻,并将所得溶液在55℃下加热15分钟。将反应混合物冷却至室温,并通过3 cm硅胶层过滤,随后用50 ml四氢呋喃冲洗硅胶层。真空浓缩滤液,将所得固体溶于100 ml热乙醇中。将溶液冷却至室温,直到形成悬浮液。过滤悬浮液,用80 ml乙醇洗涤固体。通过MPLC,使用CombiFlash Companion(硅胶,庚烷/0-40%梯度的甲苯)纯化产物,得到作为白色固体的20.3g (73%产率)中间体17-2。
ESI-MS (阳性,m/z):C
中间体17-3
将20.0g (42.0 mmol)中间体17-2和33.0g (126 mmol)三苯基膦在100 mL1,2-二氯苯中在174℃加热三小时。将反应混合物在真空下浓缩。将产物在100 mL庚烷中搅拌一小时。过滤悬浮液,将固体用庚烷洗涤。将滤液真空浓缩,将固体溶于二氯甲烷,然后通过4 cm硅胶层过滤,接着用150 mL二氯甲烷冲洗硅胶层。将合并的洗脱液在真空下浓缩,将产物通过MPLC,使用CombiFlash Companion (硅胶,庚烷/二氯甲烷)纯化。将产物溶解在30 mL二氯甲烷中,用50 mL庚烷稀释。将溶液真空浓缩至50 mL体积,直到形成悬浮液。将悬浮液过滤,并用庚烷洗涤固体。将固体悬浮于70 mL叔丁基甲基醚中。过滤悬浮液,用叔丁基甲基醚洗涤固体。将来自叔丁基甲基醚洗涤的合并的滤液在真空下浓缩,得到作为固体的6.9g(37%产率)中间体17-3。
ESI-MS (阳性,m/z):C
中间体17-4
将1.53g (4.50 mmol)中间体7-1、2.00g (4.50 mmol)中间体17-3、86 mg (0.45mmol)碘化铜(I)、154 mg (1.35 mmol)环己-1,2-二胺和2.86g (13.5 mmol)磷酸三钾悬浮于50 mL1,4-二氧杂环己烷中,在91℃加热12小时。将悬浮液冷却至室温,并通过3 cm硅胶层过滤,接着用50 mL二氧杂环己烷冲洗硅胶层。真空浓缩洗脱液,将所得固体溶于30 mL二氯甲烷和50 mL乙醇中。将溶液真空浓缩至40 mL体积。过滤悬浮液,用乙醇洗涤固体,得到作为白色固体的2.56g (87%产率)中间体17-4。
ESI-MS (阳性,m/z):C
中间体17-5
将2.50g (3.81 mmol)中间体17-4、1.70g (4.19 mmol)中间体2-3、17 mg (0.08mmol)乙酸钯(II)、188 mg (0.46 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和3.24g (15.3 mmol)磷酸三钾溶于40 mL甲苯、20 mL1,4-二氧杂环己烷和10 mL水的混合物中。将溶液抽真空三次并用氩气回填,并在82℃下加热90分钟。用50 mL甲苯和100 mL水稀释反应混合物。分离有机相,用水(3×50 mL)洗涤,用硫酸钠干燥,并在3 cm硅胶层上过滤。用甲苯冲洗硅胶层,并在真空下浓缩合并的洗脱液。将产物通过MPLC,使用
ESI-MS (阳性,m/z):C
化合物17
将2.30g (2.69 mmol)中间体17-5溶解在46 mL1,2-二氯苯中。滴加1.9 mL (10.8mmol) N,N-二异丙基乙胺和5.4 mL三溴硼烷(1.0M,在庚烷中)。将该褐色溶液在174℃下加热90分钟,并冷却至36℃。滴加5.4 mL三溴硼烷(1.0M,在庚烷中),并在174℃下继续加热90分钟。将反应混合物冷却至室温,加入100 mL甲醇。真空浓缩混合物,将残余物溶解在100mL庚烷和100 mL水中。将有机相用水(3×50 mL)洗涤,用硫酸钠干燥,然后过滤并真空浓缩。将得到的固体溶解在20 mL二氯甲烷和60 mL乙醇中。将溶液在真空下浓缩至50 ml的体积,直到形成悬浮液。过滤悬浮液,将固体用乙醇洗涤。将产物通过MPLC,使用
ESI-MS (阳性,m/z):C
化合物18
将163 mg (0.249 mmol)中间体15-3、0.152 mg (1.0 mmol) (4-甲氧基苯基)硼酸、0.325 mg (1.0 mmol)碳酸铯悬浮于甲苯/乙醇/水(6/2/1mL)的混合物中。用Ar将悬浮液脱气,并将3.4 mg (6mol%)乙酸钯和12.3 mg (12mol%) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到反应混合物中。将反应混合物加热至80℃持续2小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/二氯甲烷作为洗脱剂纯化,得到作为黄色固体的123 mg (62%产率)化合物18。
ESI-MS: 797.5 [M+H]
化合物19
将200 mg (0.306 mmol)中间体15-3、0.171 mg (1.22 mmol) (4-氟苯基)硼酸、0.399mg (1.22 mmol)碳酸铯悬浮于甲苯/乙醇/水(6/2/1mL)的混合物中。用Ar将悬浮液脱气,并将4.1 mg (6mol%)乙酸钯和15.1 mg (12mol%) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到反应混合物中。将反应混合物加热至80℃持续24小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为黄色固体的166 mg (70%产率)化合物19。
ESI-MS: 773.4 [M+H]
化合物20
中间体20-1
将10.0g (37.0 mmol) 1,3-二溴-5-氯苯、21.3g (76.0 mmol)双(4- (叔丁基)苯基)胺、703 mg (0.77 mmol)三(二亚苄基丙酮)二钯(0)、892 mg (3.07 mmol)四氟硼酸三叔丁基鏻和8.89g (92.0 mmol)叔丁醇钠悬浮于200 mL甲苯中。将悬浮液抽真空三次,并用氩气回填,并在72℃下加热90分钟。将深色悬浮液冷却至室温,用水(2×100 mL)洗涤。将有机相用硫酸钠干燥并真空浓缩。将固体从300 mL乙醇中重结晶,然后用冷乙醇洗涤,得到作为白色固体的18.8g (76%产率)中间体20-1。
ESI-MS (阳性,m/z):C
中间体20-2
将8.00g (11.9 mmol)中间体20-1、4.50g (13.1 mmol)中间体5-1、54 mg (0.24mmol)乙酸钯(II)、587 mg (1.43 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和10.1g (47.7 mmol)磷酸三钾溶解在100 mL邻二甲苯、50 mL1,4-二氧杂环己烷和30 mL水的混合物中。将反应混合物抽真空三次并用氩气回填,并在82℃下加热五小时。将反应混合物冷却至室温,用200 mL甲苯和100 mL水稀释。将有机相用水(3×100 mL)洗涤,用硫酸钠干燥,然后加入二氯甲烷。过滤混合物,真空浓缩滤液。将产物在30 mL二氯甲烷和150 mL乙醇中搅拌直至形成悬浮液。过滤悬浮液,并用100 mL乙醇和100 mL庚烷洗涤固体,得到作为白色固体的6.9g (68%产率)中间体20-2。
ESI-MS (阴性,m/z):C
化合物20
将3.00g (3.52 mmol)中间体20-2悬浮于50 mL1,2-二氯苯中。滴加2.5 mL (14mmol) N,N-二异丙基乙胺和7 mL三溴硼烷(1.0M,在庚烷中)。将黄色悬浮液在181℃下加热4小时。冷却反应混合物,加入100 mL甲醇。将悬浮液搅拌15分钟,然后过滤。将悬浮液搅拌15分钟,然后过滤。用50 mL甲醇,然后用30 mL水,接着用50 mL甲醇和30 mL庚烷洗涤固体。将固体进一步通过MPLC,使用
ESI-MS (阳性,m/z):C
化合物21
中间体21-1
将5.00g (10.8 mmol)中间体1-3、4.07g (11.9 mmol)中间体5-1、48 mg (0.22mmol)乙酸钯(II)、531 mg (1.29 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和9.15g (43.1 mmol)磷酸三钾溶于100 mL邻二甲苯、50 mL1,4-二氧杂环己烷和30 mL水的混合物中。将乳液抽真空三次并用氩气回填,并在86℃下加热26小时。冷却反应混合物,加入50 mL甲苯和50 mL水。将有机相用水(3×50 mL)洗涤,然后用硫酸钠干燥,并真空浓缩。将固体产物悬浮于100 mL庚烷中,然后过滤,并用庚烷洗涤固体。将产物通过MPLC,使用
ESI-MS (阴性,m/z):C
化合物21
将3.50g (5.43 mmol)中间体21-1溶解在200 mL1,2-二氯苯中。滴加3.8 mL(21.7 mmol) N,N-二异丙基乙胺和8.1 mL三溴硼烷(1.0M,在庚烷中)。在142℃下加热悬浮液18小时。冷却反应混合物,加入300 mL甲醇。过滤悬浮液,用100 mL甲醇、50 mL水和50 mL甲醇洗涤固体。将产物通过MPLC,使用
ESI-MS (阳性,m/z):C
化合物22
中间体22-1
将7.18g (25.50 mmol)双(4- (叔丁基)苯基)胺、10.67g (25.50 mmol)中间体7-1和3.43g (35.70 mmol)叔丁醇钠悬浮于102 mL甲苯中。在使用3个冷冻-抽吸-解冻循环使悬浮液脱气之后,将295 mg (0.51 mmol) xantphos和117 mg (0.13 mmol)三(二亚苄基丙酮)二钯(0)加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物搅拌至100℃持续14.5小时。将反应物冷却至室温,用甲苯和水稀释。将水层用甲苯萃取。将有机萃取物用盐水洗涤,用硫酸钠干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用环己烷作为洗脱剂纯化,得到作为米黄色泡沫的10.8g (79%产率)中间体22-1。
ESI-MS: 494.6 [M-H]
中间体22-2
将2.96g (6.01 mmol)中间体22-1、1.98g (7.81 mmol)双(频哪醇合)二硼和1.18g (12.02 mmol)乙酸钠悬浮在30 mL甲苯中。使用3个冷冻-抽吸-解冻循环使悬浮液脱气,并将110 mg (0.12 mmol)三(二亚苄基丙酮)二钯(0)和229 mg (0.48 mmol) 2-二环己基膦基-2 ',4 ',6 ' -三异丙基联苯加入到混合物中。在两个另外的冷冻-抽吸-解冻循环之后,将反应混合物加热至110℃持续16小时。将反应物冷却至室温,并用甲苯和水稀释。将水层用甲苯萃取,将有机层用盐水洗涤,用硫酸镁干燥,过滤,浓缩溶液。将粗产物用二氯甲烷和乙腈重结晶,得到作为白色固体的2.56g (79%产率)中间体22-2。
ESI-MS: 540.7 [M+H]
中间体22-3
将1.50g (2.68 mmol)中间体14-4、2.16g (4.01 mmol)中间体22-2和2.28g(10.70 mmol)磷酸钾溶于22 mL甲苯、11 mL二氧杂环己烷和7 mL水中。在使用3个冷冻-抽吸-解冻循环将溶液脱气之后,将21 mg (0.09 mmol)乙酸钯和231 mg (0.56 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯加入到混合物中。然后,在两个另外的冷冻-抽吸-解冻循环之后,将混合物搅拌至85℃持续21小时。将反应物冷却至室温,用甲苯稀释。将有机萃取物用水洗涤,用硫酸钠干燥,过滤,并将溶液浓缩。将残余物通过硅胶柱色谱法,使用庚烷和甲苯的混合溶剂作为洗脱液纯化,得到作为白色固体的2.87g (产率80%)的中间体22-3。
ESI-MS: 893.8 [M-H]
化合物22
将2.87g (3.21 mmol)中间体22-3溶解在46 mL二氯苯中。然后,向溶液中加入6.59 mL (6.59 mmol) 1.0M三溴化硼的庚烷溶液,接着加入2.3 mL (10.28 mmol) N,N-二异丙基乙胺,并将混合物在180℃搅拌20小时。将反应物冷却至室温,并用甲醇稀释。通过过滤收集沉淀物,用乙醇和水洗涤。将粗产物通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合溶剂作为洗脱剂纯化。将产物溶解在二氯甲烷中,加入乙腈沉淀,然后过滤,得到2.19g (产率76%)化合物22。
ESI-MS: 901.2 [M+H]
化合物23
中间体23-1
将27.2g (86.0 mmol) 2-溴-4-氯-1-碘苯、20.0g (82.0 mmol) N-苯基-2-联苯胺和11.0g (114 mmol)叔丁醇钠加入到250 mL甲苯中。通过N
中间体23-2
在惰性气氛下,将35 mL正丁基锂(2.5M,在己烷中)滴加到34.0g (78.2mmol)中间体23-1在360 mL四氢呋喃中的溶液中,同时使用丙酮-干冰浴将温度保持低于-60℃。添加完成后,将反应物在-78℃下搅拌1.5小时。然后缓慢加入30 mL (268 mmol)硼酸三甲酯,同时保持温度低于-60℃。在添加完成后,将反应物在-78℃下搅拌15分钟,然后缓慢温热至室温,并搅拌20分钟,得到乳状溶液。将400 ml10% HCl溶液加入到反应物中,并将两相混合物搅拌1小时。在旋转蒸发仪上除去有机溶剂,并过滤所得悬浮液。将固体在500mL庚烷中在回流下研磨1小时。然后在旋转蒸发仪上将白色悬浮液浓缩至一半体积,并在0℃搅拌1小时,然后过滤,得到作为白色固体的22.3g (71.3%产率)中间体23-2。
ESI-MS: 400.2 [M+H]
中间体23-3
将4.88g (18.9 mmol) 2-溴-4- (叔丁基) -1-硝基苯、6.3g (15.8 mmol)中间体23-2和1.87g (81.3 mmol)氢氧化钠溶于75 mL二氧杂环己烷和30 mL水的混合物中,通过鼓泡N
ESI-MS: 533.3 [M+H]
中间体23-4
将16.2g (30.5 mmol)中间体23-3和40.0g (152 mmol)三苯基膦溶于160 mL1,2-二氯苯中,并加热回流11小时。然后在减压下蒸馏1,2-二氯苯和大部分三苯基膦,并将剩余的黑焦油冷却至室温。然后将残余物溶于回流的庚烷中,加入15g Hyflo® Super-Cel®,然后加入5g活性炭。然后将悬浮液在Hyflo® Super-Cel®垫上热过滤,用庚烷洗涤该垫,将合并的滤液在二氧化硅垫上过滤。用庚烷洗涤该垫,并弃去无色滤液。然后用甲苯洗脱产物,得到橙色滤液。在旋转蒸发仪上从滤液中除去溶剂,并将粗产物通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合物纯化两次。将所得树脂溶于庚烷和二氯甲烷的混合物中,并在旋转蒸发仪上除去二氯甲烷。将所得溶液冷却至0℃,在此期间形成沉淀物。搅拌2小时后,过滤悬浮液,得到作为白色固体的4.53g (30%产率)中间体23-4。
ESI-MS: 499.4 [M-H]
中间体23-5
将2.04g (3.77 mmol)中间体22-2、1.8g (3.59 mmol)中间体23-4和1.91g (8.98mmol)磷酸钾悬浮在35 mL甲苯、23mL二氧杂环己烷和12mL水中,通过鼓泡N
ESI-MS: 878.7 [M+H]
化合物 23
在惰性气氛下,将0.75 mL正丁基锂(2.5M,在己烷中)滴加到1.50g (1.71 mmol)中间体23-5在70 mL叔丁基苯中的溶液中,同时使用冰/氯化钠浴保持温度低于-15℃。添加完成后,将反应物加热至室温持续20分钟,然后冷却至-15℃,然后缓慢加入3.5 mL三溴化硼(1M,在己烷中),同时保持温度低于-10℃。将反应物温热至120℃持续5小时。然后将反应物冷却至室温,并用100 mL10%碳酸氢钠水溶液淬灭。将有机相用水洗涤两次,用硫酸钠干燥,用硅胶垫过滤。用甲苯洗涤该垫,并在旋转蒸发仪上浓缩滤液以除去甲苯。将黄色溶液冷却至0℃,加入300 mL乙腈。在2小时内缓慢形成沉淀物,滤出所得固体。将母液在旋转蒸发仪上浓缩至油状物并溶解在二氯甲烷中。加入70 mL乙腈,并将溶液在旋转蒸发仪上浓缩至约40 mL。将溶液冷却至室温,用来自先前沉淀的晶体作为晶种,并搅拌1小时。然后过滤得到的沉淀物,并将合并的固体通过硅胶柱色谱法,使用庚烷和二氯甲烷的混合物作为洗脱剂纯化两次。将纯化的产物溶解在50 mL二氯甲烷和75 mL乙腈中,浓缩溶液直至形成沉淀物。将该悬浮液在室温下搅拌30分钟,过滤,得到作为亮黄色固体的870 mg (58%产率)化合物23。
ESI-MS: 886.7 [M+H]
化合物24
中间体24-1
将4.07g (12.0 mmol)中间体7-1、4.13g (10.2 mmol)中间体2-3、0.96g (24.0mmol)氢氧化钠悬浮于四氢呋喃/水(54/27 mL)的混合物中。用Ar将该悬浮液脱气,并向反应混合物中加入277 mg (2mol%)四(三苯基膦)钯(0)。将反应混合物回流1.5小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,经硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为白色泡沫的4.30g (73%产率)中间体24-1。
ESI-MS:490.2 [M-H]
中间体24-2
将1.74g (4.97 mmol) 6-溴-2,3-二苯基苯并呋喃、1.00g (4.87 mmol) 3,5-二叔丁基苯胺、1.17g (12.17 mmol)叔丁醇钠悬浮于24 mL甲苯中。用Ar将悬浮液脱气,并将166 mg (6mol%) 4,5-双(二苯基膦基) -9,9-二甲基呫吨和134 mg (3mol%)三(二亚苄基丙酮)二钯(0)加入到反应混合物中。将反应混合物加热至90℃持续45分钟。将反应物冷却至室温,用甲苯/水稀释,并经硅藻土过滤。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为白色固体的1.4g (61%产率)中间体24-2。
ESI-MS [M+H] 474.4。
中间体24-3
将1.35g (2.75 mmol)中间体24-1、1.30g (2.75 mmol)中间体24-2、661 mg(6.88mmol)叔丁醇钠悬浮于35 mL甲苯中。用Ar将悬浮液脱气,并向反应混合物中加入127mg (8mol%) 4,5-双(二苯基膦基) -9,9-二甲基呫吨和101 mg (4mol%)三(二亚苄基丙酮)二钯(0)。将反应混合物加热至90℃持续2.5小时。将反应物冷却至室温,用甲苯/水稀释,并经硅藻土过滤。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为米黄色泡沫的2.28g (94%产率)中间体24-3。
ESI-MS: 883.7 [M+H]
化合物 24
将1.86g (2.1 mmol)中间体24-3溶解在40 mL叔丁基苯中,用Ar脱气并冷却到0℃。滴加4.07 mL (6.51 mmol)叔丁基锂(1.6M戊烷溶液),然后在相同温度下搅拌5分钟。然后,将反应物在室温下搅拌2小时。然后,滴加4.20 mL (4.20 mmol)三溴硼烷(1M庚烷溶液),搅拌反应物5分钟,然后加入1.44 mL (8.40 mmol) N-乙基-N-异丙基丙-2-胺。将反应混合物在室温下搅拌3小时,然后用水/甲苯淬灭。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,用庚烷/甲苯作为洗脱剂纯化,得到作为黄色固体的1.25g (66%产率)化合物24。
ESI-MS [M+H] 891.6。
化合物25
中间体25-1
将20.0g (0.11 mol) 2,5-二氯苯-1,4-二胺、48.2g (0.23 mol) 1-溴-4- (叔丁基)苯、517 mg (0.57 mmol)三(二亚苄基丙酮)二钯(0)、1.06g (0.6 mmol) 2,2' -双(二苯基膦基) -1,1' -联萘(BINAP)和32.6g (0.34 mol)叔丁醇钠悬浮于400 mL邻二甲苯中。将悬浮液在126℃下加热四小时。将反应混合物冷却至室温,加入100 ml的5%氰化钠水溶液。将混合物剧烈搅拌30分钟,然后过滤。用300 mL乙酸乙酯洗涤剩余的固体。将收集的滤液用水(3×100 mL)洗涤,用硫酸镁干燥并真空浓缩。将所得固体悬浮于400 mL乙醇中,搅拌悬浮液一小时。将悬浮液冷却,然后过滤,并用冷乙醇洗涤固体,得到作为白色固体的31.6g (63%产率)中间体25-1。
ESI-MS (阳性,m/z):C
中间体25-2
将30.0g (68.0 mmol)中间体25-1、517 mg (0.57 mmol)乙酸钯(II)、789 mg(0.6 mmol)四氟硼酸三叔丁基鏻、5.2g (51 mmol)新戊酸和47g (0.34 mol)碳酸钾悬浮于300 mL N,N-二甲基乙酰胺中。将悬浮液在152℃下加热八小时。将反应混合物冷却至室温并倒入1000 mL水中。将悬浮液搅拌一小时,然后过滤,用400 mL水洗涤固体。将该固体溶解在300 mL二氯甲烷中,并通过3 cm硅胶层过滤,然后用600 mL二氯甲烷和1000 mL乙酸乙酯冲洗硅胶层。将收集的洗脱液真空浓缩至100 mL体积,加入200 mL庚烷。搅拌混合物直至形成悬浮液。过滤悬浮液,用庚烷洗涤白色固体,得到25.0g (定量产率)中间体25-2。
ESI-MS (阳性,m/z):C
中间体25-3
将25.0g (67.8 mmol)中间体25-2和32.6g (0.15 mol)二碳酸二叔丁酯溶解在700 mL四氢呋喃中。加入1.82g (14.9 mmol) 4- (二甲基氨基)吡啶,并在室温下搅拌悬浮液两小时。过滤悬浮液,将固体用100 mL四氢呋喃和200 mL乙酸乙酯洗涤,得到作为白色固体的29.4g (76%产率)中间体25-3。
中间体25-4
将29.0g (51.0 mmol)中间体25-3悬浮于600 mL叔丁基苯中,在164℃加热三小时。将溶液冷却并在室温下搅拌18小时。过滤所得悬浮液。将滤液在真空下浓缩,得到作为白色固体的10.2g (43%产率)中间体25-4。
ESI-MS (阴性,m/z):C
中间体25-5
将16.0g (34.1 mmol)中间体25-4、10.7g (41 mmol) 1- (叔丁基) -4-碘苯、650mg (3.41 mmol)碘化铜(I)、1.12g (10.2 mmol)环己-1,2-二胺和21.7g (102 mmol)磷酸三钾悬浮在350 mL1,4-二氧杂环己烷中,在91℃加热四小时。加入1.00g (3.8 mmol) 1-(叔丁基) -4-碘苯,并在91℃下持续加热四小时。将悬浮液通过3 cm硅胶层过滤,并用200mL二氧杂环己烷冲洗硅胶层。将收集的洗脱液真空浓缩,将产物溶解在50 ml二氯甲烷中。加入200 mL乙醇,将溶液浓缩至200 mL,直到形成悬浮液。过滤悬浮液,并用乙醇洗涤固体,得到13.8g (67%产率)中间体25-5。
ESI-MS (阳性,m/z):C
中间体25-6
将13.5g (22.5 mmol)中间体25-5在230℃加热90分钟。冷却熔融的固体,通过MPLC,使用
ESI-MS (阳性,m/z):C
中间体25-7
将2.4g (7.0 mmol)中间体7-1、2.70g (5.39 mmol)中间体25-6、103 mg (0.54mmol)碘化铜(I)、185 mg (1.62 mmol)环己-1,2-二胺和3.43g (16.2 mmol)磷酸三钾悬浮于100 mL1,4-二氧杂环己烷中,在91℃加热八小时。将悬浮液冷却至室温,并通过3 cm硅胶层过滤,随后用30 mL二氧杂环己烷冲洗硅胶层。将收集的洗脱液在真空下浓缩,并将产物通过MPLC使用
ESI-MS (阳性,m/z):C
中间体25-8
将30.0g (0.13 mol) 2-溴- (叔丁基)苯胺、34.2g (0.13 mol) 1- (叔丁基) -4-碘苯、295 mg (1.32 mmol)乙酸钯(II)、729 mg (1.32 mmol) 1,1' -双(二苯基膦基)二茂铁(dppf)和19.0g (0.20 mol)叔丁醇钠悬浮于300 mL甲苯中。将悬浮液在108℃下加热18小时。加入148 mg (0.66 mmol)乙酸钯(II)和365 mg (0.66 mmol) dppf并在108℃下继续加热八小时。将反应混合物冷却至室温,加入1g氰化钠和100 mL水。将混合物搅拌一小时,然后用水(3×100 mL)洗涤。将有机相用硫酸钠干燥,并在真空下浓缩。将产物溶解在300 mL热甲醇中,并将溶液在室温下搅拌18小时。过滤所得悬浮液,并用冷甲醇洗涤固体,得到作为灰色固体的24.3g (51%产率)中间体25-8。
ESI-MS (阳性,m/z):C
中间体25-9
将5.80g (16.1 mmol)中间体25-8、6.13g (24.1 mmol)双(频哪醇合)二硼、394mg (0.48 mmol) 1,1' -双(二苯基膦基)二茂铁-二氯化钯(II)二氯甲烷配合物和6.32g(64.4 mmol)乙酸钾悬浮于150 mL1,4-二氧杂环己烷中。将反应混合物在89℃加热八小时。将所得悬浮液冷却至室温,用100 mL水和100 mL乙酸乙酯稀释。将混合物用水(3×50mL)洗涤,将有机相经硫酸钠干燥并真空浓缩。将所得固体溶于50mL二氯甲烷和100 mL乙醇中,并在真空下浓缩至100 mL体积。过滤所得悬浮液,并用50mL乙醇洗涤固体,得到3.8g (58%产率)中间体25-9。
ESI-MS (阳性,m/z):C
中间体25-10
将1.51g (3.71 mmol)中间体25-9、2.40g (3.37 mmol)中间体25-7、151 mg(0.67 mmol)乙酸钯(II)、166 mg (0.41 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和2.86g (13.5 mmol)磷酸三钾溶于60 mL甲苯、30 mL1,4-二氧杂环己烷和20 mL水的混合物中。将反应混合物在83℃下加热一小时,然后冷却至室温。加入200 mL甲苯和100 mL水。将有机相用水(3×100 mL)洗涤,用硫酸钠干燥并真空浓缩。将产物溶解在20 mL二氯甲烷和70 mL乙醇中。将溶液在真空下浓缩至60 mL体积,直到形成悬浮液。过滤悬浮液,用50mL乙醇洗涤固体。将产物进一步通过MPLC,使用
ESI-MS (阳性,m/z):C
化合物25
将1.50g (1.64 mmol)中间体25-10溶解在45 mL1,2-二氯苯中。滴加1.15 mL(6.6 mmol) N,N-二异丙基乙胺和3.3 mL三溴硼烷(1.0M,在庚烷中)。将黄色溶液在174℃下加热2.5小时。将溶液冷却至室温。加入3.3 ml三溴硼烷(1.0M,在庚烷中),并在174℃下持续加热25小时。将反应混合物冷却至室温,用200 mL乙醇稀释,并搅拌一小时。过滤悬浮液,将固体进一步通过MPLC,使用
ESI-MS (阳性,m/z):C
化合物26
中间体26-1
将10.0g (46.9 mmol) 3-溴苯并[ b ]噻吩、7.00g (46.9 mmol) 4- (叔丁基)苯胺、540 mg (0.94 mmol)三(二亚苄基丙酮)二钯(0)、876 mg (3.07 mmol) 2-二环己基膦基-2 ',6 ' -二异丙氧基联苯(RuPhos)和9.02g (94.0 mmol)叔丁醇钠悬浮于120 mL甲苯中。将悬浮液抽真空三次,并用氩气回填,并在105℃下加热18小时。将该深色悬浮液溶解,冷却至室温,并用100 mL甲苯和100 mL水稀释。将水相用水(3×50mL)洗涤,用硫酸钠干燥,并真空浓缩。将产物进一步通过MPLC,使用
ESI-MS (阳性,m/z):C
中间体26-2
将3.61g (10.7 mmol)中间体7-1、3.00g (10.7 mmol)中间体26-1、24 mg (0.11mmol)乙酸钯(II)、63 mg (0.11 mmol) 4,5-双(二苯基膦基) -9,9-二甲基呫吨(Xantphos)和1.54g (16.0 mmol)叔丁醇钠悬浮于60 mL甲苯中。将悬浮液在77℃下加热四小时。将反应混合物冷却至室温,用100 mL水和100 mL甲苯稀释。分离有机相,用水(3×100mL)洗涤,用硫酸钠干燥。将混合物通过3 cm硅胶层过滤,并用50mL甲苯冲洗硅胶层。将收集的洗脱液在真空下浓缩,将产物通过MPLC使用
ESI-MS (阳性,m/z):C
中间体26-3
将3.00g (6.09 mmol)中间体26-2、2.30 (6.70 mmol)中间体5-1、27 mg (0.12mmol)乙酸钯(II)、300 mg (0.73 mmol) 2-二环己基膦基-2 ',6 ' -二甲氧基联苯(SPhos)和5.17g (24.4 mmol)磷酸三钾溶于60 mL甲苯、30 mL1,4-二氧杂环己烷和20 mL水的混合物中。将溶液抽真空三次并用氩气回填,并在84℃下加热七小时。将反应混合物冷却至室温,用200 mL甲苯和100 mL水稀释。将有机相用水(3×100 mL)洗涤,用硫酸钠干燥并真空浓缩。将产物通过MPLC使用
ESI-MS (阳性,m/z):C
化合物26
将1.50g (2.39 mmol)中间体26-3悬浮于25 mL1,2-二氯苯中。滴加1.7 mL (9.5mmol) N,N-二异丙基乙胺和4.8 mL三溴硼烷(1.0M,在庚烷中)。将黄色悬浮液在176℃下加热三小时。将反应混合物冷却至室温,用100 mL乙醇稀释。将悬浮液搅拌15分钟,然后过滤。真空浓缩滤液,在100 mL庚烷中搅拌残余物。过滤悬浮液,将固体用50mL庚烷洗涤,得到作为黄色固体的142 mg (9%产率)化合物26。
ESI-MS (阳性,m/z):C
化合物27
中间体 27-1
将10.32g (50.0mmol) 2-溴-5-氯苯胺、9.13 mL (51.5 mmol) 1- (叔丁基) -4-碘苯、6.73g (70.0 mmol)叔丁醇钠悬浮于250 mL甲苯中。用Ar将悬浮液脱气,并将289 mg(1mol%) 4,5-双(二苯基膦基) -9,9-二甲基呫吨和429 mg (0.5mol%)三(二亚苄基丙酮)二钯(0)加入到反应混合物中。将反应混合物加热至105℃持续50分钟。将反应物冷却至室温,用甲苯/水稀释,并经硅藻土过滤。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷作为洗脱剂纯化,得到作为透明油的15.1g (89%产率)的中间体27-1。
中间体27-2
将15.0g (44.3mmol)中间体27-1、13.35 mL (89.0 mmol) 1,8-二氮杂双环[5.4.0]十一碳-7-烯悬浮于221mL二甲基甲酰胺中。用Ar将悬浮液脱气,向反应混合物中加入451 mg (1.5mol%)的双(三苯基膦)氯化钯(II)。将反应混合物加热至120℃持续30小时。将反应物冷却至室温,用甲苯/水稀释,并经硅藻土过滤。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作为洗脱剂纯化,得到作为白色固体的6.43g (56%产率)中间体27-2。通过LC-MS [M-H]
中间体 27-3
将4.00g (15.52 mmol)中间体27-2、4.59g (23.28 mmol) 3-溴苯并呋喃、6.43g(46.6 mmol)碳酸钾和986 mg (15.52 mmol)铜悬浮于52 mL硝基苯中。用Ar将悬浮液脱气,然后加热至195℃持续3天。将反应物冷却至室温,用甲苯稀释,并经硅藻土过滤。用10%的3-氨基-1-丙醇溶液洗涤有机层,直到蓝色消失。将水层进一步用甲苯萃取。将合并的有机萃取物用盐水洗涤,用硫酸钠干燥,过滤,并蒸发。将残余物通过硅胶柱色谱法,使用庚烷作为洗脱剂纯化,得到作为白色泡沫的2.38g (41%收率)中间体27-3。通过LC-MS [M + H]
中间体27-4
将2.30g (6.15mmol)中间体27-3、2.74g (6.77mmol)中间体2-3、4.01g(12.3mmol)碳酸铯悬浮于甲苯/乙醇/水(28/9/5mL)的混合物中。用Ar将悬浮液脱气,并将55mg (4mol%)乙酸钯和235mg (8mol%) 2-二环己基膦基-2 ',4 ',6 ' -三异丙基联苯加入到反应混合物中。将反应混合物加热至85℃持续3小时。将反应物冷却至室温,用甲苯稀释,然后经硅藻土垫过滤。向滤液中加入水,然后分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法使用庚烷/甲苯纯化,然后使用庚烷/二氯甲烷作为洗脱剂进行第二次色谱法纯化,得到作为白色固体的2.1g (55%收率)中间体27-4。通过LC-MS [M-H]
化合物 27
将200mg (0.324mmol)中间体27-4溶于32mL叔丁基苯,用Ar脱气并冷却至0℃。滴加0.51 mL (0.973mmol)叔丁基锂(1.9M戊烷溶液),然后在相同温度下搅拌5分钟。然后,将反应物在85℃下搅拌2小时。然后,在0℃下滴加0.81 mL (0.81mmol)三溴硼烷(1M庚烷溶液),使反应物在45分钟内升至室温,随后加入1.44 mL (8.40 mmol) N-乙基-N-异丙基丙-2-胺。将反应混合物在155℃搅拌16小时。将反应物冷却至室温,并用甲苯/水稀释。分离各层,将水层进一步用甲苯萃取。将有机萃取物用水、盐水洗涤,用硫酸钠干燥,过滤并蒸发。将残余物通过硅胶柱色谱法,使用庚烷/甲苯作洗脱剂纯化,得到作为黄色固体的5mg (2%收率)化合物27。通过LC-MS [M + H]
Ⅱ化合物的评估
接着,测量实施例中使用的化合物的性质。测量和计算方法如下所示。
1.1光致发光应用数据
表中所列化合物的甲苯溶液是通过将相应化合物以10
已经测定了本发明化合物1-27在甲苯溶液中的光致发光(PL)数据,并总结于下表中。作为对比例,根据US 2019/0067577A1 [0122]的对比化合物1在甲苯溶液中的PL数据公开于表中:
1)光致发光
2)半峰全宽。
这些结果证明,本发明化合物1至6、8至27产生比对比化合物1更窄的光谱(更小的FWHM),即更好的色纯度。本发明化合物7的PL在比对比化合物1更长的波长处。
1.2器件应用数据(本发明化合物作为发光体掺杂剂)
有机EL器件的制备和评估
如下制备和评估有机EL器件:
应用实施例1
首先,用N
表1
这些结果表明,本发明的化合物在用作OLED中的荧光发光材料时,给出良好的EQE和窄的光谱(较小的FWHM),即良好的色纯度。
1.3另外的应用实施例
重复应用实施例1,除了化合物3-6、8、11、12、15-17、20、21和23代替化合物2用作荧光发光层中的发光体。
表2
- 杂环化合物和包括该杂环化合物的有机电致发光装置
- 有机电致发光器件用材料及包含其的有机电致发光器件
- 一种有机电致发光材料及包含其的有机电致发光器件
- 一种含喹唑啉杂环化合物及其在有机光电器件中的应用
- 用于有机电致发光显示器件的盖板及其制作方法、有机电致发光显示器件以及显示设备
- 一种杂环化合物和包含该杂环化合物的有机电致发光器件
- 一种杂环化合物及包含该杂环化合物的有机电致发光器件